


Microbial dysbiosis in the skin has been implicated in the pathogenesis of atopic dermatitis (AD); however, whether and how changes in the skin microbiome initiate skin inflammation, or vice versa, remains poorly understood. Here, we report that the levels of sebum and its microbial metabolite, propionate, were lower on the skin surface of AD patients compared with those of healthy individuals. Topical propionate application attenuated skin inflammation in mice with MC903-induced AD-like dermatitis by inhibiting IL-33 production in keratinocytes, an effect that was mediated through inhibition of HDAC and regulation of the AhR signaling pathway. Mice lacking sebum spontaneously developed AD-like dermatitis, which was improved by topical propionate application. A proof-of-concept clinical study further demonstrated the beneficial therapeutic effects of topical propionate application in AD patients. In summary, we have uncovered that the dysregulated sebum–microbial metabolite–IL-33 axis might play an initiating role in AD-related skin inflammation, thereby highlighting novel therapeutic strategies for the treatment of AD.

Introduction
Atopic dermatitis (AD) is a common inflammatory skin disease with a lifetime prevalence of ≤20% and manifested as intensive itch, eczematous dermatitis, and dry skin (Kennedy et al., 2018; Weidinger and Novak, 2016). Research to date has demonstrated that AD is a complex, multifactorial disease characterized by type 2 inflammatory cell infiltration, which is often induced by IL-33 and thymic stromal lymphopoietin (TSLP) produced by keratinocytes (Weidinger and Novak, 2016; Kennedy et al., 2018; Brunner et al., 2018; Werfel et al., 2016); however, the exact mechanism underlying the pathogenesis of AD, especially the initiation step, remains unclear. It is increasingly recognized that the skin microbiota are involved in the pathogenesis of AD. In AD patients, skin dysbiosis mainly manifests as decreased microbial diversity, increased abundance of the genus Staphylococcus, and decreased abundance of the genus Cutibacterium (formerly known as Propionibacterium; Paller et al., 2019; Li et al., 2019; Dainichi et al., 2018). Significant alterations in the metabolism of skin microbiota, such as attenuated tryptophan metabolism, are also observed in AD patients (Yu et al., 2019; Chng et al., 2016). Whether skin inflammation is induced by skin dysbiosis, or vice versa, has long been debated. Recent studies have shown that, in children with AD, skin microbiome dysregulation precedes the appearance of skin lesions (Kennedy et al., 2017; Meylan et al., 2017), suggesting that it is skin dysbiosis that induces skin inflammation and barrier damage in AD, and not the other way around. However, the initiating factors for skin dysbiosis and the subsequent interactions between the skin microbiota and the host in the pathogenesis of AD remain unclear.
The composition and function of the skin microbiome are primarily determined by the physical and chemical characteristics of the skin, in which the folliculosebaceous unit plays a central role (Chen et al., 2018; Oh et al., 2014; Kobayashi et al., 2019). The skin microbiome is heavily influenced by the function of sebaceous glands (Baurecht et al., 2018; Kobayashi et al., 2019). The abundance of lipophilic microbes, such as Corynebacteria and Cutibacteria, is significantly correlated with the level of skin sebum, especially fatty acids (Baurecht et al., 2018). Early studies reported that AD patients have fewer and smaller sebaceous glands (Shi et al., 2015) and lower levels of sebum production than healthy individuals. Moreover, these effects have been associated with decreased skin hydration (Firooz et al., 2007) and attenuated skin barrier function (Dahlhoff et al., 2016), suggesting a link between sebaceous gland dysfunction and the development of AD. Studies investigating the AD-associated skin microbiota have consistently demonstrated that, compared with healthy people, AD patients exhibit a markedly lower abundance of Cutibacterium species and significant alterations in lipid-related metabolism, including fatty acid metabolism and biosynthesis (Li et al., 2019). These observations suggest that the sebaceous gland–skin microbiota axis might play an unanticipated role in the pathogenesis of AD.
