


Mucosal-associated invariant T (MAIT) cells detect microbial infection via recognition of riboflavin-based antigens presented by the major histocompatibility complex class I (MHC-I)–related protein 1 (MR1). Most MAIT cells in human peripheral blood express CD8αα or CD8αβ coreceptors, and the binding site for CD8 on MHC-I molecules is relatively conserved in MR1. Yet, there is no direct evidence of CD8 interacting with MR1 or the functional consequences thereof. Similarly, the role of CD8αα in lymphocyte function remains ill-defined. Here, using newly developed MR1 tetramers, mutated at the CD8 binding site, and by determining the crystal structure of MR1–CD8αα, we show that CD8 engaged MR1, analogous to how it engages MHC-I molecules. CD8αα and CD8αβ enhanced MR1 binding and cytokine production by MAIT cells. Moreover, the CD8–MR1 interaction was critical for the recognition of folate-derived antigens by other MR1-reactive T cells. Together, our findings suggest that both CD8αα and CD8αβ act as functional coreceptors for MAIT and other MR1-reactive T cells.

Introduction
Mucosal-associated invariant T (MAIT) cells are a subset of unconventional T cells that recognize small molecules presented by the monomorphic MHC class I (MHC-I)–like, MHC-I–related protein 1 (MR1) via their TCR (Corbett et al., 2014; Tilloy et al., 1999; Treiner et al., 2003; Kjer-Nielsen et al., 2012). The most potent MAIT cell antigen identified to date is the riboflavin biosynthesis precursor derivative 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU; Corbett et al., 2014; Kjer-Nielsen et al., 2018). In humans, the MAIT TCR is comprised of an “invariant” TCRα chain, involving the gene segment TRAV1-2 joined to either TRAJ33, TRAJ20, or TRAJ12, which is paired typically with a TCRβ chain consisting of TRBV6-1, TRBV6-4, or TRBV20 gene segments (Porcelli et al., 1993; Reantragoon et al., 2013; Tilloy et al., 1999; Lepore et al., 2014). MAIT cells have been identified within most tissues and constitute ∼3% of T cells in adult peripheral blood (Gherardin et al., 2018). Stimulation of MAIT cells by microbial antigens such as 5-OP-RU induces the rapid secretion of proinflammatory cytokines TNF and IFNγ, and cytotoxic granules (Dusseaux et al., 2011; Kurioka et al., 2015); under certain conditions, MAIT cells also produce IL-17A, IL-21, and IL-13 (Dusseaux et al., 2011; Kurioka et al., 2015; Bennett et al., 2017; Kelly et al., 2019). Accordingly, MAIT cells can contribute to anti-microbial immunity in an antigen-dependent manner, as demonstrated for the lung pathogens Mycobacterium bovis Bacillus Calmette–Guérin, Klebsiella pneumoniae, Francisella tularensis, and Legionella longbeachae (Chua et al., 2012; Georgel et al., 2011; Meierovics et al., 2013; Wang et al., 2018; Zhao et al., 2021) and urinary tract infection by Escherichia coli (Cui et al., 2015). MAIT cells can also exhibit a tissue repair signature (Hinks et al., 2019; Lamichhane et al., 2019; Leng et al., 2019) and can contribute to skin wound healing (Constantinides et al., 2019). MAIT cells in humans can be identified based on the expression of surrogate markers CD161 and TRAV1-2 or more accurately using MR1 tetramers bound with microbial antigens (Corbett et al., 2014; Reantragoon et al., 2013; Gherardin et al., 2018). Phenotypic characterization revealed that MAIT cells in peripheral blood can vary in coreceptor expression. MAIT cells can be CD4+, CD8αα+, CD8αβ+, double positive (DP), or double negative (DN) for CD4 and CD8 coreceptors (Corbett et al., 2014; Reantragoon et al., 2013; Martin et al., 2009). In humans, the majority of MAIT cells in adult blood express CD8 (Reantragoon et al., 2013; Gherardin et al., 2018; Dias et al., 2018; Corbett et al., 2014).
Previous work by us and others identified other MR1-reactive T cells, as recently reviewed (Souter and Eckle, 2020), which exhibit antigen reactivity patterns distinct from the 5-OP-RU specificity of MAIT cells. This includes reactivity to MR1 independent of antigen (MR1-centered/MR1-autoreactivity), bound to folate derivatives 6-formylpterin (6-FP) and acetyl-6-formylpterin (Ac-6-FP; Gherardin et al., 2016; Koay et al., 2019), drug-like metabolites (Keller et al., 2017; Salio et al., 2020), endogenous or cancer antigens (Lepore et al., 2017; Crowther et al., 2020), or undefined antigens derived from a microbe (Streptococcus pyogenes) deficient in the riboflavin biosynthesis pathway (Meermeier et al., 2016). Whilst some of these other MR1-reactive T cells are MAIT-like in their phenotype, most are phenotypically heterogenous compared to MAIT cells, including their expression of distinct, TRAV1-2− αβ TCRs (Gherardin et al., 2016; Gherardin et al., 2018; Koay et al., 2019; Crowther et al., 2020; Lepore et al., 2017; Meermeier et al., 2016; Harriff et al., 2018) or γδ TCRs (Le Nours et al., 2019; Rice et al., 2021). Many of these other MR1-reactive T cells express CD8, including those that are reactive to folate-derived antigens (Gherardin et al., 2016; Koay et al., 2019).
For CTLs, the function of the CD8 coreceptor and underlying mechanisms have been well-characterized. CD8 is expressed on the surface of CTLs as an αβ heterodimer, where it improves recognition of antigen (Gao et al., 1997; Leahy et al., 1992; Wyer et al., 1999; Sewell et al., 1999; Wooldridge et al., 2005; Laugel et al., 2007). CD8 binds MHC-I via two Ig-like ectodomains, one from each CD8 subunit (Wang et al., 2009; Gao et al., 1997; Kern et al., 1998; Shi et al., 2011; Agea et al., 2005; Leahy et al., 1992) and predominantly contacts the conserved α3-domain of MHC-I (Wang et al., 2009; Gao et al., 1997; Kern et al., 1998; Shi et al., 2011; Agea et al., 2005). The α3-domain of MHC-I is spatially distinct from the TCR binding site, enabling CD8 and TCR to engage MHC-I simultaneously (Wyer et al., 1999), thereby increasing the stability of the overall complex (Wooldridge et al., 2005; Laugel et al., 2007). Furthermore, the CD8α subunit binds the early signaling kinase, lymphocyte-specific protein tyrosine kinase (Lck). Although both CD8αα and CD8αβ bind to MHC-I with a comparable affinity, greater activation of CTLs is observed with CD8αβ (Kern et al., 1999; Bosselut et al., 2000). Some evidence suggest that CD8β is important for colocalization of CD8 with CD3 complexes within the membrane, thus CD8αβ, but not CD8αα, is capable of enhancing TCR signaling by bringing Lck into close proximity to CD3 (Veillette et al., 1988; Pang et al., 2007; Zareie et al., 2021). Accordingly, CD8αβ improves CTL antigen recognition by increasing the overall stability of the TCR–MHC-I complex and by enhancing TCR signaling.
Whilst CD8αα is essentially absent from conventional T cells, it is present on other lymphocyte subsets, including some MAIT cells, yet its role remains ill-defined (Leishman et al., 2001; Reantragoon et al., 2013; Moebius et al., 1991; Geng and Raghavan, 2019; Goodall et al., 2020; Goodall et al., 2019; Cheroutre and Lambolez, 2008; Sarrabayrouse et al., 2015; Zhu et al., 2013). CD8αα has also been shown to bind to various MHC-Ib molecules (Agea et al., 2005; Clements et al., 2005; Leishman et al., 2001; Olivares-Villagomez et al., 2008; Pardigon et al., 2004; Teitell et al., 1991; Gao and Jakobsen, 2000; Goodall et al., 2019; Goodall et al., 2020; Huang et al., 2011), but the functional consequences for the interaction of CD8αα with most of these MHC-Ib molecules are unknown or controversial. While many unconventional T cell subsets express coreceptors, their role in modulating antigen responsiveness is also poorly understood.
Previous observations suggest that CD8 may contribute to MAIT cell responsiveness, whereby CD8 blocking antibodies have been shown to reduce or abrogate activation of CD8+ MAIT cells (Gold et al., 2013; Kurioka et al., 2017; Dias et al., 2018). However, anti-CD8 antibodies are known to alter T cell responses independently of CD8 function (Wooldridge et al., 2003). Thus, an interaction between CD8 and MR1 has not been formally established. Considering these data and the abundance of CD8+ MAIT cells in adult blood, we sought to determine the role of CD8 on these cells. Here, we formally demonstrate an interaction between CD8 and MR1 and reveal the role of CD8 for the function of MAIT and other MR1-reactive T cells.
Results
CD8+ MAIT cells are highly abundant in adult blood
Using MR1-5-OP-RU tetramers, we determined the frequency of each MAIT cell coreceptor subset in peripheral blood mononuclear cells (PBMCs) from 12 healthy adult donors by flow cytometry. As previously published (Reantragoon et al., 2013; Gherardin et al., 2018; Dias et al., 2018; Corbett et al., 2014), the majority of MAIT cells expressed CD8, with a mean frequency of 83% (Fig. 1, A and B), followed by DN, CD4+, and DP subsets with mean values of 10, 4.0, and 3.5%, respectively (Fig. 1 B). In the thymus, CD8+ MAIT thymocytes predominantly express CD8αβ (Koay et al., 2016); however, CD8+ MAIT cells acquire a CD8αα+ phenotype after birth (Ben Youssef et al., 2018), and this phenotype persists into adulthood such that on average half of the CD8+ MAIT cells are CD8α+β− (Martin et al., 2009; Gherardin et al., 2018; Reantragoon et al., 2013; Walker et al., 2012). Similarly, in our adult donor cohort, CD8αα and CD8αβ expression among CD8+ MAIT cells was evenly split, with mean values of 49 and 51%, respectively (Fig. 1 C). A similar trend was observed for DP MAIT cells, with mean values of 57% for CD8αα expression and 43% for CD8αβ expression but was variable between individuals (Fig. 1 D). In contrast, non-MAIT CD8+ T cells were predominantly CD8αβ+ (Fig. 1 E). Notably, MAIT cells typically expressed lower levels of CD8α and CD8β on the cell surface compared to non-MAIT CD8αβ+ T cells, defined as MR1-5-OP-RU tetramer−, as previously shown (Gherardin et al., 2018; Martin et al., 2009; Walker et al., 2012; Fig. 1, F and G).

Adult peripheral blood MAIT cells predominately express CD8, and the canonical CD8 binding site is conserved between MHC-I and MR1. (A) Gating strategy for assessing coreceptor usage by MAIT and non-MAIT T cells from peripheral blood identified using MR1-5-OP-RU tetramer. (B) Coreceptor usage by MAIT cells among 11 healthy donors showing the frequency of each subset (CD4, DN, DP and CD8) as a percentage of total MAIT cells. (C–E) The frequency of CD8αα and CD8αβ usage as a percentage of CD8 SP MAIT cells, DP MAIT cells, or CD8+ non-MAIT T cells, respectively. (F and G) Geometric MFI (gMFI) of CD8α and CD8β antibody staining of CD8αα+ and CD8αβ+ MAIT cells compared to non-MAIT CD8αβ+ T cells. (B–G) Data from 11 healthy blood donors were assessed in two independent experiments. (H) Alignment of residues 211–235 (Q223 highlighted in red) of the α3-domains of human and mouse MHC-Ia/b molecules with human MR1, annotated with residues engaged in hydrogen bonds (highlighted in blue) between both the T cell proximal (CD8β or CD8α1) and distal (CD8α2) CD8 subunits, respectively. Indicated residue numbers apply to MR1, whereby HLA-A*02:01 residue numbers are those of MR1 plus 3. CD8 subunit positions are highlighted in red on cartoons of CD8–MHC-I. Interactions of CD8 with MHC-I molecules were identified with PDBsum (Laskowski et al., 2018) using published crystal structures with PDB IDs; 1AKJ (Gao et al., 1997), 3QZW (Shi et al., 2011), 1BQH (Kern et al., 1998), 3DMM (Wang et al., 2009), and 1NEZ (Liu et al., 2003). Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (B and F) or Wilcoxon signed-rank test (G).
Adult peripheral blood MAIT cells predominately express CD8, and the canonical CD8 binding site is conserved between MHC-I and MR1. (A) Gating strategy for assessing coreceptor usage by MAIT and non-MAIT T cells from peripheral blood identified using MR1-5-OP-RU tetramer. (B) Coreceptor usage by MAIT cells among 11 healthy donors showing the frequency of each subset (CD4, DN, DP and CD8) as a percentage of total MAIT cells. (C–E) The frequency of CD8αα and CD8αβ usage as a percentage of CD8 SP MAIT cells, DP MAIT cells, or CD8+ non-MAIT T cells, respectively. (F and G) Geometric MFI (gMFI) of CD8α and CD8β antibody staining of CD8αα+ and CD8αβ+ MAIT cells compared to non-MAIT CD8αβ+ T cells. (B–G) Data from 11 healthy blood donors were assessed in two independent experiments. (H) Alignment of residues 211–235 (Q223 highlighted in red) of the α3-domains of human and mouse MHC-Ia/b molecules with human MR1, annotated with residues engaged in hydrogen bonds (highlighted in blue) between both the T cell proximal (CD8β or CD8α1) and distal (CD8α2) CD8 subunits, respectively. Indicated residue numbers apply to MR1, whereby HLA-A*02:01 residue numbers are those of MR1 plus 3. CD8 subunit positions are highlighted in red on cartoons of CD8–MHC-I. Interactions of CD8 with MHC-I molecules were identified with PDBsum (Laskowski et al., 2018) using published crystal structures with PDB IDs; 1AKJ (Gao et al., 1997), 3QZW (Shi et al., 2011), 1BQH (Kern et al., 1998), 3DMM (Wang et al., 2009), and 1NEZ (Liu et al., 2003). Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (B and F) or Wilcoxon signed-rank test (G).
The putative CD8 binding site is conserved between MR1 and MHC-I
Because CD8+ MAIT cells constitute the majority of MAIT cells in most individuals, we hypothesized that CD8 may play a role in the recognition of MR1 and aid in the function of MAIT cells. Crystal structures of complexes between the Ig-like ectodomains of CD8αα or CD8αβ and human or mouse MHC-I molecules (HLA-A*02:01 [Gao et al., 1997], HLA-A*24:02 [Shi et al., 2011], H-2Kb [Kern et al., 1998], H-2Db [Wang et al., 2009]), or the mouse MHC-Ib molecule thymus leukemia antigen (TL; Liu et al., 2003) have previously been determined. CD8 primarily contacts the flexible CD loop within the α3-domain of the MHC-I heavy chain (Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009; Fig. 1 H). Both CD8 subunits bind to the CD loop, although the molecular contacts are unevenly distributed, such that one CD8 subunit dominates the interaction (Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009). Upon MHC-I ligation, CD8α1 (or CD8β in CD8αβ interactions), is positioned proximal to the T cell surface and, within the CD8αα–MHC-I crystal structures (or CD8αβ–MHC-I crystal structures), CD8α1 (or CD8β) makes most of the contacts with the MHC-I α3-domain, as well as all of the contacts with the MHC-I α2-domain and β2-microglobulin (β2m; Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009). Within the CD loop is a highly conserved glutamine residue (Gln226) that forms multiple side- and main-chain contacts with both CD8 subunits (Fig. 1 H), which are crucial for CD8 engagement (Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009; Liu et al., 2003). An alignment of human MR1 with various mouse and human MHC-I molecules and the MHC-Ib molecule TL highlights the conservation of the CD8 contact residues within and adjacent to the CD loop (MHC-I: Gln226 and Asp/Glu227, MR1: Gln223 and Glu224; Fig. 1 H). These residues are also mostly conserved in MR1 from different species (Fig. S1 A), including non-human primates (Juno et al., 2019) and cattle (Edmans et al., 2021), where MAIT cells are almost exclusively CD8+. This suggested a priori that CD8 may be capable of engaging MR1 in a similar manner to how it engages MHC-I, and that this interaction could also be important for MAIT cell function.