Abundant sebum production provides the skin microbiota with the substrates necessary for lipid metabolism, producing short-chain fatty acids (SCFAs; Chen et al., 2018; James et al., 2004). Studies have reported that SCFAs have contradictory roles in the regulation of skin homeostasis, both promoting keratinocyte inflammation through the regulation of histone deacetylase (HDAC) and attenuating skin inflammation in mouse models via the induction of regulatory T cells (Sanford et al., 2016; Schwarz et al., 2017; Keshari et al., 2019). Thus, the exact role played by SCFAs under both physiological and pathological conditions remains unclear. Additionally, no study to date has reported on the levels of SCFAs on the skin surface. In the present study, using liquid chromatography–tandem mass spectrometry (LC-MS/MS), we found that the level of propionate on the skin surface was relatively higher than that of other SCFAs and was significantly lower in AD patients than in healthy individuals, which was consistent with the observed decrease in sebum production in these patients. We further found that topical propionate application attenuated skin inflammation in mice with MC903-induced AD-like dermatitis by inhibiting IL-33 production in keratinocytes. These effects were achieved by regulating the aryl hydrocarbon receptor (AhR) signaling pathway via the inhibition of HDAC2 and HDAC3. Cidea KO mice that displayed decreased sebum production spontaneously developed AD-like dermatitis, which was ameliorated by topical propionate treatment. Moreover, in a proof-of-concept clinical trial, topical propionate administration alleviated skin inflammation in AD patients. Our results indicate that a sebum–microbial metabolite–IL-33 axis is involved in the pathogenesis of AD, possibly by playing an initiating role in the induction of skin inflammation.
Results
AD patients exhibit decreased production of sebum and its microbial metabolite, propionate
We first analyzed the sebum excretion rate (SER) in nonlesional skin of different body sites of AD patients and healthy individuals from different age groups using Sebutape patches. Associations between the SER and age were visualized using locally weighted scatterplot smoothing (LOESS) curves. The results showed that the SERs of AD patients were significantly lower than those of healthy individuals across all four sites assessed (Fig. 1, A–D). Detailed information regarding the differences in SERs between AD patients and healthy controls according to age is presented in Table 1. The SER was also assessed using the Delfin SebumScale in a separate cohort. The results were consistent with those obtained using Sebutape patches, i.e., we found that SERs were lower in lesional and nonlesional skin of the antecubital fossa (Af) and forehead of AD patients than in the corresponding normal skin of healthy controls (Fig. 1, E and F). Moreover, the SERs of AD patients were negatively correlated with transepidermal water loss (TEWL) and disease severity (scoring atopic dermatitis [SCORAD]) and positively correlated with epidermal hydration (Fig. 1, G–I).

The production of sebum and microbial metabolites is decreased in the skin of AD patients. (A–D) SERs (pixels/cm2/h) measured using a Sebutape patch in nonlesional skin of different body sites of AD patients (n = 95) and healthy controls (n = 64) from different age groups. The solid lines represent LOESS fit lines. Gray shading around each line denotes 95% confidence intervals. (E) SERs (AU/h) measured by Delfin SebumScale in lesional (AD-L) and nonlesional (AD-NL) skin of AD patients and healthy individuals at the Af (n = 40 per group). (F) SERs (AU/h) measured by Delfin SebumScale in the nonlesional skin of forehead of AD patients and healthy controls (n = 40 per group). (G–I) Correlations between SERs and TEWL, epidermal hydration, and SCORAD of AD patients. The solid lines represent linear regression fit lines. Gray shading around each line denotes 95% confidence intervals. (J) SCFA levels on the nonlesional skin of the Af and the back of AD patients and healthy controls as measured by LC–MS/MS (n = 30 per group). (K) Correlations between the propionate level in the skin of Af/back and SCORAD of AD patients. (L) Correlations between the propionate level in the skin of Af/back and pruritus scores of AD patients. Data are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test (E), unpaired t tests with Welch’s correction (F and J), and Spearman’s correlation test (G–I, K, and L). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The production of sebum and microbial metabolites is decreased in the skin of AD patients. (A–D) SERs (pixels/cm2/h) measured using a Sebutape patch in nonlesional skin of different body sites of AD patients (n = 95) and healthy controls (n = 64) from different age groups. The solid lines represent LOESS fit lines. Gray shading around each line denotes 95% confidence intervals. (E) SERs (AU/h) measured by Delfin SebumScale in lesional (AD-L) and nonlesional (AD-NL) skin of AD patients and healthy individuals at the Af (n = 40 per group). (F) SERs (AU/h) measured by Delfin SebumScale in the nonlesional skin of forehead of AD patients and healthy controls (n = 40 per group). (G–I) Correlations between SERs and TEWL, epidermal hydration, and SCORAD of AD patients. The solid lines represent linear regression fit lines. Gray shading around each line denotes 95% confidence intervals. (J) SCFA levels on the nonlesional skin of the Af and the back of AD patients and healthy controls as measured by LC–MS/MS (n = 30 per group). (K) Correlations between the propionate level in the skin of Af/back and SCORAD of AD patients. (L) Correlations between the propionate level in the skin of Af/back and pruritus scores of AD patients. Data are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test (E), unpaired t tests with Welch’s correction (F and J), and Spearman’s correlation test (G–I, K, and L). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
SERs in different body sites of AD patients and healthy controls from different age groups
| Age (yr) | Group | Forehead | Chest | Abdomen | Forearm | Total |
|---|---|---|---|---|---|---|
| 0–2 | AD (n = 31) | 372.42 ± 133.71 | 96.13 ± 24.72 | 113.48 ± 22.93 | 88.87 ± 18.20 | 670.90 ± 147.59 |
| Control (n = 23) | 1,344.52 ± 381.41 | 350.13 ± 111.83 | 158.13 ± 36.46 | 180.00 ± 30.17 | 2,032.78 ± 516.88 | |
| P value | 0.023* | 0.036* | 0.306 | 0.014* | 0.018* | |
| 2–8 | AD (n = 35) | 386.60 ± 206.03 | 75.57 ± 19.36 | 68.46 ± 15.19 | 65.86 ± 10.77 | 596.49 ± 215.02 |
| Control (n = 20) | 1,217.15 ± 565.01 | 90.55 ± 28.41 | 54.75 ± 6.45 | 135.70 ± 30.12 | 1,498.15 ± 569.01 | |
| P value | 0.180 | 0.666 | 0.411 | 0.039* | 0.151 | |
| 8–45 | AD (n = 29) | 20,666.59 ± 5,191.97 | 1,215.897 ± 314.13 | 206.7931 ± 44.89 | 104.069 ± 18.78 | 22,193.34 ± 5,309.12 |
| Control (n = 21) | 46,844.33 ± 9,408.71 | 4,883.952 ± 973.13 | 699.4286 ± 216.26 | 326.381 ± 86.95 | 52,754.1 ± 10,299.26 | |
| P value | 0.026* | 0.001** | 0.036* | 0.020* | 0.013* |
Data are expressed as means ± SEM. Statistical significance was analyzed by unpaired t test with Welch’s correction. *, P < 0.05; **, P < 0.01.
Next, we measured the levels of SCFAs, essential microbial metabolites of skin sebum (Chen et al., 2018; James et al., 2004), on the surface of nonlesional skin in the Af and back of AD patients and the corresponding normal skin of healthy controls using targeted metabolomics (Han et al., 2015). The results showed that propionate presented the highest level on the skin surface among the seven SCFAs analyzed. Additionally, the propionate level was significantly lower on the skin surface of AD patients than on that of healthy individuals in both the Af and back (Fig. 1 J), and the level of propionate was negatively correlated with the SCORAD and pruritus scores of the patients (Fig. 1, K and L). The level of caproate was also lower in the skin of AD patients, whereas that of isovalerate was higher (Fig. 1 J). However, there were no correlations between SCORAD or pruritus score and the level of isovalerate or caproate (data not shown). The metabolite isovalerate is usually associated with Staphylococcus spp. (Lam et al., 2018; Bos et al., 2013), suggesting that the higher level of this metabolite observed on the skin of AD patients might be due to the increased abundance of staphylococci (Yamazaki et al., 2017). These results were in line with those of our previous report, where we demonstrated a decreased abundance of lipophilic microbes, such as Cutibacterium species, and a significant alteration in skin microbiota-associated fatty acid metabolism and biosynthesis in AD patients (Li et al., 2019). These and our current findings indicated that a sebum–microbial metabolite axis might be involved in the pathogenesis of AD.