High sequence conservation of MR1 in the putative CD8 binding site and validation of reporter cell lines and recombinant proteins. (A) Protein sequence alignment of a segment of the MR1 α3-domain from common mammals, including human (Homo sapiens), monkey (Macaca fascicularis), pig (Sus scrofa), cattle (Bos taurus), rat (Rattus norvegicus), and mouse (Mus musculus) using UniProt accession numbers Q95460, A0A2K5W2L6, A0A5G2R2T2, C1ITJ8, O19477, and Q8HWB0, respectively. The conserved residue Q223 is highlighted in red, and residues not conserved with human MR1 are highlighted in black. (B) Histograms comparing gMFI of parental (CD8 deficient), CD8αα transduced (+CD8αα), and CD8α- and CD8β-transduced (+CD8αβ) cells stained with anti-CD8α/β conjugated antibodies. (C) MR1-5-OP-RU tetramer staining of parental or CD8 transduced SKW-3.β2mnull cells described above. (D) As above, comparing MR1 and HLA tetramer staining and displaying gMFI. Data are representative of two experiments. (E) WT and CD8-null MR1 monomers folded with 5-OP-RU or 6-FP (5 μg each) analyzed by SDS-PAGE (15% polyacrylamide) under reducing conditions using 1 mM dithiothreitol (DTT) alongside a molecular weight marker (BM) with a protein range of 10–220 kD. Proteins were stained using Coomassie Blue R-250 dye. (F) WT and CD8-null MR1 monomers folded with 5-OP-RU or 6-FP (5 μg each) mixed with streptavidin (SAv; 5 μg) and analyzed by SDS-PAGE (12% polyacrylamide) under non-reducing conditions with SAv alone, or MR1-6-FP and MR1-5-OP-RU monomers alone alongside a molecular weight marker. (G) WT and CD8-null MR1-5-OP-RU (black) or -6-FP (gray) tetramer staining of a MAIT TCR (A-F7) expressing Jurkat cell line. Data are representative of two experiments. (H) Soluble CD8αα (2 μg) analyzed by SDS-PAGE (12% polyacrylamide) under reducing (1 mM DTT, +DTT) and non-reducing (−DTT) conditions alongside a molecular weight marker (BM).
High sequence conservation of MR1 in the putative CD8 binding site and validation of reporter cell lines and recombinant proteins. (A) Protein sequence alignment of a segment of the MR1 α3-domain from common mammals, including human (Homo sapiens), monkey (Macaca fascicularis), pig (Sus scrofa), cattle (Bos taurus), rat (Rattus norvegicus), and mouse (Mus musculus) using UniProt accession numbers Q95460, A0A2K5W2L6, A0A5G2R2T2, C1ITJ8, O19477, and Q8HWB0, respectively. The conserved residue Q223 is highlighted in red, and residues not conserved with human MR1 are highlighted in black. (B) Histograms comparing gMFI of parental (CD8 deficient), CD8αα transduced (+CD8αα), and CD8α- and CD8β-transduced (+CD8αβ) cells stained with anti-CD8α/β conjugated antibodies. (C) MR1-5-OP-RU tetramer staining of parental or CD8 transduced SKW-3.β2mnull cells described above. (D) As above, comparing MR1 and HLA tetramer staining and displaying gMFI. Data are representative of two experiments. (E) WT and CD8-null MR1 monomers folded with 5-OP-RU or 6-FP (5 μg each) analyzed by SDS-PAGE (15% polyacrylamide) under reducing conditions using 1 mM dithiothreitol (DTT) alongside a molecular weight marker (BM) with a protein range of 10–220 kD. Proteins were stained using Coomassie Blue R-250 dye. (F) WT and CD8-null MR1 monomers folded with 5-OP-RU or 6-FP (5 μg each) mixed with streptavidin (SAv; 5 μg) and analyzed by SDS-PAGE (12% polyacrylamide) under non-reducing conditions with SAv alone, or MR1-6-FP and MR1-5-OP-RU monomers alone alongside a molecular weight marker. (G) WT and CD8-null MR1-5-OP-RU (black) or -6-FP (gray) tetramer staining of a MAIT TCR (A-F7) expressing Jurkat cell line. Data are representative of two experiments. (H) Soluble CD8αα (2 μg) analyzed by SDS-PAGE (12% polyacrylamide) under reducing (1 mM DTT, +DTT) and non-reducing (−DTT) conditions alongside a molecular weight marker (BM).
CD8 binds MR1 in a similar manner as MHC-I
To examine whether CD8 can bind MR1, we stained human CD8-overexpressing T cell lines with MR1 and MHC-I tetramers. For this purpose, we transduced β2m deficient SKW-3 cells (SKW-3.β2mnull) with high levels of either CD8αα (SKW-3.β2mnull.CD8αα) or CD8αβ (SKW-3.β2mnull.CD8αβ; Fig. S1 B) and stained parental and CD8 transduced cell lines with MR1-5-OP-RU tetramers (Fig. S1 C). Intriguingly, despite the lack of cognate TCR, MR1-5-OP-RU tetramers could stain the CD8αα and CD8αβ overexpressing cell lines (Fig. S1 C). Next, we assessed the ability of MHC-I tetramers, including HLA-A*02:01-NLV (Peggs et al., 2002), HLA-B*08:01-FLR (Argaet et al., 1994; Callan et al., 1995; Kjer-Nielsen et al., 2003), HLA-C*06:02-TRAT (Rist et al., 2009), and HLA-G*01:01-RII (Allan et al., 1999; Diehl et al., 1996; Lee et al., 1995) to bind to these CD8 overexpressing cell lines (Fig. 2 A and Fig. S1 D). All MHC-I tetramers bound to both CD8 overexpressing cell lines to varying degrees in the absence of TCR (Fig. 2 A). Generally, all tetramers stained the CD8αα cell line with a higher intensity relative to the CD8αβ cell line (Fig. 2 A), likely in part due to the disparate expression levels of CD8α between the cell lines (Fig. S1 B). Interestingly, the hierarchy of CD8 binding by tetramers differed when engaging CD8αα or CD8αβ (Fig. 2 A). For instance, MR1 and HLA-B*08:01 tetramers bound more strongly to the CD8αβ cell line than HLA-A*02:01 and HLA-G*01:01 tetramers, whereas the opposite was observed for the CD8αα cell line (Fig. 2 A). Thus, although largely conserved in the CD8 binding site, different MHC-I molecules appear to engage the two CD8 dimers differentially. To further elucidate differences in MR1 tetramer binding to CD8, we stained the CD8αα and CD8αβ overexpressing cell lines with MR1-5-OP-RU or HLA-A*02:01-NLV tetramers and assessed their dissociation over time (Fig. 2 B). MR1 tetramers dissociated from the CD8αα overexpressing cell line approximately twice as rapidly as HLA-A*02:01 tetramers, with rate constants (k) of 0.13 min−1 (95% confidence interval [CI], 0.095–0.20) and 0.061 min−1 (95% CI, 0.036–0.11), respectively (Fig. 2 B). In contrast, for the CD8αβ overexpressing cell line, tetramer dissociation was nearly identical for MR1 and HLA-A*02:01, with k values of 0.087 min−1 (95% CI, 0.074–0.10) and 0.088 min−1 (95% CI, 0.074–0.10), respectively (Fig. 2 B). Notably, the amount of tetramer bound to each cell line at equilibrium as a percentage of maximum tetramer fluorescence (time zero) also varied between the tetramers (Fig. 2 B) with the pattern matching that of the dissociation rates. Thus, from these two assays (Fig. 2, A and B), the dissociation rate was higher, and overall avidity of MR1 tetramers was lower than those of HLA-A*02:01 tetramers for CD8αα, while the dissociation rate of MR1 tetramers were similar and the avidity higher compared to those of HLA-A*02:01 tetramers for CD8αβ.

MR1 binds to CD8 in a manner concordant with MHC-I. (A) gMFI of CD8αα or CD8αβ expressing cells stained with titrating doses of MR1 (MR1-5-OP-RU) or MHC-I (HLA-A*02:01-NLV, HLA-B*08:01-FLR, HLA-C*06:02-TRAT and HLA-G*01:01-RII) tetramers or streptavidin (SAv) control as determined by flow cytometry. (B) Dissociation of MR1-5-OP-RU and HLA-A*02:01-NLV tetramers from CD8αα or CD8αβ expressing cells over 120 min, measured by flow cytometry. Data points are mean values fitted with a nonlinear regression line (least squares) and 95% CI bands. (C) Binding of α3-domain MR1-Ac-6-FP mutant tetramers to CD8αα (left) and CD8αβ (right) expressing cell lines, displayed as fold change compared to WT MR1-Ac-6-FP tetramer (gMFI). Green underlay defines a ±0.5-fold change from baseline. Schematic representation of MR1-5-OP-RU (PDB ID; 6PUC; Awad et al., 2020) with a color-coded α3-domain Connolly surface overlay of key residues. (D) Histograms depicting 5-OP-RU–, 6-FP–, or Ac-6-FP–folded WT or Q223A, E224K mutant (MT) MR1 tetramer binding to CD8αα and CD8αβ expressing cells. (E) Affinity plot (top right panel) and sensorgrams (all other panels) of the WT or CD8-null MR1-Ac-6-FP (left panels), HLA-A*02:01-NLV (middle panels), and CD1d (bottom right panel) interactions with immobilized CD8αα, determined by SPR, where the response is measured in resonance units (RU). Data are representative of two (A, C, and E) or three (B and D) independent experiments.
MR1 binds to CD8 in a manner concordant with MHC-I. (A) gMFI of CD8αα or CD8αβ expressing cells stained with titrating doses of MR1 (MR1-5-OP-RU) or MHC-I (HLA-A*02:01-NLV, HLA-B*08:01-FLR, HLA-C*06:02-TRAT and HLA-G*01:01-RII) tetramers or streptavidin (SAv) control as determined by flow cytometry. (B) Dissociation of MR1-5-OP-RU and HLA-A*02:01-NLV tetramers from CD8αα or CD8αβ expressing cells over 120 min, measured by flow cytometry. Data points are mean values fitted with a nonlinear regression line (least squares) and 95% CI bands. (C) Binding of α3-domain MR1-Ac-6-FP mutant tetramers to CD8αα (left) and CD8αβ (right) expressing cell lines, displayed as fold change compared to WT MR1-Ac-6-FP tetramer (gMFI). Green underlay defines a ±0.5-fold change from baseline. Schematic representation of MR1-5-OP-RU (PDB ID; 6PUC; Awad et al., 2020) with a color-coded α3-domain Connolly surface overlay of key residues. (D) Histograms depicting 5-OP-RU–, 6-FP–, or Ac-6-FP–folded WT or Q223A, E224K mutant (MT) MR1 tetramer binding to CD8αα and CD8αβ expressing cells. (E) Affinity plot (top right panel) and sensorgrams (all other panels) of the WT or CD8-null MR1-Ac-6-FP (left panels), HLA-A*02:01-NLV (middle panels), and CD1d (bottom right panel) interactions with immobilized CD8αα, determined by SPR, where the response is measured in resonance units (RU). Data are representative of two (A, C, and E) or three (B and D) independent experiments.
To interrogate which residues in the α3-domain of MR1 contributed to the CD8–MR1 interaction, we stained the CD8-expressing cell lines with a panel of 16-point-mutated MR1 tetramers loaded with the MAIT cell non-stimulatory MR1 ligand Ac-6-FP, described previously (Le Nours et al., 2019). Overall, the mutant MR1-Ac-6-FP tetramers bound similarly to both CD8 cell lines, suggesting MR1 is bound by both CD8 dimers in a largely conserved manner (Fig. 2 C). The MR1 mutants Ile210Ala, Tyr211Ala, Tyr211Arg, Thr213Ala, and Glu259Ala all abrogated or substantially reduced tetramer staining of the CD8αα and CD8αβ overexpressing cell lines (>50% reduction in binding, Fig. 2 C). These residues map to equivalent positions within various MHC-I alleles that form contacts with CD8, except for the residues Ile210 and Thr213 (Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009; Liu et al., 2003). The mutations Glu99Ala, Glu209Ala, Met215Ala, His260Ala, Gly262Ala, and His264Ala had no discernible or mild effects on CD8 binding. Although the mutation Glu102Ala substantially reduced binding to CD8αα, it had no discernible effect on CD8αβ binding (Fig. 2 C). Surprisingly, residue Val222, located adjacent to the critical CD8-binding residue Gln223, enhanced tetramer binding to the CD8αα cell line while mildly reducing tetramer binding to the CD8αβ cell line when mutated to alanine (Val222Ala). In contrast, mutation to arginine (Val222Arg) had no discernible impact on staining of either cell line (Fig. 2 C). Interestingly, the human MR1 Val222Ala substitution occurs in the native sequence of murine MR1 (Fig. S1 A). Similarly for residue Asp229, mutation to alanine had no discernible effect, whilst mutation to arginine abolished binding on both cell lines. Collectively, these data suggest that there are subtle differences in MR1 engagement by CD8αα and CD8αβ; however, the overall footprint on MR1 is largely comparable (Fig. 2C). Given the large overlap in the residues involved in CD8 binding of MR1 and MHC-I (Fig. 1, H and I), CD8 likely engages MR1 in a manner analogous to how it engages MHC-I.
Based on our mutational analysis of MR1 (Fig. 2 C) and previously described CD8-null MHC-I mutations (Choi et al., 2003; Laugel et al., 2007; Purbhoo et al., 2001), we hypothesized that generating an MR1 double mutant would totally abrogate the CD8–MR1 interaction on both the CD8αα and CD8αβ lines. We generated “CD8-null” (Q223A and E224K) MR1 tetramers folded with 5-OP-RU, 6-FP, and Ac-6-FP (Fig. S1, E and F), analogous to previously described “CD8-null” MHC-I tetramers (Laugel et al., 2007), and tested their ability to stain the CD8 cell lines against equivalently produced WT MR1 tetramers. All three WT tetramers stained CD8αα and CD8αβ lines; the lower level of staining with 5-OP-RU–loaded compared to 6-FP– and Ac-6-FP–loaded tetramers was likely due to differences in tetramer concentrations (Fig. 2 D). We observed no staining with either CD8 cell line using CD8-null MR1 tetramers (Fig. 2 D), indicative of abrogation of the CD8–MR1 interaction (Gao et al., 1997; Shi et al., 2011; Kern et al., 1998; Wang et al., 2009; Wooldridge et al., 2005; Hutchinson et al., 2003). Importantly, these MR1 mutations did not impact staining of a Jurkat MAIT TCR reporter cell line (Fig. S1 G), validating their use as CD8-null MR1 tetramers. We next determined the relative binding strength of the CD8–MR1 interaction using surface plasmon resonance (SPR; Fig. 2 E). We measured the binding affinity of WT or CD8-null mutant MR1 and HLA-A*02:01 monomers to soluble CD8αα (Fig. 2 E and Fig. S1 H). MR1 and HLA-A*02:01 bound to CD8αα with an estimated equilibrium dissociation constant (KD) of 177 and 228 μM, respectively, indicating that they have similar affinities for CD8αα (Fig. 2 E). Therefore, the affinity of the CD8–MR1 interaction is similar to what has been reported for CD8-MHC-I (Wyer et al., 1999; Hutchinson et al., 2003; Gao et al., 2000; Cole et al., 2007; Cole et al., 2008; Iglesias et al., 2011).
The crystal structure confirms CD8αα interactions with MR1 and MHC-I are largely conserved
We next determined the structure of the human CD8αα homodimer in complex with MR1-Ac-6-FP at 2.4 Å resolution (Fig. 3 and Table S1). The electron densities of the ligand Ac-6-FP and at the interfaces of the CD8αα/MR1-Ac-6-FP complex were unambiguous (Fig. S2). Overall, CD8αα engaged MR1-β2m in a manner conserved with that of the known CD8–MHC-Ia complexes (Gao et al., 1997; Kern et al., 1998; Liu et al., 2003; Shi et al., 2011; Wang et al., 2009), where the CD8αα dimer binds to the underside of the MR1 antigen-binding cleft (Fig. 3 A and Fig. S3). However, when interacting with MR1-β2m compared with HLA-A*02:01-β2m, CD8αα buried a larger surface area (total buried surface area [BSA]: 1330 Å2 versus 1070 Å2), which correlated with the slightly higher affinity measured by SPR (Fig. 2 E). The contribution of each CD8 subunit was comparable, with the CD8α1 subunit dominating the BSA in each case (68.3% versus 71.3%; Fig. S3, B and E).

Crystal structure of the CD8αα–MR1-Ac-6-FP ternary complex. (A) Ribbon diagram of the X-ray crystal structure of the CD8αα–MR1-Ac-6-FP complex. The MR1 and β2m molecules are colored white and pale cyan, respectively, and Ac-6-FP is shown as green sticks. The CD8α1 and CD8α2 subunits are colored pale green and wheat, respectively. Displayed are two orientations of the complexes, involving a 45° rotation along the y axis. (B) Surface representation of the CD8αα–MR1-Ac-6-FP complex in the same colors and orientation (right panel) as in A. The lower left panel displays the footprint of CD8αα on MR1-β2m, rotated clockwise by 90° along the y axis; the lower right panel displays the footprint of MR1-β2m on CD8αα, rotated counter-clockwise by 90° along the y axis. The interaction regions are highlighted with exchanged colors and the H-bond/salt bridge/vdw forming residues are indicated, with H-bond or salt bridge forming residues bolded and underlined. Residues that contact both CD8α1 and CD8α2 subunits are in red. Residues mutated in CD8-null MR1 are highlighted as black dotted lines. (C–F) Close-up presentation of the molecular contacts at the interface between CD8αα and MR1-Ac-6-FP. Selected hydrogen bonds (black dashed lines), salt bridges (red dashed lines), and vdw interactions (orange dashed lines) between the β-sheet base of the MR1 antigen presentation cleft, β2m and the CD8α1 subunit (C), and between the MR1-α3 domain and the CD81α subunit (D) or the CD8α2 subunit (E), as well as between the MR1 CD loop with residues of both subunits of CD8αα (F) are shown. The residues of MR1 and β2m are presented as white and pale cyan sticks respectively, whereas the interacting residues of CD8α1 and CD8α2 are displayed as pale green and wheat sticks, respectively.
Crystal structure of the CD8αα–MR1-Ac-6-FP ternary complex. (A) Ribbon diagram of the X-ray crystal structure of the CD8αα–MR1-Ac-6-FP complex. The MR1 and β2m molecules are colored white and pale cyan, respectively, and Ac-6-FP is shown as green sticks. The CD8α1 and CD8α2 subunits are colored pale green and wheat, respectively. Displayed are two orientations of the complexes, involving a 45° rotation along the y axis. (B) Surface representation of the CD8αα–MR1-Ac-6-FP complex in the same colors and orientation (right panel) as in A. The lower left panel displays the footprint of CD8αα on MR1-β2m, rotated clockwise by 90° along the y axis; the lower right panel displays the footprint of MR1-β2m on CD8αα, rotated counter-clockwise by 90° along the y axis. The interaction regions are highlighted with exchanged colors and the H-bond/salt bridge/vdw forming residues are indicated, with H-bond or salt bridge forming residues bolded and underlined. Residues that contact both CD8α1 and CD8α2 subunits are in red. Residues mutated in CD8-null MR1 are highlighted as black dotted lines. (C–F) Close-up presentation of the molecular contacts at the interface between CD8αα and MR1-Ac-6-FP. Selected hydrogen bonds (black dashed lines), salt bridges (red dashed lines), and vdw interactions (orange dashed lines) between the β-sheet base of the MR1 antigen presentation cleft, β2m and the CD8α1 subunit (C), and between the MR1-α3 domain and the CD81α subunit (D) or the CD8α2 subunit (E), as well as between the MR1 CD loop with residues of both subunits of CD8αα (F) are shown. The residues of MR1 and β2m are presented as white and pale cyan sticks respectively, whereas the interacting residues of CD8α1 and CD8α2 are displayed as pale green and wheat sticks, respectively.

Electron density maps of the ligand Ac-6-FP and important interfaces in the crystal structure of the CD8αα–MR1-Ac-6-FP ternary complex. (A) Ribbon diagram of the X-ray crystal structure of the CD8αα–MR1-Ac-6-FP complex. (B–F) Electron density maps (2Fo-Fc; blue mesh contoured at 1σ) of selected regions of the MR1-Ac-6-FP interface with CD8αα, each highlighted with a differently colored box in panel A: the MR1-β2m interface with the CD8α1 subunit (B), the MR1 interacting regions of the CD8α1 subunit (C), the MR1 CD loop (D), the MR1 interacting regions of the CD8α2 subunit (E), and Ac-6-FP (F).
Electron density maps of the ligand Ac-6-FP and important interfaces in the crystal structure of the CD8αα–MR1-Ac-6-FP ternary complex. (A) Ribbon diagram of the X-ray crystal structure of the CD8αα–MR1-Ac-6-FP complex. (B–F) Electron density maps (2Fo-Fc; blue mesh contoured at 1σ) of selected regions of the MR1-Ac-6-FP interface with CD8αα, each highlighted with a differently colored box in panel A: the MR1-β2m interface with the CD8α1 subunit (B), the MR1 interacting regions of the CD8α1 subunit (C), the MR1 CD loop (D), the MR1 interacting regions of the CD8α2 subunit (E), and Ac-6-FP (F).
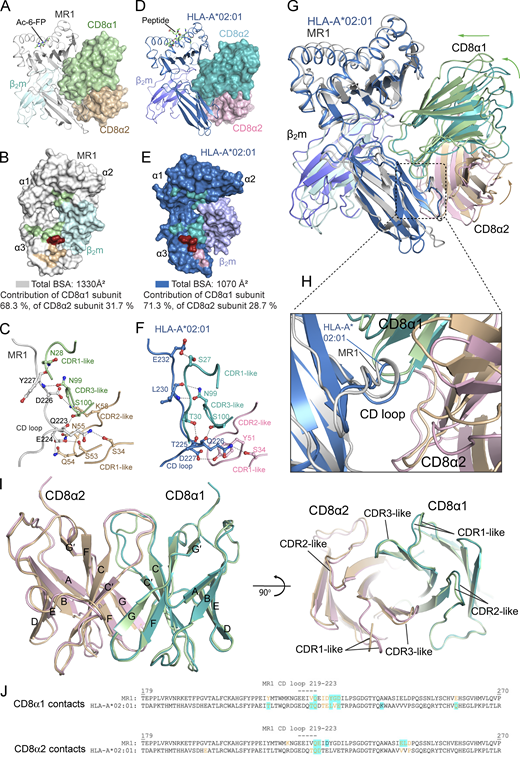
Structural comparison of the ternary complexes of CD8αα–MR1-Ac-6-FP and CD8αα–HLA-A*02:01. (A and D) Docking of CD8αα (surface presentation) on the side of MR1-Ac-6-FP (A) and HLA-A*02:01-peptide (PDB: 1AKJ) (D; ribbon presentation). (B and E) Surface presentation showing the footprint of CD8αα on MR1-Ac-6-FP (B) and HLA-A*02:01-peptide (E). (C and F) Selected H-bond and salt-bridge interactions (black dashed lines) between CD8αα and the CD loops of MR1 (C) and HLA-A*02:01 (analysis of the crystal structure with PDB ID 1AKJ [Gao et al., 1997] as per the criteria in Table S2; F), respectively. The two complexes were aligned via the α1/α2 domains of the MHC-I–like/MHC-I heavy chains in PyMOL. The CD8αα–MR1-Ac-6-FP complex is colored as in Fig. 4. The CD8αα–HLA-A*02:01-peptide complex is colored as follows: HLA-A*02:01, sky blue; β2m, slate blue; CD8α1, teal; CD8α2, light pink. (G) Superposition of the CD8αα–HLA-A*02:01-peptide and CD8αα–MR1-Ac-6-FP structures. Arrows illustrate the CD8αα rotation around the center of mass of the MR1/HLA-A*02:01 molecules. (H) Zoomed view of the interaction between CD8αα and the CD loops in the α3 domains of MR1 and HLA-A*02:01. (I) Superposition of the CD8αα molecules (ribbon presentation) in both MR1-Ac-6-FP and HLA-A*02:01-peptide complex structures. The right panel shows the bottom view of various CD8αα–CDR-like loops. The CD8αα molecules in panel G were aligned using PyMOL. (J) Alignment of residues 179–270 of the α3-domains of human MR1 and HLA-A*02:01, annotated with residues engaged in hydrogen bonds (highlighted in blue) between both the T cell proximal (CD8α1) and distal (CD8α2) CD8 subunits. Indicated residue numbers apply to MR1, whereby HLA-A*02:01 residue numbers are those of MR1 plus 3. Interactions of CD8 with the HLA-A*02:01 molecule in the published crystal structure with PDB ID 1AKJ (Gao et al., 1997) were identified as per the criteria in Table S2.
Structural comparison of the ternary complexes of CD8αα–MR1-Ac-6-FP and CD8αα–HLA-A*02:01. (A and D) Docking of CD8αα (surface presentation) on the side of MR1-Ac-6-FP (A) and HLA-A*02:01-peptide (PDB: 1AKJ) (D; ribbon presentation). (B and E) Surface presentation showing the footprint of CD8αα on MR1-Ac-6-FP (B) and HLA-A*02:01-peptide (E). (C and F) Selected H-bond and salt-bridge interactions (black dashed lines) between CD8αα and the CD loops of MR1 (C) and HLA-A*02:01 (analysis of the crystal structure with PDB ID 1AKJ [Gao et al., 1997] as per the criteria in Table S2; F), respectively. The two complexes were aligned via the α1/α2 domains of the MHC-I–like/MHC-I heavy chains in PyMOL. The CD8αα–MR1-Ac-6-FP complex is colored as in Fig. 4. The CD8αα–HLA-A*02:01-peptide complex is colored as follows: HLA-A*02:01, sky blue; β2m, slate blue; CD8α1, teal; CD8α2, light pink. (G) Superposition of the CD8αα–HLA-A*02:01-peptide and CD8αα–MR1-Ac-6-FP structures. Arrows illustrate the CD8αα rotation around the center of mass of the MR1/HLA-A*02:01 molecules. (H) Zoomed view of the interaction between CD8αα and the CD loops in the α3 domains of MR1 and HLA-A*02:01. (I) Superposition of the CD8αα molecules (ribbon presentation) in both MR1-Ac-6-FP and HLA-A*02:01-peptide complex structures. The right panel shows the bottom view of various CD8αα–CDR-like loops. The CD8αα molecules in panel G were aligned using PyMOL. (J) Alignment of residues 179–270 of the α3-domains of human MR1 and HLA-A*02:01, annotated with residues engaged in hydrogen bonds (highlighted in blue) between both the T cell proximal (CD8α1) and distal (CD8α2) CD8 subunits. Indicated residue numbers apply to MR1, whereby HLA-A*02:01 residue numbers are those of MR1 plus 3. Interactions of CD8 with the HLA-A*02:01 molecule in the published crystal structure with PDB ID 1AKJ (Gao et al., 1997) were identified as per the criteria in Table S2.
The majority of CD8–MR1 interactions involved the MR1 α3-domain, in particular the CD loop, and, to a much lesser extent, the α2-domain and β2m (Fig. 3, C–F; and Table S2). Namely, the N-terminal Arg4 of the CD8α1 subunit was buried between β2m and the α2-domain of MR1, forming H-bond interactions with the β2m-Lys58 and MR1-Asp118 residues (Fig. 3 C). When contacted by CD8αα, the MR1 CD loop adopted a similar conformation as that of HLA-A*02:01 (Fig. S3, C, F, and H), and so did both subunits of the CD8αα molecules (Fig. S3 I). The MR1 CD loop projected deeply into the interface between the two subunits of CD8αα, and a network of H-bonds and van der Waals interactions formed between the MR1-Gln223, and -Glu224, the CD8α1-Leu97, and -Ser100 residues (Table S2 and Fig. 3, D–F), as well as the CD8α2-Ser34, -Tyr51, -Ser53, -Gln54, and -Asn55 residues (Fig. 3, E and F; and Table S2). In addition, a broad pattern of interactions formed between the MR1-α3 domain with both the CDR1-like loop of the CD8α1 subunit and the CDR2-like loop of the CD8α2 subunit (Fig. 3, B–F; and Table S2). Even though, based on cellular assays, mutations of the MR1 residues Ile210 and Thr213 impacted CD8 binding, they do not participate in direct contacts based on the crystal structure. Ile210 and Thr213 are in the β-sheet before the CD loop so that their mutagenesis could indirectly affect CD8αα binding by impacting the conformation of the CD loop and/or the neighboring β-sheet (225–229). In addition, mutation of Ile210 could impact the adjacent Tyr211 residue, which interacts with Gln54 of the CD8α1 subunit (Table S2). In summary, we formally demonstrate that CD8αα binds MR1 in an analogous manner as it binds to MHC-I.