Topical propionate application attenuates MC903-induced AD-like dermatitis in mice by inhibiting IL-33 production
We next explored the role of propionate in AD, using an MC903-induced AD-like mouse model. Topical application with MC903 on the ears of BALB/c mice for 9 d resulted in apparent skin inflammation; however, when 1 mmol/liter propionate was applied 30 min after the MC903 application, considerably milder symptoms of skin inflammation were observed, as evidenced by the presence of fewer visible scales, less redness, and reduced ear thickness, compared with the AD model control (Fig. 2, A and B). H&E staining of ear sections on day 9 indicated decreased epidermal thickness and less inflammatory cell infiltration in the ears of AD mice administered propionate compared with that in ears of the AD model control (Fig. 2, C and D). The scratching frequency and level of total serum IgE were much lower in mice of the MC903 plus propionate group than those in the MC903 group (Fig. 2, E and F). We further found that the expression of various inflammatory cytokines, including IL-4, IL-5, IL-13, IL-6, IL-22, and IL-25, was significantly lower in the ears of AD mice treated with propionate compared with that in the ears of AD model control (Fig. 2 G). As the cytokine TSLP has been implicated as the initiating factor for the skin inflammation observed in MC903-induced AD, we analyzed its expression in lesional skin using both quantitative real-time PCR (RT-qPCR) and Western blotting. We found no significant differences in TSLP expression between the propionate-treated AD mice and the AD model control (Fig. 2, H and I), indicating that TSLP might not be involved in the attenuating effects of propionate in mice with MC903-induced dermatitis.

Topical propionate application attenuates MC903-induced AD-like dermatitis in mice by inhibiting IL-33 production. MC903 or MC903 plus propionate (Prop) was applied topically on the ears of BALB/c mice once a day for 9 d (n = 3–5 per group). (A) Representative gross appearance of the ears. (B) Dynamic changes in ear thickness on days 0, 3, 6, and 9. (C) H&E staining of ear sections. (D) Epidermal (EM) thickness of ear sections under high-power magnification. (E) Total serum IgE. (F) Scratching frequency. (G) mRNA expression of various cytokines in the ears of mice from each group. (H and I) Expression of TSLP mRNA and protein in the ears. (J–L) The expression of IL-33 in the ears of mice with MC903-induced AD-like dermatitis as determined by immunohistochemistry (J), RT-PCR (K), and Western blotting (L). IL-33FL, the full-length form of IL-33; IL-33cle, the cleaved form of IL-33. (M) Western blotting showing the protein expression of IL-33 in cultured JB6 cells treated with MC903 only or MC903 plus propionate. (N) The mRNA expression of IL-25 in cultured JB6 cells treated with MC903 only or MC903 plus propionate. Scale bar = 1 cm (A); 100 μm (C and J). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F2.