CD8 binding enhances the avidity and slows the decay kinetics of the TCR–MR1 tetramer complex
To determine whether CD8 on primary MAIT cells could influence MR1 tetramer recognition, we stained PBMCs from 11 healthy adult blood donors using WT and CD8-null MR1-5-OP-RU tetramers and assessed tetramer fluorescence by flow cytometry. In most donors, a discernible population of MR1-5-OP-RU tetramer+ cells was identified for each of the MAIT cell coreceptor subsets (CD4, DN, DP, CD8αα, and CD8αβ; Fig. 4 A). Notably, the CD8+ MAIT cell subsets exhibited the highest level of tetramer staining across all donors stained with WT MR1-5-OP-RU, with an average mean fluorescence intensity (MFI) of 20,413, 18,922, and 21,109 for DP, CD8αα, and CD8αβ expressing MAIT cells, respectively, compared to 12,220 and 13,784 for CD4 and DN subsets, respectively (Fig. 4 B). Additionally, within individual donors, CD8+ MAIT cells stained with WT MR1-5-OP-RU tetramer were significantly brighter compared to the other subsets (Fig. 4 C). Notably, differences in tetramer staining of each MAIT cell coreceptor subset were not due to differences in TCR expression levels, which were consistent based on CD3 expression levels (Fig. S4 A). We also found that the amount of surface-expressed CD8 correlated with tetramer fluorescence, consistently among donors, by examining CD8+ MAIT cell populations based on low, intermediate, or high CD8 expression (Fig. 4 D). Again, tetramer fluorescence did not correlate with CD3 expression levels (Fig. S4 B). These data support the notion that CD8 contributes to MR1 recognition by MAIT cells. To verify whether the increase in MR1-5-OP-RU tetramer staining of CD8+ MAIT cells was due to CD8 cooperatively engaging MR1 with the TCR, in another 12 PBMC donors we compared the staining level of WT and CD8-null MR1-5-OP-RU tetramers from each donor across a wide range of tetramer concentrations (Fig. 4 E and F; and Fig. S4 C). We observed a consistent and significant reduction in tetramer fluorescence intensity on all three subsets of CD8+ MAIT cells (DP, CD8αα, and CD8αβ) at all but the highest tetramer dilution when comparing the staining with the CD8-null MR1-5-OP-RU tetramers to WT MR1-5-OP-RU tetramers (Fig. 4 F). In contrast, when staining the CD4 and DN subsets of MAIT cells, only for the highest concentration of tetramer, there was a tendency or significant difference, respectively, between the two tetramers (Fig. 4 F). No significant difference was observed between MAIT cell subsets when comparing CD8-null MR1-5-OP-RU tetramer staining (Fig. S4 C), indicating CD8 is a major contributor to the observed increase in binding with WT MR1-5-OP-RU tetramers by CD8+ MAIT cells (Fig. 4 B). To examine this interaction further, we measured the dissociation of WT and CD8-null MR1 tetramers from CD8 single-positive (SP) or DN MAIT cells over time (Fig. 4 G). MHC tetramer dissociation from T cells occurs in a biphasic manner (Wang and Altman, 2003), therefore we used a two-phase (fast and slow) decay model for our analysis (Fig. 4 G). Although, as expected, there were some donor-specific differences in the tetramer dissociation kinetics, there was a 2.5-fold increase in the rate of tetramer dissociation among CD8 SP MAIT cells in the absence of CD8 engagement, when comparing the fast rate constant between WT and CD8-null MR1-5-OP-RU tetramers of 0.064 min−1 and 0.16 min−1, respectively (Fig. 4 G). Comparatively, minimal differences in tetramer dissociation were evident among DN MAIT cells (Fig. 4 G). Accordingly, based on assays with tetrameric MR1, CD8 contributes to the overall avidity of MR1 binding by MAIT TCRs and slows the decay kinetics of the TCR–MR1 complex.

CD8–MR1 interactions enhance MR1 tetramer binding to MAIT cells and slow MR1 tetramer dissociation kinetics. (A) MAIT cells identified using WT MR1-5-OP-RU tetramers from PBMCs of human healthy donors and gated based on coreceptor usage. (B) Cumulative data for WT tetramer staining intensity of MAIT cell coreceptor subsets (10 donors for CD4+, 11 donors for all other subsets). (C) Comparison of WT tetramer staining intensity of CD8− and CD8+ MAIT cells in individual donors. (D) Gating strategy for defining low, intermediate, and high CD8α expression by CD8+ MAIT cells and cumulative data comparing WT tetramer staining intensity of CD8α+ MAIT cells with mean and SD value. (E) MAIT cells stained with titrating amounts of WT or CD8-null MR1-5-OP-RU or MR1-6-FP tetramers. (F) Cumulative data of WT and CD8-null tetramer staining intensity for MAIT cell coreceptor subsets. (G) Cumulative data (in triplicate) of WT and CD8-null tetramer dissociation over time from CD8 SP or DN MAIT cells from healthy blood donors. A nonlinear regression line (least squares) and 95% CI interval bands are fitted. (A–D) Data are from the same 11 healthy blood donors in Fig. 1, recorded from two independent experiments. kfast, fast rate constant; kslow, slow rate constant. (E–G) Data are from 12 additional healthy blood donors from three independent experiments. Statistical significance was determined using a Kruskal–Wallis test (B), Wilcoxon signed-rank test (C), Friedman test with Dunn’s multiple comparison (D), or a two-way ANOVA with Sidak's multiple comparisons test (F).
CD8–MR1 interactions enhance MR1 tetramer binding to MAIT cells and slow MR1 tetramer dissociation kinetics. (A) MAIT cells identified using WT MR1-5-OP-RU tetramers from PBMCs of human healthy donors and gated based on coreceptor usage. (B) Cumulative data for WT tetramer staining intensity of MAIT cell coreceptor subsets (10 donors for CD4+, 11 donors for all other subsets). (C) Comparison of WT tetramer staining intensity of CD8− and CD8+ MAIT cells in individual donors. (D) Gating strategy for defining low, intermediate, and high CD8α expression by CD8+ MAIT cells and cumulative data comparing WT tetramer staining intensity of CD8α+ MAIT cells with mean and SD value. (E) MAIT cells stained with titrating amounts of WT or CD8-null MR1-5-OP-RU or MR1-6-FP tetramers. (F) Cumulative data of WT and CD8-null tetramer staining intensity for MAIT cell coreceptor subsets. (G) Cumulative data (in triplicate) of WT and CD8-null tetramer dissociation over time from CD8 SP or DN MAIT cells from healthy blood donors. A nonlinear regression line (least squares) and 95% CI interval bands are fitted. (A–D) Data are from the same 11 healthy blood donors in Fig. 1, recorded from two independent experiments. kfast, fast rate constant; kslow, slow rate constant. (E–G) Data are from 12 additional healthy blood donors from three independent experiments. Statistical significance was determined using a Kruskal–Wallis test (B), Wilcoxon signed-rank test (C), Friedman test with Dunn’s multiple comparison (D), or a two-way ANOVA with Sidak's multiple comparisons test (F).

MAIT cell coreceptor subsets stain similarly with anti-CD3 and CD8-null MR1-5-OP-RU tetramer, validation of MR1 expression by reporter cell lines and readout for MAIT cell responses, and CD8 dependency of MAIT cell coreceptor subset responses. (A) CD3 expression (gMFI) of MAIT cells identified using MR1-5-OP-RU tetramer and segregated based on coreceptor expression as part of experiments shown in Figs. 1 and 4. (B) CD3 expression (gMFI) of CD8+ MAIT cells identified using MR1-5-OP-RU tetramer and segregated based on anti-CD8α antibody fluorescence (low, intermediate, high) as part of experiments shown in Fig. 4. (C) Cumulative data for CD8-null MR1-5-OP-RU tetramer staining intensity of MAIT cell coreceptor subsets (10–11 donors) shown in Fig. 4. Data are from two independent experiments. Statistical significance was determined using a Kruskal–Wallis test. (D) Histograms comparing the gMFI of MR1-deficient (MR1null), WT MR1 (MR1null+MR1), mutant CD8-null MR1 (MR1null+CD8-null MR1), and mutant MR1-K43A (MR1null+MR1-K43A) overexpressing C1R cells. (E) Percentage of IL-17A–producing MAIT cells in response to 10 nM 5-OP-RU in the presence of MR1 deficient (C1R.MR1null) cells or WT MR1 expressing (C1R.MR1null+MR1) cells. Mean and SD are displayed. (F and G) Percentage of TNF- or IFNγ-producing MAIT cells by individual donors in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with 1,000 pM 5-OP-RU. (H and I) Percentage of TNF- or IFNγ-producing MAIT cell coreceptor subsets in response to WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of 5-OP-RU. (J) Comparison of TNF- and IFNγ-producing DN, CD8αα+ and CD8αβ+ MAIT cells in response to CD8-null MR1 expressing C1R cells pulsed with titrating doses of 5-OP-RU. Data are normalized to the maximum response for each MAIT cell subset. (H–J) Mean, SD and nonlinear regression line (least squares) are displayed. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (F and G).
MAIT cell coreceptor subsets stain similarly with anti-CD3 and CD8-null MR1-5-OP-RU tetramer, validation of MR1 expression by reporter cell lines and readout for MAIT cell responses, and CD8 dependency of MAIT cell coreceptor subset responses. (A) CD3 expression (gMFI) of MAIT cells identified using MR1-5-OP-RU tetramer and segregated based on coreceptor expression as part of experiments shown in Figs. 1 and 4. (B) CD3 expression (gMFI) of CD8+ MAIT cells identified using MR1-5-OP-RU tetramer and segregated based on anti-CD8α antibody fluorescence (low, intermediate, high) as part of experiments shown in Fig. 4. (C) Cumulative data for CD8-null MR1-5-OP-RU tetramer staining intensity of MAIT cell coreceptor subsets (10–11 donors) shown in Fig. 4. Data are from two independent experiments. Statistical significance was determined using a Kruskal–Wallis test. (D) Histograms comparing the gMFI of MR1-deficient (MR1null), WT MR1 (MR1null+MR1), mutant CD8-null MR1 (MR1null+CD8-null MR1), and mutant MR1-K43A (MR1null+MR1-K43A) overexpressing C1R cells. (E) Percentage of IL-17A–producing MAIT cells in response to 10 nM 5-OP-RU in the presence of MR1 deficient (C1R.MR1null) cells or WT MR1 expressing (C1R.MR1null+MR1) cells. Mean and SD are displayed. (F and G) Percentage of TNF- or IFNγ-producing MAIT cells by individual donors in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with 1,000 pM 5-OP-RU. (H and I) Percentage of TNF- or IFNγ-producing MAIT cell coreceptor subsets in response to WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of 5-OP-RU. (J) Comparison of TNF- and IFNγ-producing DN, CD8αα+ and CD8αβ+ MAIT cells in response to CD8-null MR1 expressing C1R cells pulsed with titrating doses of 5-OP-RU. Data are normalized to the maximum response for each MAIT cell subset. (H–J) Mean, SD and nonlinear regression line (least squares) are displayed. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (F and G).
CD8–MR1 interactions enhance antigen-dependent MAIT cell responses
Having established that CD8 expressed by MAIT cells can bind MR1, we next addressed whether CD8–MR1 engagement could contribute to the functional potential of MAIT cells. We assessed the production of TNF, IFNγ, and IL-17A cytokines upon stimulation, as a measure of MAIT cell activation in vitro (Kjer-Nielsen et al., 2012; Dusseaux et al., 2011). To examine the impact of CD8 ligation on MAIT cell cytokine production, we generated C1R cells expressing similar levels of either WT MR1 or CD8-null (Q223K, E224A) MR1, or C1R cells deficient in MR1 (Fig. S4 D). We pulsed these cells with titrating amounts of 5-OP-RU and co-cultured them with TRAV1-2+ cells, enriched from PBMCs of 12 healthy donors, and then assessed the cytokine production by TRAV1-2+CD161++ MAIT cells (Fig. 5, A–C; and Fig. S4 E). As expected, MAIT cells incubated with MR1-deficient C1R cells pulsed with 10 nM 5-OP-RU did not produce any detectable cytokines (Fig. 5, B and C; and Fig. S4 E). Interestingly, a small proportion of MAIT cells cultured in the absence of C1R cells, but in the presence of 5-OP-RU antigen, produced TNF (Fig. 5 B), suggesting that MAIT cells are capable of weakly auto-presenting antigen. Notably, MR1-expressing C1R cells (C1R.MR1null+MR1) pulsed with 5-OP-RU elicited potent cytokine production by MAIT cells from all donors, with on average ∼70% of cells producing TNF and ∼35% of cells producing IFNγ (Fig. 5, B and C). As we detected very few IL-17A+ MAIT cells overall (Fig. S4 E), we focused on TNF and IFNγ cytokine production for further analysis. Following stimulation with titrating amounts of 5-OP-RU, the proportions of TNF- and IFNγ-producing MAIT cells were substantially greater within the CD8 SP expressing subsets, particularly at the 100 pM dose; there were no significant differences in the capacity to produce cytokine between CD8αα+ and CD8αβ+ MAIT cells (Fig. 5, D and E). In contrast, the fractions of TNF- and IFNγ-producing CD4+ MAIT cells were the smallest of all subsets (Fig. 5, D and E), as previously reported in response to E. coli stimulus (Kurioka et al., 2017), although not to PMA/ionomycin (Gherardin et al., 2018). Given the inter-donor variability in cytokine production by MAIT cell coreceptor subsets, we next examined the cytokine response at the 100 and 1,000 pM antigen dose within individuals based on MAIT cell coreceptor usage (Fig. 5, F and G; and Fig. S4, F and G). A greater frequency of CD8+ MAIT cells tended to produce both TNF and IFNγ cytokines than DN MAIT cells; however, statistical significance was reached only for CD8αβ+ MAIT cells. Thus, MAIT cells expressing CD8 may have a functional advantage in responding to cognate antigen compared to other MAIT cell subsets.

CD8–MR1 interactions enhance antigen-dependent MAIT cell responses. (A) MAIT cells identified using surrogate markers CD161 and TRAV1-2 (left plots), and analysis of coreceptor usage and cytokine production (TNF and IFNγ) of unstimulated and 5-OP-RU stimulated MAIT cells (middle and right plots). (B and C) Percentage of TNF- or IFNγ-producing MAIT cells in response to 10 nM 5-OP-RU in the absence of C1R cells, or in the presence of MR1 deficient (C1R.MR1null) or WT MR1 expressing (C1R.MR1null+MR1) C1R cells. Mean and SD are displayed. (D and E) Percentage of TNF- or IFNγ-producing MAIT cell coreceptor subsets in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with titrating doses of 5-OP-RU. Mean, SD, and nonlinear regression line (least squares) are displayed. (F and G) Percentage of TNF- or IFNγ-producing MAIT cells by individual donors in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with 100 pM 5-OP-RU (∼EC50 dose). (H and I) Percentage of TNF- or IFNγ-producing MAIT cells, comparing the response in individual donors to WT or CD8-null MR1 expressing C1R cells (C1R.MR1null+MR1 or C1R.MR1null+MR1 CD8-null) pulsed with titrating doses of 5-OP-RU. (J and K) As above, comparing the percentage of TNF- or IFNγ-producing DN or CD8 SP MAIT cells. (B–J) Data are from 12 healthy blood donors from three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (F and G) or a two-way ANOVA with Sidak’s multiple comparisons test (H–K).
CD8–MR1 interactions enhance antigen-dependent MAIT cell responses. (A) MAIT cells identified using surrogate markers CD161 and TRAV1-2 (left plots), and analysis of coreceptor usage and cytokine production (TNF and IFNγ) of unstimulated and 5-OP-RU stimulated MAIT cells (middle and right plots). (B and C) Percentage of TNF- or IFNγ-producing MAIT cells in response to 10 nM 5-OP-RU in the absence of C1R cells, or in the presence of MR1 deficient (C1R.MR1null) or WT MR1 expressing (C1R.MR1null+MR1) C1R cells. Mean and SD are displayed. (D and E) Percentage of TNF- or IFNγ-producing MAIT cell coreceptor subsets in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with titrating doses of 5-OP-RU. Mean, SD, and nonlinear regression line (least squares) are displayed. (F and G) Percentage of TNF- or IFNγ-producing MAIT cells by individual donors in response to WT MR1 expressing C1R cells (C1R.MR1null+MR1) pulsed with 100 pM 5-OP-RU (∼EC50 dose). (H and I) Percentage of TNF- or IFNγ-producing MAIT cells, comparing the response in individual donors to WT or CD8-null MR1 expressing C1R cells (C1R.MR1null+MR1 or C1R.MR1null+MR1 CD8-null) pulsed with titrating doses of 5-OP-RU. (J and K) As above, comparing the percentage of TNF- or IFNγ-producing DN or CD8 SP MAIT cells. (B–J) Data are from 12 healthy blood donors from three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (F and G) or a two-way ANOVA with Sidak’s multiple comparisons test (H–K).
We next examined cytokine production by MAIT cell subsets from individual donors stimulated with titrating amounts of 5-OP-RU in the presence of C1R cells expressing either WT or CD8-null MR1. A consistent and significant reduction in the percentage of TNF- and IFNγ-producing MAIT cells was observed for both CD8αα+ and CD8αβ+ MAIT cells in the absence of CD8 binding (Fig. 5, H and I). Interestingly, for DP MAIT cells, which generally contained smaller fractions of TNF- and IFNγ-producing cells than CD8 SP MAIT cells (Fig. 5, D–G), the effect of CD8 binding was less consistent, particularly at the highest and lowest antigen doses (Fig. 5, H and I). For CD4+ and DN MAIT cells, CD8 binding did not impact cytokine production (Fig. 5, H and I). This was expected and is consistent with CD4 not affecting MR1 binding (Fig. 4, B and F), although expression of CD4 may alter T cell activation by competing for Lck, as both CD8α and CD4 possess a conserved Lck binding motif (Shaw et al., 1990; Turner et al., 1990). To address whether the reduction in the fraction of CD8+ cytokine-producing cells was entirely due to the lack of CD8 engagement, we directly compared CD8 SP and DN MAIT cells stimulated with WT or CD8-null MR1 (Fig. 5, J and K). In the presence of WT MR1, at all but the lowest 5-OP-RU doses tested, the fractions of both TNF- and IFNγ-producing cells were significantly higher among CD8 SP MAIT cells than DN MAIT cells (Fig. 5, J and K). In contrast, no significant difference between CD8 SP and DN MAIT cell subsets was observed in the presence of CD8-null MR1 (Fig. 5, J and K). Accordingly, CD8 engagement appears to be the primary contributor to the greater fraction of cytokine production by CD8+ MAIT cells observed. Nevertheless, among individual donors, differences between CD8 SP and DN subsets were observed that were not accounted for by CD8 engagement (Fig. 5, J and K).
Interestingly, at low antigen doses (1–10 pM), the loss of CD8 engagement had no discernible impact on CD8αα+ MAIT cell function (Fig. S4, H and I). However, the effect of CD8-binding loss was detectable at higher antigen doses as a consistent reduction in the total frequency of responding cells (∼10%; Fig. S4, H and I), comparable to that of responding DN MAIT cells (Fig. S4 J). In comparison, loss of CD8 engagement by CD8αβ+ MAIT cells impacted functionally across all but the lowest antigen doses (Fig. S4, H and I), increasing with antigen dose, most notably at the half maximum dose (∼40% reduction in the total frequency of responding cells), with the effect less pronounced at the highest doses. In contrast to CD8αα+ MAIT cells, in the absence of CD8 binding, the response by CD8αβ+ MAIT cells was reduced compared to that by DN MAIT cells, particularly at the antigen dose consistent with half maximum activation measured based on % TNF production (∼25% reduction; Fig. S4 J). Accordingly, CD8 engagement increased the responses by CD8 SP MAIT cells in general, and especially CD8αβ+ MAIT cells, as well as the sensitivity of CD8αβ+ MAIT cells at low doses of antigen. Of note, both the differences between CD8 SP and DN subsets among individual donors (Fig. 5, J and K) and the reduced responses by CD8αβ+ relative to CD8αα+ (and DN) MAIT cells in the absence of CD8 binding to MR1 (Fig. S4 J), speak to other cell-intrinsic factors, as described previously (Dias et al., 2018; Gherardin et al., 2018; Vorkas et al., 2022), influencing cytokine secretion within and between coreceptor subsets. Collectively, these data strongly support that the CD8–MR1 interaction contributes to the functional potential of MAIT cells in the context of TCR-dependent stimulation.
T cell recognition of MR1-6-FP tetramers is dependent on CD8–MR1 interactions
Our observations thus far revealed that CD8 engagement enhanced MAIT cell responses to potent stimuli such as 5-OP-RU. However, CD8 is known to be crucial for fine-tuning T cell responses in the presence of weakly stimulating antigens characterized by TCR–pMHC-I interactions of low affinity (Hutchinson et al., 2003; Laugel et al., 2007). Therefore, we sought to understand whether CD8 also plays a role in fine-tuning reactivity by non-MAIT, MR1-reactive T cells, many of which express CD8 and likely produce low-affinity TCR–MR1 interactions, including almost all described that are reactive to folate-derived antigens (Gherardin et al., 2016; Koay et al., 2019). To examine this, we selected the folate degradation product 6-FP as our model antigen as it induces strong upregulation of MR1 surface expression and is recognized by some MR1-reactive T cells (Kjer-Nielsen et al., 2012; Eckle et al., 2014; Gherardin et al., 2016). Using MR1-6-FP tetramer, we enriched T cells from PBMCs of 12 healthy donors, segregating them based on TRAV1-2 expression, and expanded them in vitro using nonspecific TCR stimulation (Fig. S5 A).

Expanded MR1-6-FP–reactive T cells from human PBMCs respond in a CD8 dependent manner, and splenic CD8 + MR1-reactive T cells are reliant on CD8 engagement for recognition of MR1 tetramers. (A) Gating strategy for sorting of enriched MR1-6-FP tetramer+ T cells (Post-enrichment) and verification of antigen reactivity after in vitro expansion (Post-expansion). (B) CD8α expression of expanded MR1-6-FP–reactive T cells from up to 12 healthy donors examined in Fig. 6. (C and D) Frequencies of expanded TRAV1-2− or TRAV1-2+ T cells that retain MR1-6-FP or -5-OP-RU tetramer reactivity post-expansion as part of experiments shown in Fig. 6. (E) Concentrations of cytokines secreted into culture supernatant by mixed TRAV1-2+/− expanded T cells from four healthy donors after stimulation with PMA/ionomycin (18 h). (F) CD8α expression of expanded MR1-6-FP–reactive T cells from nine healthy donors. (G and H) Frequencies of expanded TRAV1-2− or TRAV1-2+ T cells that retain MR1-6-FP or -5-OP-RU tetramer reactivity post-expansion as part of experiments shown in Fig. 7. (I and K) Percentages of IFNγ-producing expanded TRAV1-2+ or TRAV1-2− cells cultured in the absence or presence of MR1 deficient (C1R.MR1null), WT MR1 expressing (C1R.MR1null+MR1), or mutant (C1R.MR1null+MR1-K43A) expressing C1R cells pulsed with 10 nM 5-OP-RU, 10 μM 6-FP, or no antigen. Mean and SD are displayed. (J and L) Percentages of IFNγ-producing expanded TRAV1-2+ or TRAV1-2− cells cultured with WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of antigen. Data are from the same six (TRAV1-2+) or nine (TRAV1-2−) healthy blood donors as in Fig. 7, representing three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (I and K) or a two-way ANOVA with Sidak’s multiple comparisons test (J and L). (M) Top panels display dot plots of splenic T cells from a single donor stained directly ex vivo with WT or CD8-null MR1-5-OP-RU tetramers, gated on MAIT cells (elliptical gate) and other MR1-reactive T cells (polygon gate) and showing the frequency of total T cells. Bottom panels are dot plots of gated populations in top panels (Tetlow and Tethigh[MAIT]) displaying CD3 and TRAV1-2 expression. Data are from one experiment.
Expanded MR1-6-FP–reactive T cells from human PBMCs respond in a CD8 dependent manner, and splenic CD8 + MR1-reactive T cells are reliant on CD8 engagement for recognition of MR1 tetramers. (A) Gating strategy for sorting of enriched MR1-6-FP tetramer+ T cells (Post-enrichment) and verification of antigen reactivity after in vitro expansion (Post-expansion). (B) CD8α expression of expanded MR1-6-FP–reactive T cells from up to 12 healthy donors examined in Fig. 6. (C and D) Frequencies of expanded TRAV1-2− or TRAV1-2+ T cells that retain MR1-6-FP or -5-OP-RU tetramer reactivity post-expansion as part of experiments shown in Fig. 6. (E) Concentrations of cytokines secreted into culture supernatant by mixed TRAV1-2+/− expanded T cells from four healthy donors after stimulation with PMA/ionomycin (18 h). (F) CD8α expression of expanded MR1-6-FP–reactive T cells from nine healthy donors. (G and H) Frequencies of expanded TRAV1-2− or TRAV1-2+ T cells that retain MR1-6-FP or -5-OP-RU tetramer reactivity post-expansion as part of experiments shown in Fig. 7. (I and K) Percentages of IFNγ-producing expanded TRAV1-2+ or TRAV1-2− cells cultured in the absence or presence of MR1 deficient (C1R.MR1null), WT MR1 expressing (C1R.MR1null+MR1), or mutant (C1R.MR1null+MR1-K43A) expressing C1R cells pulsed with 10 nM 5-OP-RU, 10 μM 6-FP, or no antigen. Mean and SD are displayed. (J and L) Percentages of IFNγ-producing expanded TRAV1-2+ or TRAV1-2− cells cultured with WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of antigen. Data are from the same six (TRAV1-2+) or nine (TRAV1-2−) healthy blood donors as in Fig. 7, representing three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (I and K) or a two-way ANOVA with Sidak’s multiple comparisons test (J and L). (M) Top panels display dot plots of splenic T cells from a single donor stained directly ex vivo with WT or CD8-null MR1-5-OP-RU tetramers, gated on MAIT cells (elliptical gate) and other MR1-reactive T cells (polygon gate) and showing the frequency of total T cells. Bottom panels are dot plots of gated populations in top panels (Tetlow and Tethigh[MAIT]) displaying CD3 and TRAV1-2 expression. Data are from one experiment.
Within the TRAV1-2− subset, amongst donors, a mean of 84.6% of cells were CD8α+ and 6-FP reactivity was retained, with a mean of 64% of cells binding MR1-6-FP tetramer. A proportion of the TRAV1-2− cells displayed cross-reactivity to MR1-5-OP-RU tetramer with a mean average of 37% amongst donors (Fig. S5, B and C), akin to our previously published study (Gherardin et al., 2016). Strikingly, when we stained the TRAV1-2− subset with CD8-null MR1 tetramers, the majority of cells in all donors could not recognize MR1-6-FP or MR1-5-OP-RU tetramers (Fig. 6, A and B), indicating that these cells relied on CD8 binding for recognition of MR1 tetramers, regardless of antigen specificity.

MR1-6-FP–reactive T cells are dependent on CD8 for MR1-6-FP tetramer recognition. (A) Expanded TRAV1-2− or TRAV1-2+ T cells stained with WT or CD8-null MR1-6-FP and MR1-5-OP-RU tetramers from a single healthy blood donor. (B) Comparison of WT and CD8-null MR1-6-FP and MR1-5-OP-RU tetramer staining of expanded TRAV1-2− cells from 12 donors. (C) Same format as B but of TRAV1-2+ T cells from six donors. (D) Comparison of WT and CD8-null MR1-5-OP-RU tetramer fluorescence of expanded TRAV1-2+ cells. Data are from three independent experiments. Statistical significance was determined using a two-way ANOVA with Sidak’s multiple comparisons test (B and C) or Wilcoxon signed-rank test (D).
MR1-6-FP–reactive T cells are dependent on CD8 for MR1-6-FP tetramer recognition. (A) Expanded TRAV1-2− or TRAV1-2+ T cells stained with WT or CD8-null MR1-6-FP and MR1-5-OP-RU tetramers from a single healthy blood donor. (B) Comparison of WT and CD8-null MR1-6-FP and MR1-5-OP-RU tetramer staining of expanded TRAV1-2− cells from 12 donors. (C) Same format as B but of TRAV1-2+ T cells from six donors. (D) Comparison of WT and CD8-null MR1-5-OP-RU tetramer fluorescence of expanded TRAV1-2+ cells. Data are from three independent experiments. Statistical significance was determined using a two-way ANOVA with Sidak’s multiple comparisons test (B and C) or Wilcoxon signed-rank test (D).
Amongst donors, an average of 87% of TRAV1-2+ cells were CD8α+ and 66% of cells retained MR1-6-FP tetramer reactivity (Fig. S5, B and D). Consistent with the classical MAIT TCRα chain (TRAV1-2+) usage, more (78%) of these cells amongst donors recognized MR1-5-OP-RU tetramer (Fig. S5 D). Similar to the TRAV1-2− subset, most TRAV1-2+ cells failed to bind the CD8-null MR1-6-FP tetramer, but interestingly retained the ability to bind the CD8-null MR1-5-OP-RU tetramer, suggesting that a component of the TCR interaction is intrinsically due to weak antigen cross-reactivity and/or autoreactivity to MR1 (Fig. 6, A and C). Like the tetramer staining analysis in Fig. 4, the fluorescence intensity of the CD8-null MR1-5-OP-RU tetramer was significantly reduced compared to WT tetramer (Fig. 6 D), indicating that CD8 plays a role in MR1-5-OP-RU recognition for this subset of MR1-reactive T cells.
In line with a potential MR1-centric or -autoreactive binding interaction between TCR–MR1 that is mediated by CD8, we identified a substantial population of MR1-5-OP-RU tetramer+ T cells (5.5% of T cells) in addition to MAIT cells (4.7% of T cells) in lymphocyte preparations of human spleen directly ex vivo (Fig. S5 M). This novel population was TRAV1-2−, bound weakly to MR1-5-OP-RU tetramers, and was only detected amongst CD8α+ Τ cells. Akin to MR1-6-FP–reactive T cells, the CD8-null mutation largely abrogated MR1 tetramer binding (Fig. S5 M), suggesting these cells are also reliant on CD8 for recognition of MR1.