Topical propionate application attenuates MC903-induced AD-like dermatitis in mice by inhibiting IL-33 production. MC903 or MC903 plus propionate (Prop) was applied topically on the ears of BALB/c mice once a day for 9 d (n = 3–5 per group). (A) Representative gross appearance of the ears. (B) Dynamic changes in ear thickness on days 0, 3, 6, and 9. (C) H&E staining of ear sections. (D) Epidermal (EM) thickness of ear sections under high-power magnification. (E) Total serum IgE. (F) Scratching frequency. (G) mRNA expression of various cytokines in the ears of mice from each group. (H and I) Expression of TSLP mRNA and protein in the ears. (J–L) The expression of IL-33 in the ears of mice with MC903-induced AD-like dermatitis as determined by immunohistochemistry (J), RT-PCR (K), and Western blotting (L). IL-33FL, the full-length form of IL-33; IL-33cle, the cleaved form of IL-33. (M) Western blotting showing the protein expression of IL-33 in cultured JB6 cells treated with MC903 only or MC903 plus propionate. (N) The mRNA expression of IL-25 in cultured JB6 cells treated with MC903 only or MC903 plus propionate. Scale bar = 1 cm (A); 100 μm (C and J). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F2.
We then analyzed the expression of IL-33, as studies have shown that this cytokine is also required for the MC903-mediated induction of AD (Li et al., 2017; Imai et al., 2013; Molofsky et al., 2015). Immunohistochemical staining showed that IL-33 was mainly expressed in keratinocytes of the epidermis, and that IL-33 expression was markedly weaker in the propionate-treated AD mice than in the AD model control (Fig. 2 J). RT-qPCR results demonstrated that the expression level of IL-33 was significantly lower in the ears of AD mice administered propionate than in the AD model control; this result was confirmed by Western blotting (Fig. 2, K and L). We further found that propionate significantly inhibited the MC903-induced production of IL-33 in the mouse epidermal cell line JB6 (Fig. 2 M). As IL-25 from keratinocytes is also regarded as one of the initiating factors of AD inflammation, we examined the effect of propionate on IL-25 expression in JB6 cells and found that propionate had no effect on the MC903-induced production of IL-25 (Fig. 2 N), which suggested that the decreased expression of IL-25 in the ears of AD mice treated with propionate might be a downstream consequence of the overall reduced inflammation rather than a direct effect of propionate on keratinocytes.
Furthermore, we used IL-33 KO mice and ST2 KO mice to verify the mechanism for the antiinflammatory effect of propionate on AD mice. The results showed that AD-like inflammation induced by MC903 was significantly milder in ST2 KO mice than in WT BALB/c mice, while propionate failed to inhibit the MC903-induced AD-like dermatitis in ST2 KO mice (Fig. S1, A–D). When IL-33 KO mice were used, similar phenomena were observed. AD-like inflammation was significantly milder in IL-33 KO mice than in WT C57BL/6 mice, and propionate failed to inhibit MC903-induced AD in IL-33 KO mice (Fig. S1, E–H). These results demonstrated the critical role of IL-33 signaling via ST2 in MC903-induced AD-like dermatitis, and the absence of IL-33 or ST2 abolished the propionate-mediated attenuation in skin inflammation of the MC903-treated mice. These results suggested that the inhibitory effect of propionate on AD was mainly through down-regulation of IL-33 expression. We also assessed the possibility that propionate exerts antiinflammatory effects through skin-resident immune cells such as Langerhans cells (LCs) and γδT cells. We analyzed the number of LCs and γδT cells from WT mice and IL-33 KO mice using flow cytometry. The results showed that for WT mice, the increased numbers of both LCs and γδT cells in the MC903-induced AD model recovered after propionate application (Fig. S1, I and J), while in IL-33 KO mice, there was no difference between the MC903 group and the MC903 plus propionate group in the numbers of LCs and γδT cells (Fig. S1, K and L). These data suggested that the changes in the numbers of LCs and γδT cells in the WT mice group upon propionate application might be a downstream response to the changes of inflammation rather than the direct effect of propionate. Collectively, our results demonstrated that topical application of propionate attenuated MC903-induced AD-like dermatitis in mice, which might be achieved by inhibiting IL-33 expression in keratinocytes. The antiinflammatory effects of propionate were also observed in mouse models of imiquimod (IMQ)-induced psoriatic dermatitis (Fig. S2, A–D) and oxazolone (OXA)-induced contact hypersensitivity (Fig. S2, E–H).