CD8–MR1 interactions are critical for MR1-reactive T cell responses to 6-FP
Analogous to MHC-Ia restricted T cell responses, the threshold for MR1-antigen reactivity based on tetramer staining may be lower than that based on cellular activity, although it can also be higher (Wooldridge et al., 2009). Accordingly, we next sought to determine whether MR1 tetramer binding correlated with cellular activity and examined to what extent the interaction between CD8 and MR1 impacted the function of MR1-6-FP reactive T cells. MR1-6-FP tetramer-reactive T cells were enriched from eight healthy PBMC donors and expanded in vitro as described above. We confirmed that the reactivity of the expanded T cells to MR1-6-FP tetramers was largely retained (Fig. S5, F–H). Due to the heterogeneous phenotype of TRAV1-2− MR1-reactive T cells (Gherardin et al., 2016; Koay et al., 2019), we determined the dominant cytokines produced by our expanded cells by measuring the production of various T helper (Th)1, Th2, and Th17 cytokines after PMA/ionomycin stimulation. We identified IFNγ, followed by TNF, as the most abundant cytokines secreted in all donors tested (Fig. S5 E), and included these as activation markers in subsequent assays.
We stimulated expanded MR1-6-FP tetramer-binding T cells in the presence or absence of MR1 deficient, WT, mutant CD8-null, or MR1–K43A MR1 overexpressing C1R cells, the latter three matched for similar MR1 expression levels (Fig. S4 D), pulsed with titrating amounts of 5-OP-RU, 6-FP, or no exogenous antigen. MR1–K43A lacks the ability to form a Schiff base with MR1 ligand, yet is reasonably stable, and expressed at the cell surface in the absence of exogenous ligand (Eckle et al., 2014; McWilliam et al., 2016; Reantragoon et al., 2013; Corbett et al., 2014). Thus, MR1–K43A may be expressed without ligand or bound with endogenous ligands that are not dependent on Schiff base formation, allowing us to probe for MR1 reactivity that is ligand independent (or permissive). As expected, TRAV1-2+ cells, which generally bound strongly to MR1-5-OP-RU tetramer (Fig. 6, A and C), were most responsive to 5-OP-RU, involving a higher fraction of TNF- than IFNγ-producing cells (Fig. 7 A and Fig. S5 I), like MAIT cells (Fig. 5, B and C). A small proportion of TRAV1-2+ cells produced TNF and IFNγ in response to 6-FP, yet similar percentages of cytokine-producing TRAV1-2+ cells were detected in the absence of exogenous antigen or when stimulated by MR1-K43A (Fig. 7 A and Fig. S5 I), suggesting ligand-independent MR1 responsiveness. Indeed, when assessing TRAV1-2+ cell responsiveness to titrating amounts of antigens, two distinct patterns of reactivity emerged in donors, those that responded to 5-OP-RU in a dose-dependent manner, exhibiting weak MR1-reactivity in the absence of 5-OP-RU, or those that were moderately responsive to MR1 and not augmented by exogenous antigen (Fig. 7 B and Fig. S5 J). Strikingly, unlike 5-OP-RU–specific responses, MR1-reactivity by TRAV1-2+ cells appeared almost entirely CD8 dependent (Fig. 7 B and Fig. S5 J), consistent with the tetramer-binding capacity of these cells (Fig. 6, A, C, and D). Among TRAV1-2− cells, a similar proportion of cells produced cytokines in response to both 5-OP-RU and 6-FP, as well as to no exogenous antigen in the context of WT MR1 and MR1-K43A, suggesting ligand-independent MR1 responsiveness (Fig. 7 C and Fig. S5 K). In the absence of CD8 binding, and consistent across titrating amounts of both antigens, cytokine production was significantly reduced or ameliorated entirely (Fig. 7 D and Fig. S5 L). Thus, the CD8–MR1 interaction is critically important for the production of cytokines by these MR1-reactive T cells, enhancing specific recognition of the potent riboflavin-based antigen 5-OP-RU, observed for TRAV1-2+ cells in some donors, and permitting reactivity to weaker stimulating MR1-antigen complexes or ligand independent MR1 reactivity, as observed for TRAV1-2+ and TRAV1-2− cells in all donors.

MR1-6-FP T cell reactivity is reliant on CD8 for cytokine production. (A and C) Percentage of TNF-producing expanded TRAV1-2+ or TRAV1-2− cells cultured in the absence or presence of MR1 deficient (C1R.MR1null), WT MR1 expressing (C1R.MR1null+MR1), or mutant (C1R.MR1null+MR1-K43A) expressing C1R cells pulsed with 10 nM 5-OP-RU, 10 μM 6-FP, or no antigen. Mean and SD values are displayed. (B and D) Percentages of TNF-producing expanded TRAV1-2+ or TRAV1-2− cells cultured with WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of antigen. Data are from six (TRAV1-2+) or nine (TRAV1-2−) healthy blood donors from three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (A and C) or a two-way ANOVA with Sidak’s multiple comparisons test (B and D).
MR1-6-FP T cell reactivity is reliant on CD8 for cytokine production. (A and C) Percentage of TNF-producing expanded TRAV1-2+ or TRAV1-2− cells cultured in the absence or presence of MR1 deficient (C1R.MR1null), WT MR1 expressing (C1R.MR1null+MR1), or mutant (C1R.MR1null+MR1-K43A) expressing C1R cells pulsed with 10 nM 5-OP-RU, 10 μM 6-FP, or no antigen. Mean and SD values are displayed. (B and D) Percentages of TNF-producing expanded TRAV1-2+ or TRAV1-2− cells cultured with WT or CD8-null MR1 expressing C1R cells pulsed with titrating doses of antigen. Data are from six (TRAV1-2+) or nine (TRAV1-2−) healthy blood donors from three independent experiments. Statistical significance was determined using a Friedman test with Dunn’s multiple comparison (A and C) or a two-way ANOVA with Sidak’s multiple comparisons test (B and D).
Discussion
Conflicting analyses on whether MR1 features a CD8 binding site (Riegert et al., 1998; Walter and Gunther, 1998; Miley et al., 2003; Hashimoto et al., 1995) affirm that the CD8–MR1 interaction cannot be predicted based on sequence homology within a putative binding site in the α3-domain of MR1. We examined the potential for a CD8–MR1 interaction by interrogating CD8 binding to MR1 directly by mutating MR1 in the putative CD8 binding site and by determining the crystal structure of the CD8αα–MR1-Ac-6-FP complex. Here, we describe that both CD8αα homodimers and CD8αβ heterodimers bind to MR1 at a site that is partially conserved with MHC-I using a relatively conserved mode of engagement. Further, we show that the MR1–CD8αα affinity is in the range reported for MHC-I–CD8αα interactions (KD∼ 200–1,000 μM; Gao and Jakobsen, 2000; Wyer et al., 1999). To our knowledge, no other β2m-associated MHC-I–like molecule has been shown to interact with CD8; however, both CD8 dimers have previously been described to interact with comparable affinities with classical MHC-Ia molecules (Kern et al., 1999; Bosselut et al., 2000; Garcia et al., 1996; Huang et al., 2007). In addition, a number of non-classical MHC-Ib molecules have been identified that bind to CD8. Namely, CD8αα is upregulated on a large proportion of intraepithelial cells (IELs) in mouse gut where it binds to the MHC-Ib molecule TL (Leishman et al., 2001), which itself does not present antigens (Liu et al., 2003); TL can also bind CD8αβ, albeit with a slightly lower affinity (Leishman et al., 2001). Similarly, CD8αα is expressed on subsets of murine liver and small intestine γδ T cells and was shown to interact with the soluble MHC-Ib molecules H2-Q10 (Goodall et al., 2019) and Qa-1b (Goodall et al., 2020), respectively. Furthermore, both HLA-G (Clements et al., 2005; Gao et al., 2000) and H2-T22 (Goodall et al., 2020) have been identified as ligands for CD8αα.
Using antigen-presenting cells that express mutant versions of MR1, we determined the functional consequences of the CD8–MR1 interactions for MAIT and other MR1-reactive T cells. We found that both CD8 dimers can engage MR1 cooperatively with the TCR, enhancing T cell activation. As such, both CD8 dimers have a functional role similar to CD8αβ on conventional T cells (Arcaro et al., 2000), and are viable coreceptors for MAIT and some other MR1-reactive T cells. In fact, comparing MAIT cell coreceptor subsets (CD4+, DN, DP, CD8αα+, and CD8αβ+), we observed that CD8αα+ and CD8αβ+ MAIT cells were consistently the strongest cytokine producers in response to stimulation with 5-OP-RU. This was primarily due to CD8 engagement of MR1, where loss of binding significantly reduced MR1-antigen recognition and cytokine production. Greater activation of conventional T cells is usually only observed with CD8αβ (Kern et al., 1999; Bosselut et al., 2000), whilst CD8αα is non-functional (Pang et al., 2007) or perhaps acts as a repressor of activation (reviewed in Cheroutre and Lambolez, 2008). CD8αα expression by CD8α+CD8βlow CTLs coincides with enhanced function only in patients with chronic viral infections, but it is unclear whether this was dependent on an interaction between CD8αα+ and MHC-I molecules (Walker et al., 2013). The α3-domain of MR1 is also the primary contact site of some MR1-reactive γδ T cells (Le Nours et al., 2019), and the crystal structure of CD8αα–MR1-Ac-6-FP revealed overlapping binding sites between CD8αα and G7 γδTCR complexes with MR1, therefore γδ TCRs that adopt this binding mode would be expected to compete with CD8 for MR1 binding.
MHC-I engagement by CD8αβ and TCR increases the avidity of the TCR–MHC-I interaction and brings CD8αβ-bound Lck into close proximity to the CD3 complex, enhancing TCR signaling (Delon et al., 1998; Renard et al., 1996; Zareie et al., 2021), whereas the role of CD8αα in lymphocyte function is poorly defined. For MAIT and other MR1-reactive T cells, binding of MR1 by either dimer (CD8αα or CD8αβ) increased the tetramer avidity, slowed down the decay kinetics of the TCR–MR1 complex, and enhanced cytokine production. Thus, for these T cells, CD8αα possibly functions the same way as CD8αβ, bringing CD8α-bound Lck into close proximity to the CD3 complex, such that both CD8 dimers can enhance TCR signaling. Alternatively, CD8 may function by primarily enhancing the avidity (and decay kinetics) of the TCR–MR1 complex whilst not contributing to enhancing TCR signaling, with the MAIT–MR1 axis potentially being less dependent on the latter. Interestingly, CD8αα was also shown to function as a coreceptor on a subset of CD8αα+ natural killer (NK) cells, whereby CD8αα bound to MHC-I concurrently with the KIR3DL1 receptor to fine-tune NK cell inhibitory signals and cytolytic activity (Geng and Raghavan, 2019). However, the functional consequences of CD8αα binding to most of the MHC-Ib molecules, described above, is unknown, except for some studies that have investigated the CD8αα–TL interaction. Namely, whilst independent of TCR, the CD8αα–TL interaction has been proposed to occur alongside TCR–MHC-I interactions to regulate the activation (Agea et al., 2005; Leishman et al., 2001; Pardigon et al., 2004; Olivares-Villagomez et al., 2008) and trafficking (Takei et al., 2020; Pardigon et al., 2004) of IELs, but these findings are controversial. TL interacting with CD8αα expressed by activated CD8αβ+ T cells has also been shown to mediate affinity-based selection of intestinal mucosa resident memory T cells (CD8αβ+TEM; Huang et al., 2011). Altogether, our finding that CD8αα acts as a coreceptor for MAIT and possibly other MR1-reactive T cells contributes to the accumulating evidence of a role for CD8αα in lymphocyte function.
In the absence of CD8 binding, most CD8+ MAIT cells remained clearly identifiable in blood using MR1 tetramers. This implies that for most CD8+ MAIT cells, CD8 engagement is not a strict requirement for recognition of MR1 presenting the strongly agonistic antigen 5-OP-RU, to which classical human MAIT TCRs bind with high affinity (KD ∼ 1–10 μM; Eckle et al., 2014; Patel et al., 2013). Furthermore, we noted modest differences in the capacity of DN and CD8+ MAIT cells to produce cytokines upon stimulation, as previously described (Brozova et al., 2016; Kurioka et al., 2017; Dias et al., 2018; Gherardin et al., 2018; Booth et al., 2015). This included a consistent reduction in the proportion of cytokine-producing cells within DN as compared to CD8+ MAIT cells, as noted previously for E. coli (Kurioka et al., 2017; Dias et al., 2018) and Helicobacter pylori (Booth et al., 2015). Importantly, the cytokine response to 5-OP-RU by CD8 SP MAIT cells was significantly reduced in the absence of CD8 binding to levels that were similar or diminished, compared to DN MAIT cells, for CD8αα+ and CD8αβ+ MAIT cells, respectively. Accordingly, CD8 appears to enhance the responsiveness of MAIT cells, and in the case of CD8αβ+ MAIT cells, possibly compensates for subset intrinsic features that reduce its functional capacity. Based on the lack of differences in CD8-null MR1 tetramer fluorescence between coreceptor subsets, CD8αβ+ MAIT TCRs are unlikely to be of lower affinity.
In support of CD8 playing a role in the fine-tuning of MAIT cell responsiveness, Dias et al. (2018) revealed that the TCR repertoire of DN MAIT cells is less diverse and shared within the TCR repertoire of CD8+ MAIT cells, suggesting that many DN MAIT cells may have previously downregulated CD8. Indeed, CD8 expression is regulated transcriptionally (Bosselut et al., 2003; Park et al., 2007) and by modulation at the cell surface (Maile et al., 2005; Xiao et al., 2007). In addition, post-translational modifications of CD8αα and CD8αβ proteins can alter their ability to bind MHC-Ia molecules (Daniels et al., 2001; Moody et al., 2001; Kao et al., 2006; Lischke et al., 2013) and MHC-Ib molecules, as recently demonstrated for H2-Q10 (Goodall et al., 2021). It is also known that CD8αα can be induced on IELs, conventional T cells, and immature thymocytes in response to microenvironmental cues and TCR stimulation (Reis et al., 2013; Gangadharan and Cheroutre, 2004). Thus, one could speculate that MAIT cell coreceptor expression is similarly modulated in response to infection or the microenvironment. Comparatively, in the case of low-affinity TCR–MR1-ligand interactions, as exemplified here by the recognition of folate-derived antigens by MR1-reactive T cells, the CD8–MR1 interaction is crucial. Notably, our study analyzed populations of cells that were enriched for MR1-6-FP tetramer reactivity without discerning the details of antigen specificity or preference at the clonal T cell level. Indeed, previous studies characterized non-MAIT MR1-reactive primary T cell clones (Crowther et al., 2020; Harriff et al., 2018; Lepore et al., 2017; Meermeier et al., 2016) and TCR reporter lines (Gherardin et al., 2016; Koay et al., 2019) that displayed specificity or preference for non-riboflavin-based antigens or antigen-loaded MR1 tetramers, respectively. Broadly, most of these T cells identified express CD8 (Gherardin et al., 2016; Koay et al., 2019; Crowther et al., 2020; Lepore et al., 2017), similar to CTLs, and it is to be determined whether CD8 is important for the function of these cells. Indeed, the MR1-reactive T cell response to both Mycobacterium tuberculosis and M. smegmatis appears to be largely composed of CD8+ T cells, and CD8 was indispensable for detection of M. tuberculosis infection in vitro (Gold et al., 2013; Sharma et al., 2015).
Overall, the dependence on CD8 for TCR recognition of MR1 is similar to that observed for TCR recognition of pMHC-I by CTLs (Daniels and Jameson, 2000; Laugel et al., 2007; Clement et al., 2021; Clement et al., 2016; Holler and Kranz, 2003), where MR1 ligands of sufficient potency do not require CD8 engagement, while responses to weaker MR1 ligands and MR1 autoreactivity are reduced or abrogated in the absence of CD8 binding. CD8 thus appears to play a crucial role in expanding the antigen repertoire detected by MAIT and other MR1-reactive T cells and in this way grants greater antigenic promiscuity and autoreactivity to MR1. Recognition of a wider range of MR1 ligands may involve allergen antigens, leading to the hypothetical involvement of MR1-reactive T cells in hypersensitivities (De Lima Moreira et al., 2020), and analogous to previous reports of CD8-dependent cross-reactivities mediated by CTLs (Blok et al., 1992; Kasprowicz et al., 2008; Wooldridge et al., 2010). Considering the accumulating diversity of the MR1-reactive TCR repertoire and the discovery of novel MR1 ligands (Souter and Eckle, 2020), both related and unrelated to the riboflavin biosynthesis pathway (Gherardin et al., 2016; Keller et al., 2017; Meermeier et al., 2016; Lepore et al., 2017; Harriff et al., 2018; Crowther et al., 2020), we foresee the importance of CD8 will become increasingly evident. The use of CD8-null MR1 tetramers, which we describe here, alongside WT tetramers, will serve as powerful experimental tools to assess CD8 dependence and distinguish between TCR-mediated interactions with MR1 of high and low affinity.
Materials and methods
MR1 ligands
6-FP and Ac-6-FP (Schircks Laboratories) were dissolved at 5 mM in water supplemented with 17 mM NaOH. 5-OP-RU was synthesized in-house as a 1 mM stock solution in DMSO (Mak et al., 2017; Mak et al., 2021). For cellular assays, the stock solutions of 6-FP and 5-OP-RU were diluted into PBS.
Production of soluble MHC-I and CD1d molecules
Soluble peptide MHC-I heterodimers (HLA-A*02:01-NLVPMVATV, HLA-B*08:01-FLRGRAYGL, HLA-C*06:02-TRATKMQVI and HLA-G*01:01-RIIPRHLQL) were prepared similarly to previously described (Reid et al., 1996; Clements et al., 2005; Gao et al., 1997) based on Garboczi et al. (1992). Peptides were purchased from Genscript. Briefly, 30 μg/ml of peptide, 24 mg/ml of β2m, and 93 mg/ml of HLA heavy chain from E. coli inclusion bodies were refolded in buffer containing 10 mM Tris, pH 8, 2 mM EDTA, pH 8, 1 M L-arginine (#A5006; Sigma-Aldrich), 5 mM L-glutathione reduced (#G4251; Sigma-Aldrich), and 0.5 mM L-glutathione oxidized (#G4376; Sigma-Aldrich). Following dialysis, refolded monomers were then purified using sequential anion exchange, size exclusion, anion exchange, and hydrophobic interaction chromatography. Soluble human CD1d loaded with mammalian endogenous lipid antigens (CD1d-endo) and expressing a C-terminal His-tag was generated in Expi293F cells using ExpiFectamine (#A14525; Thermo Fisher Scientific) and purified similarly as described previously (Rigau et al., 2020). For SPR, CD1d-endo without a His-tag was generated as above but purified by anion exchange and size exclusion chromatography. All MHC-I/CD1d monomers displayed >95% purity based on characterization by SDS-PAGE.
Production of soluble WT and CD8-null (Q223A, E224K) MR1 molecules
Soluble human WT MR1 and CD8-null (Q223A, E224K) monomers were generated in-house, as described previously for the generation of WT MR1 (Reantragoon et al., 2013; Corbett et al., 2014). In brief, MR1 monomers were folded from E. coli inclusion bodies in the presence of MR1 ligands and, following dialysis, purified using sequential anion exchange, size exclusion, and anion exchange chromatography. MR1 monomers were analyzed for purity by SDS-PAGE (Fig. S1 E). For the generation of MR1 tetramers, a version of soluble MR1 with a C-terminal cysteine was produced and biotinylated using maleimide-PEG2-biotin (#21901BID; Thermo Fisher Scientific), followed by an additional anion exchange chromatography purification step. Biotinylation of MR1 monomers was assessed by SDS-PAGE with streptavidin (#S0677; Sigma-Aldrich; Fig. S1 F). To generate tetramers, biotinylated MR1 monomers were incubated with streptavidin-PE (#554061; BD Biosciences) at a 5:1 mass ratio by sequentially adding equal amounts of streptavidin-PE every 10 min at room temperature in the dark. Tetramers were diluted to a final monomer concentration of 0.25 mg/ml with TBS, pH 8, and used at a 1:200 dilution in all experiments unless stated otherwise.
Production of soluble CD8αα
Soluble CD8αα was produced in vitro by refolding from E. coli inclusion bodies. In brief, a truncated gene encoding the extracellular Ig-like domain of CD8α with a Cys75Ser mutation and a C-terminal His-tag was purchased from Genscript (5′-HMSQFRVSPLDRTWNLGETVELKCQVLLSNPTSGSSWLFQPRGAAASPTFLLYLSQNKPKAAEGLDTQRFSGKRLGDTFVLTLSDFRRENEGYYFCSALSNSIMYFSHFVPVFLPAKPTTTPHHHHHH-3′) and cloned into the bacterial vector pET30. E. coli were transformed with the pET30 vector and induced to produce CD8α using 1 mM isopropyl β-D-1-thiogalactopyranoside. Inclusion bodies were harvested from cultured bacteria, purified, and refolded similarly as described previously (Goodall et al., 2019; Cole et al., 2008). CD8αα dimers were purified by sequential cation exchange, size exclusion, and cation exchange chromatography, and purity was assessed by SDS-PAGE (Fig. S1 H; Goodall et al., 2019).
Culture of cell lines, human PBMCs, and spleen tissue samples
All cell lines and PBMCs were cultured in RPMI1640 (#11875-093; Gibco) supplemented with 10% FBS (JRH Biosciences), 2% penicillin (100 U/ml), streptomycin (100 μg/ml), Glutamax (2 mmol/liter), sodium pyruvate (1 mmol/liter), nonessential amino acids (0.1 mmol/liter), Hepes buffer (15 mmol/liter), pH 7.2–7.5 (all from Thermo Fisher Scientific, Life Technologies), and 2-mercaptoethanol (50 μmol/liter; Sigma-Aldrich). PBMCs were obtained from the Australian Red Cross Blood Service (authorized by the Australian Red Cross Blood Service Material Supply Agreement with The University of Melbourne and approved by The University of Melbourne STEMM 1 Human Ethics Committee; ID: 12540-23422) and isolated as described previously (Reantragoon et al., 2013). Spleen tissue samples were obtained from Austin Health of Austin Hospital through the Australian Donation and Transplantation Biobank (approved by the Austin Health Human Research Ethics Committee; ID HREC/48184/Austin-2019 and The University of Melbourne STEMM 1 Human Ethics Committee; ID: 13009). Spleen tissue was processed by first removing any visible splenic capsule and chopping the tissue into 1–5 mm pieces. Dissected tissue was incubated in digestion media consisting of supplemented RPMI1640 with DNAse I (10 μg/ml) and Collagenase D (1 mg/ml) at 37°C for 1 h while shaking. Digested tissue was mashed successively through 300 and 100 μm sieves. Splenocytes were then isolated by centrifugation in the presence of 44% Percoll (#17-0891-01; Cytiva) in PBS. All experiments involving human PBMCs and spleen tissue samples were conducted in compliance with the Australian National Statement on Ethical Conduct in Human Research (2007, Updated 2018).
Generation of cell lines
The Jurkat MAIT TCR reporter cell line expresses the A-F7 MAIT TCR (Tilloy et al., 1999) and was previously generated (Kjer-Nielsen et al., 2012). Parental SKW-3.β2mnull cells, generated previously from parental SKW-3 cells (McWilliam et al., 2020), were transduced with CD8αα (SKW-3.β2mnull.CD8αα) or CD8αβ (SKW-3.β2mnull.CD8αβ) by retroviral transduction using polybrene, similarly as described previously (Holst et al., 2006; Herold et al., 2008). Briefly, gene segments encoding full-length human CD8α and CD8β were cloned into a self-cleaving 2A-peptide-based (MSCV)-IRES-GFP (pMIG) vector as CD8α alone (pMIG.CD8α-IRES-GFP) or together with CD8β (pMIG.CD8β-IRES-CD8α) and co-transfected into HEK293T cells with the plasmids pVSV-G and pEQ.PAM (-E) using Fugene 6 to produce retrovirus (Holst et al., 2006). Transduced SKW-3 cells were cloned based on CD8 expression by single-cell sorting using a BD FACSAriaIII. While SKW-3 cells are listed on the database of cross-contaminated or misidentified cell lines, where they are described as being contaminated with the KE-37 line, we have specifically transduced these cells with CD8 and recloned these cells by single-cell sorting. MR1-deficient C1R cells (C1R.MR1null) were generated with CRISPR-Cas9 RNPs as previously described (Seki and Rutz, 2018). Two custom single guide RNAs (sgRNAs) targeting genomic MR1 at regions 5′-TGGAACTGAAGCGCCTACAG-3′ and 3′-ACCATTAACACAATGATGAG-5′ were purchased from IDT. Briefly, sgRNAs were duplexed with Alt-R trans-activating CRISPR RNA (#1072533; IDT) and complexed with Alt-R S.p. Cas9 (#1081058; IDT). 106 C1R cells were washed twice with PBS, resuspended in supplemented nucleofector solution (#V4XC-2032; Lonza), and the two MR1-specific RNPs were then transferred to a Nucleocuvette strip (#V4XC-2032; Lonza) for electroporation. C1R cells were electroporated using a 4D-nucleofector (Lonza; pulse code CM130). After nucleofection, C1R cells were resuspended into warmed supplemented RPMI media and cultured for 7 d. CRISPR-Cas9 treated C1R cells were pulsed with 50 μM 6-FP for 4 h to induce MR1 upregulation on the cell surface. Subsequently, C1R cells were stained with anti-MR1-PE (#361106, 26.5; Biolegend) for 20 min at 4°C, in the dark prior to single cell sorting on MR1 deficient C1R cells. MR1-deficient C1R clones were then further validated by measuring MR1 surface expression as described above and via activation of a MAIT TCR reporter cell line. MR1 deficient C1R clones were subsequently transduced with WT MR1 (C1R.MR1null+MR1), CD8-null MR1 (C1R.MR1null+CD8-null MR1), or MR1-K43A (C1R.MR1null+MR1-K43A) by retroviral transduction. Gene segments encoding full-length MR1A, CD8-null (Q223A, E224K) MR1A, or MR1A-K43A were cloned into pMIG (pMIG.MR1A-IRES-GFP), and retrovirus was generated as described above. C1R.MR1null cells were transduced and single-cell sorted for similar expression of MR1 based on staining with the anti-MR1 antibody (26.5; Fig. S4 D).
Staining of cell lines and PBMCs with tetramers for flow cytometric analysis
SKW-3.β2mnull.CD8αα or SKW-3.β2m null.CD8αβ (105 per sample) were stained with MR1 or MHC-I tetramers in PBS + 2% FBS for 20 min at 4°C in the dark. Cells were washed with PBS + 2% FBS and resuspended in a surface antibody stain consisting of anti-CD3-BV421 (#562426, UCHT1; BD Horizon), anti-CD8α-BUV805 (#564912, SK1; BD Horizon), anti-CD8β-APC (#641058, 2ST8.5H7; BD FastImmune), and LIVE/DEAD fixable Near-IR dead cell stain (#L10119; Thermo Fisher Scientific) for a further 20 min at 4°C in the dark. Cells were washed twice with PBS + 2% FBS and data were acquired using a BD LSR Fortessa (BD Biosciences). PBMCs were stained with MR1 tetramers as described in (Souter et al., 2019). In brief, PBMCs (107 per sample) were stained with MR1 tetramer in PBS + 2% FBS for 30 min at room temperature in the dark, washed with PBS + 2% FBS, and stained with surface antibodies anti-CD3-BV421, anti-CD19-APC-Cy7 (#302218, HIB19; Biolegend), anti-CD14-APC-Cy7 (#557831, MφP9; BD Pharmingen), anti-CD8α-BUV805, anti-CD8β-APC, anti-CD161-PE-Vio770 (#130-113-597, REA631; Miltenyi Biotec), anti-CD4-AF700 (#557922, RPA-T4; BD Pharmingen), and LIVE/DEAD fixable Near-IR dead cell stain for 20 min at 4°C. Cells were washed twice and resuspended in PBS + 2% paraformaldehyde (PFA) before data acquisition on a BD LSR Fortessa.
Cell line and PBMC tetramer dissociation assays
SKW-3.β2mnull cells transduced with CD8αα (SKW-3.β2mnull.CD8αα) or CD8αβ (SKW-3.β2m null.CD8αβ) were stained with MR1-5-OP-RU or HLA-A*02:01-NLV tetramers and LIVE/DEAD fixable Near-IR dead cell stain in PBS for 30 min at 4°C in the dark. Cells were washed once with PBS and resuspended in PBS containing 10 μg/ml of purified anti-MR1 (26.5) or anti-pan-HLA-A, -B, -C (W6/32) for MR1-5-OP-RU and HLA-A*02:01-NLV tetramers, respectively. Aliquots were taken periodically over 120 min and fixed using 2% PFA in PBS. PBMC samples were stained similarly with WT or CD8-null MR1-5-OP-RU tetramers for 45 min at 4°C in the dark. Cells were washed twice with ice-cold PBS and resuspended in PBS + 0.5 μM anti-MR1 (26.5) and incubated on ice in the dark throughout the time course. Aliquots were taken periodically over 120 min and fixed using 2% PFA in PBS. Fixed PBMCs were subsequently stained with surface antibodies anti-CD3-BV421, anti-CD19-APC-Cy7, anti-CD14-APC-Cy7, anti-CD8α-BUV805, anti-CD161-PE-Vio770 (#130-113-597, REA631; Miltenyi Biotec), CD4-BUV496 (#564652, SK3; BD Horizon) anti-TCRγδ-FITC (#347903, 11F2; BD Biosciences), and LIVE/DEAD fixable Near-IR dead cell stain for 20 min at 4°C. Cells were washed twice and resuspended in PBS. Cell line and PBMC samples were acquired on a BD LSRFortessa.