The absence of IL-33 or ST2 abolishes the propionate-mediated attenuation in skin inflammation of the MC903-treated mice. (A–D) MC903 or MC903 plus propionate (Prop)/vehicle was applied topically on the ears of WT BALB/c mice and ST2 KO mice once daily for 9 d (n = 3 per group). Representative gross appearance (A), H&E staining of ear sections on day 9 (B), ear thickness (C), and epidermal (EM) thickness of ear sections under high-power magnification (D) of each mice group are shown. (E–L) MC903 or MC903 plus propionate/vehicle was applied topically on the ears of WT C57BL/6 mice and IL-33 KO mice once daily for 14 d (n = 3 per group). Ear thickness (E), epidermal thickness of ear sections under high-power magnification (F), representative gross appearance (G), and H&E staining of ear sections on day 14 (H) of each mice group are shown. Representative flow cytometry dot plots of LCs (I and K) and γδ T cells (J and L) in ears of each mice group are shown. Bar graphs on the right showing the percentage of LCs/γδ T cells within MHC II+ CD207+ gates (LCs) or CD3+ γδ TCR+ gates (γδ T cells). Scale bar = 1 cm (A and G); 100 μm (B and H). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The absence of IL-33 or ST2 abolishes the propionate-mediated attenuation in skin inflammation of the MC903-treated mice. (A–D) MC903 or MC903 plus propionate (Prop)/vehicle was applied topically on the ears of WT BALB/c mice and ST2 KO mice once daily for 9 d (n = 3 per group). Representative gross appearance (A), H&E staining of ear sections on day 9 (B), ear thickness (C), and epidermal (EM) thickness of ear sections under high-power magnification (D) of each mice group are shown. (E–L) MC903 or MC903 plus propionate/vehicle was applied topically on the ears of WT C57BL/6 mice and IL-33 KO mice once daily for 14 d (n = 3 per group). Ear thickness (E), epidermal thickness of ear sections under high-power magnification (F), representative gross appearance (G), and H&E staining of ear sections on day 14 (H) of each mice group are shown. Representative flow cytometry dot plots of LCs (I and K) and γδ T cells (J and L) in ears of each mice group are shown. Bar graphs on the right showing the percentage of LCs/γδ T cells within MHC II+ CD207+ gates (LCs) or CD3+ γδ TCR+ gates (γδ T cells). Scale bar = 1 cm (A and G); 100 μm (B and H). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.

Topical propionate treatment attenuates IMQ-, OXA-, or MC903-induced skin inflammation in mice. (A–D) IMQ-induced psoriatic dermatitis was produced in the ears of mice, following which the animals were topically treated with propionate once daily for 14 d (n = 3–5 per group). Gross appearance of the ears (A), ear thickness (B), H&E staining of ear sections (C), and the protein expression of IL-33 in lesional ears (D) are shown. (E–H) The mouse model of contact hypersensitivity was generated using OXA, after which propionate was topically applied during the sensitization and challenge stages (n = 3 per group). Gross appearance of the ears (E), ear thickness (F), H&E staining of ear sections (G), and the protein expression of IL-33 in lesional ears (H) are shown. (I and J) MC903 or MC903 plus propionate (Prop)/vehicle was applied topically on the ears of WT BALB/c mice once daily for 9 d (n = 4 per group). (I) Representative flow cytometry dot plots of Tregs in ears of each mice group gated on CD3+ CD4+ live cells. Bar graphs on the right showing the percentage of Tregs within CD3+ CD4+ Foxp3+ gates among CD4+ T cells. (J) The mRNA expression of Foxp3 and IL-10 in the ears of mice in each group. IL-33FL, the full-length form of IL-33; IL-33cle, the cleaved form of IL-33. Scale bar = 1 cm (A and E); 100 μm (C and G). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS2.