SPR
SPR was performed at 25°C on a Biacore T200 instrument (GE Healthcare) using 10 mM Hepes-HCl pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.05% Tween 20 buffer. Soluble CD8αα or CD1d-endo monomers with C-terminal His-tags were immobilized on a Biacore sensor chip CM5 pre-coated with an anti-His-tag monoclonal antibody. Soluble WT or CD8-null mutant MR1-Ac-6-FP, HLA-A*02:01-NLV, or control CD1d-endo monomers (without His-tags) were diluted and simultaneously injected over test and control surfaces at a rate of 30 μl/min for 30 s. After subtraction of data from the control flow cell (anti-His-tag antibody alone) and blank injections, interactions were analyzed using Scrubber 2.0 (BioLogic Software).
Complexation of soluble CD8αα with soluble MR1-Ac-6-FP and crystallization
Soluble CD8αα was mixed with soluble MR1-β2m-Ac-6-FP, generated as described above, in a 1:1 molar ratio at concentrations of 10–15 mg/ml and incubated for 2 h at 4°C in buffer (10 mM Tris-HCl, 150 mM NaCl, pH 8.0). To identify suitable crystallization conditions, sparse matrix screening was performed involving the commercially available screens PACT Premier, JCSG+, ProtComplex, Morpheus, MorpheusII, Wizard classical 1&2, JBScreen Classic HTS I, and JBScreen Classic HTS II. For this, protein (10, and 15 mg/ml) was mixed with reservoir solution in a 1:1 volume ratio (200:200 nl) and subjected to hanging-drop vapor diffusion at 20°C. Initial crystals of CD8αα–MR1-Ac-6-FP appeared after 3 d with a precipitant consisting of 100 mM Na K Phos 6.5 pH, 25% (wt/vol) polyethylene glycol (PEG) 1K, and 200 mM NaCl. After manual grid optimization around this original condition, single hexagonally shaped crystals of CD8αα–MR1-Ac-6-FP (dimensions of 0.1 × 0.15 × 0.1 mm) were grown over 3 wk against a reservoir solution of 100 mM Na K Phos 6.1 pH, 28–30% (wt/vol) PEG 1K, and 100 mM NaCl at 20°C.
X-ray diffraction data collection and structure determination
CD8αα–MR1-Ac-6-FP crystals were flash-frozen in liquid nitrogen after quick soaking in reservoir solution supplemented with 8–10% of glycerol for cryo-protection. X-ray diffraction data were collected at 100 K on the Australian Synchrotron at MX2 beamline (Aragão et al., 2018). Diffraction images were indexed, integrated, and scaled using XDS (Kabsch, 2010), and further processed and analyzed using programs from the CCP4 suite (Winn et al., 2011) and the Phenix package (Adams et al., 2010). The CD8αα–MR1-Ac-6-FP structure was determined by molecular replacement using PHASER (Mccoy, 2007), with modified CD8αα (Protein Data Bank [PDB ID]: 1AKJ; Gao et al., 1997) and MR1-β2m (PDB ID: 4L4T; Patel et al., 2013) as search models. Afterward, an initial run of rigid body refinement was performed with Phenix.refine (Adams et al., 2010), and the CDR-like loops of CD8αα were subsequently rebuilt using the program COOT (Emsley and Cowtan, 2004). Iterative rounds of model building using COOT and refinement with Phenix.refine were performed to improve the model. The Grade Webserver and Phenix tools were used to build and to generate ligand restraints (Winn et al., 2011). The structure was validated using MolProbity (Chen et al., 2010) and graphical representations were generated using PyMOL Molecular Graphics System, Version 2.2, (Schrödinger, LLC). The quality of the structure was confirmed using the Research Collaboratory for Structural Bioinformatics PDB Data Validation and Deposition Services. The total interface area was evaluated by PISA analysis (Krissinel and Henrick, 2007) and the contacts were analyzed by the Contact program, both form the CCP4 suite. Statistics on the data collection and the final model are summarized in Table S1.
Enrichment of TRAV1-2+ cells from PBMCs
Enrichment of TRAV1-2+ T cells was performed similarly as described in Souter et al. (2019). In brief, 5 × 107 PBMCs were stained in PBS + 2% FBS with anti-TRAV1-2-PE (#351702, 3C10; Biolegend) for 30 min at 4°C in the dark, washed once with cold magnetic-activated cell sorting (MACS) buffer (0.5% FBS, 2 mM EDTA in PBS), and incubated with anti-PE beads (#130-097-054; Miltenyi Biotec) diluted in MACS buffer for 20 min at 4°C. Cells were washed, resuspended, and passed through a LS column (#130-042-401; Miltenyi Biotec) under magnetic duress. TRAV1-2–enriched cells were eluted from the column and resuspended in supplemented RPMI-1640.
Isolation and expansion of 6-FP–reactive T cells from PBMCs
Enrichment of MR1-6-FP–reactive T cells was performed similarly as described in Souter et al. (2019). In brief, 3 × 107 PBMCs were stained with MR1-6-FP tetramer labeled with streptavidin-PE in PBS + 2% FBS for 30 min at room temperature in the dark and enriched using a Miltenyi LS column as described for the enrichment of TRAV1-2+ T cells. Eluted cells were then sorted based on MR1-6-FP tetramer using a BD AriaIII. Sorted cells were stimulated with plate-bound anti-CD3 antibody (#555329; BD Pharmingen), -CD28 (#555725; BD Pharmingen), and soluble phytohemagglutinin (Sigma-Aldrich) at concentrations of 10, 5 and 3 μg/ml respectively in a 1:1 mix of complete RPMI and AIM-V media (#12-055-083; Gibco) supplemented with 200 U/ml rhuIL-2 (#200-02; Peprotech), 50 ng/ml rhuIL-7 (#200-07; Peprotech) and 25 ng/ml rhuIL-15 (#200-15; Peprotech) for 48 h. Cells were washed and resuspended in a 1:1 mix of complete RPMI and AIM-V media supplemented with rhu-IL-2, -7, and -15 for 14 d.
Stimulation of T cells with C1R cells and intracellular cytokine staining
In stimulation assays, in the absence of target cells (TRAV1-2 enriched PBMCs or expanded MR1-6-FP–reactive T cells), C1R.MR1null, C1R.MR1null+MR1, C1R.MR1null+CD8-null MR1, or C1R.MR1null+MR1-K43A cells were pulsed with titrating amounts of 5-OP-RU or 6-FP for 2 h and then washed three times with PBS to remove extracellular antigen, this way preventing T cell auto-presentation. C1R cells were resuspended in complete RPMI and cultured with target cells at a 1:1 ratio for 6 h. Brefeldin A (#20350-15-6; Sigma-Aldrich) was added for the final 5 h of culture. Before intracellular staining, cells were stained with surface antibodies anti-CD3-BUV395 (#563546, UCHT1; BD Horizon), anti-CD4-BUV496, anti-CD8α-BUV805, anti-CD8β-APC, anti-CD161-PE-Vio770, anti-TRAV1-2-PE, anti-CD19-APC-Cy7, anti-CD14-APC-Cy7 and LIVE/DEAD fixable Near-IR dead cell stain for 30 min at room temperature and then without washing, fixed with PBS + 2% PFA for 20 min at room temperature. Cells were then washed with PBS + 2% FBS twice and stained with intracellular antibodies anti-TNF-BV421 (#562783, Mab11; BD Horizon), anti-IFNγ-BV650 (#563416, 4S.B3; BD Horizon), and anti-IL-17A-PE-Dazzle 594 (#512336, BL168; Biolegend) overnight in 0.3% Saponin (#8047-15-2; Sigma-Aldrich). Cells were washed with PBS the following day and acquired using a BD LSR Fortessa.
Cellular and SPR data analysis and statistics
Flow cytometry data were analyzed using the software Flowjo 10 (Tree Star, Inc.), and graphs of flow cytometry and SPR data were generated using Prism 9 (GraphPad). Statistical analyses were performed without assuming Gaussian distribution (non-parametric). Statistical significance (two-tailed, P < 0.05) was determined where appropriate using a two-way ANOVA with a Geisser-Greenhouse correction and a Sidak multiple comparisons test, Friedman test (paired data), or a Kruskal–Wallis test (unpaired data) with a Dunn multiple comparison test.
Online supplemental material
Fig. S1 shows the sequence conservation of MR1 in the putative CD8 binding site, MR1 and HLA tetramer binding to CD8 transduced cell lines, SDS-PAGE analysis of recombinant MR1 monomers and CD8αα, and the capacity of MR1 tetramers to stain a MAIT TCR reporter cell line. Fig. S2 shows electron density maps of the ligand Ac-6-FP and important interfaces in the crystal structure of the CD8αα–MR1-Ac-6-FP ternary complex. Fig. S3 depicts a structural comparison of the ternary complexes of CD8αα–MR1-Ac-6-FP and CD8αα–HLA-A*02:01. Fig. S4 demonstrates that CD3 expression is comparable between MAIT cells segregated by coreceptor usage and there are no significant differences in CD8-null MR1-5-OP-RU tetramer staining intensities between MAIT cell coreceptor subsets. It also shows the MR1 expression levels by antigen-presenting cells, IL-17A production by stimulated MAIT cells, and MAIT cell coreceptor subset responses in the presence or absence of CD8 engagement. Fig. S5 shows that expanded MR1-6-FP–reactive T cells retain MR1 tetramer reactivity and produce cytokines in a CD8-dependent manner upon stimulation. It also shows that splenic CD8+ MR1-reactive T cells are reliant on CD8 engagement for recognition of MR1 tetramers. Table S1 lists the data collection and refinement statistics for the crystal structure CD8αα–MR1-Ac-6-FP, and Table S2 lists the atomic contacts between CD8αα and MR1-Ac-6-FP.
Data availability
The coordinates of the CD8αα–MR1-Ac-6-FP complex have been deposited in the PDB under accession code 7UMG.
Acknowledgments
We would like to acknowledge Dr. Olan Dolezal (Commonwealth Scientific and Industrial Research Organisation, Manufacturing) for providing access to a Biacore T200 instrument and for his advice on SPR experimental design and analysis. We also thank Dr. Lars Kjer-Nielsen, John Waddington, and Zheng Ruan (University of Melbourne) for intellectual advice and technical assistance. We thank Dr. Ted Hansen (University of Washington, Seattle, WA) and Dr. Wei-Jen Chua Yankelevich (U.S. Food and Drug Administration, Washington, D.C.) for provision of hybridomas producing monoclonal antibody 26.5. We also would like to thank Prof L. Wooldridge (University of Bristol, Bristol, UK) for the provision of vectors encoding mutants of soluble MHC-I molecules in collaboration with co-author K. Kedzierska. We acknowledge the Melbourne Cytometry Platform (Peter Doherty Institute and Melbourne Brain Centre nodes) for provision of flow cytometry services, and we thank the staff at the Monash Macromolecular Crystallization Facility for expert assistance. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of Australian Nuclear Science and Technology Organisation, and made use of the Australian Cancer Research Foundation detector. We acknowledge Dr. Claire Gordon at Austin Health (Heidelberg, Victoria, Australia) for the provision of spleen tissue samples through the Australian Donation and Transplantation Biobank, and gratefully acknowledge the generosity of the organ donor families in providing valuable tissue samples.
We also would like to acknowledge the following funding: Australian Postgraduate Award (M.N.T. Souter), Australian Research Council (ARC) Discovery Early Career Research Award (DECRA) fellowship DE220101491 (W. Awad), ARC Future Fellowship FT160100074 (J. Le Nours), National Health and Medical Research Council (NHMRC) of Australia Leadership Investigator Grant 1173871 (K. Kedzierska), ARC Future Fellowship FT160100083 (A.J. Corbett), NHMRC Leadership Investigator Grant 1193745 (A.J. Corbett), Dame Kate Campbell Research Fellowship from The University of Melbourne (A.J. Corbett), NHMRC Leadership Investigator Grant 2009551 (D.P. Fairlie), NHMRC Leadership Investigator Grant 2008981 (J. Rossjohn), NHMRC Senior Principal Research Fellowship 1117766 and NHMRC Leadership Investigator Grant 2008913 (D.I. Godfrey), National Institutes of Health RO1 AI148407-01A1 (J. McCluskey, J. Rossjohn, D.P. Fairlie), ARC Centre of Excellence CE140100011 (J. Rossjohn, D.P. Fairlie, D.I. Godfrey), NHMRC Program Grant 1113293 (J. McCluskey, J. Rossjohn, D.I. Godfrey), CSL Centenary Fellowship (D.G. Pellicci), NHMRC Project Grant APP1144308 (D.G. Pellicci), NHMRC Project Grant APP1140126 (D.I. Godfrey, D.G.Pellicci), ARC DECRA fellowship DE170100407 (S.B.G. Eckle), NHMRC Project Grant APP1157388 (S.B.G. Eckle), 2019 Allergy and Immunology Foundation of Australasia research grant (S.B.G. Eckle), and NHMRC Emerging Leadership Investigator Grant 1196881 (S.B.G. Eckle).
Author contributions: Conceptualization: M.N.T. Souter, D.I. Godfrey, J. McCluskey, D.G. Pellicci, S.B.G. Eckle. Data curation: W. Awad, J. Rossjohn. Formal analysis: M.N.T. Souter, W. Awad. Funding acquisition: M.N.T. Souter, W. Awad, K. Kedzierska, A.J. Corbett, D.P. Fairlie, J. Rossjohn, D.I. Godfrey, J. McCluskey, D.G. Pellicci, S.B.G. Eckle. Investigation: M.N.T. Souter, W. Awad, S. Li, T.J. Pediongco, L.J. Meehan, Z. Tian, Z. Zhao, H. Wang, A. Nelson, L. Kostenko, Z. Chen, D.G. Pellicci, S.B.G. Eckle. Methodology: M.N.T. Souter, W. Awad, Y. Khandokar, L.C. Sullivan, A. Brooks, N.A. Gherardin, Z. Chen, D.G. Pellicci, S.B.G. Eckle. Project administration: M.N.T. Souter, J. Rossjohn, D.G. Pellicci, S.B.G. Eckle. Resources: M.N.T. Souter, S. Li, T.J. Pediongco, B.S. Meehan, L.J. Meehan, Z. Tian, J.L. Le Nours, Y. Khandokar, T. Praveena, J. Wubben, J. Lin, G.O. Lovrecz, J.Y.W. Mak, L. Liu, L. Kostenko, K. Kedzierska, A.J. Corbett, D.P. Fairlie, A.G. Brooks, A.P. Uldrich, Z. Chen, J. Rossjohn, D.I. Godfrey, D.G. Pellicci, S.B.G. Eckle. Supervision: D.P. Fairlie, J. Rossjohn, D.I. Godfrey, J. McCluskey, D.G. Pellicci, S.B.G. Eckle. Validation: M.N.T. Souter, N.A. Gherardin, J. Rossjohn, S.B.G. Eckle. Visualization: M.N.T. Souter, W. Awad. Writing – original draft: M.N.T. Souter, S.B.G. Eckle. Writing – review & editing: M.N.T. Souter, W. Awad, H. Wang, J.Y.W. Mak, K. Kedzierska, A.J. Corbett, D.P. Fairlie, N.A. Gherardin, A.P. Uldrich, Z. Chen, J. Rossjohn, D.I. Godfrey, J. McCluskey, D.G. Pellicci, S.B.G. Eckle.