Topical propionate treatment attenuates IMQ-, OXA-, or MC903-induced skin inflammation in mice. (A–D) IMQ-induced psoriatic dermatitis was produced in the ears of mice, following which the animals were topically treated with propionate once daily for 14 d (n = 3–5 per group). Gross appearance of the ears (A), ear thickness (B), H&E staining of ear sections (C), and the protein expression of IL-33 in lesional ears (D) are shown. (E–H) The mouse model of contact hypersensitivity was generated using OXA, after which propionate was topically applied during the sensitization and challenge stages (n = 3 per group). Gross appearance of the ears (E), ear thickness (F), H&E staining of ear sections (G), and the protein expression of IL-33 in lesional ears (H) are shown. (I and J) MC903 or MC903 plus propionate (Prop)/vehicle was applied topically on the ears of WT BALB/c mice once daily for 9 d (n = 4 per group). (I) Representative flow cytometry dot plots of Tregs in ears of each mice group gated on CD3+ CD4+ live cells. Bar graphs on the right showing the percentage of Tregs within CD3+ CD4+ Foxp3+ gates among CD4+ T cells. (J) The mRNA expression of Foxp3 and IL-10 in the ears of mice in each group. IL-33FL, the full-length form of IL-33; IL-33cle, the cleaved form of IL-33. Scale bar = 1 cm (A and E); 100 μm (C and G). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS2.
As SCFAs have been described to be able to regulate intestinal homeostasis by inducing regulatory T cells (Tregs), we also examined whether propionate reduces AD-like inflammation by promoting Treg development in the skin. The flow cytometry results showed that the number of Tregs was increased in the lesional skin of mice in the MC903-treated group compared with the normal control (NC) group and had a tendency to recover in the MC903 plus propionate group (Fig. S2 I). RT-qPCR analysis showed that the expression of Foxp3 and IL-10 in mouse ears was increased in the MC903-treated group compared with the control group and returned to nearly normal levels in the MC903 plus propionate group (Fig. S2 J). These results suggested that topical application with propionate might not promote Treg development.
We also explored whether applying lipophilic microbes topically has effects similar to those of propionate. As Cutibacterium acnes mainly produce propionate in vitro when grown under anaerobic and lipid-rich conditions (Sanford et al., 2016), we assessed the effect of applying a mixture of C. acnes (1 × 105 CFU/ml) in the medium with or without 2% glycerol to mice with MC903-induced AD. The results showed that ear inflammation was significantly attenuated when the mixture of C. acnes and 2% glycerol was applied, as supported by fewer visible scales, less redness, and reduced ear thickness (Fig. S3, A and B). H&E staining of ear sections indicated that there were reduced epidermal thickness and less inflammatory cell infiltration in the ears of AD mice treated with the mixture of C. acnes and 2% glycerol compared with that in AD mice treated with C. acnes alone or 2% glycerol alone (Fig. S3, C and D). These data indicate that when in a lipid-rich condition, C. acnes could attenuate the symptoms of MC903-induced AD in mice.

Lipophilic microbes in lipid-rich conditions attenuate MC903-induced AD-like dermatitis in mice. C. acnes (1 × 105 CFUs/ml) mixed with 2% glycerol (G) were topically applied onto MC903-treated mouse ears once daily for 9 consecutive days (n = 3 per group). (A) Gross appearance of the ears. (B) Ear thickness of mice. (C) H&E staining of ear sections. Scale bar = 100 μm. (D) Epidermal (EM) thickness of ear sections. Scale bar = 1 cm (A); 100 μm (C). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Lipophilic microbes in lipid-rich conditions attenuate MC903-induced AD-like dermatitis in mice. C. acnes (1 × 105 CFUs/ml) mixed with 2% glycerol (G) were topically applied onto MC903-treated mouse ears once daily for 9 consecutive days (n = 3 per group). (A) Gross appearance of the ears. (B) Ear thickness of mice. (C) H&E staining of ear sections. Scale bar = 100 μm. (D) Epidermal (EM) thickness of ear sections. Scale bar = 1 cm (A); 100 μm (C). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Propionate inhibits IL-33 production in keratinocytes
Next, we sought to determine the effects of propionate on the production of IL-33 in human primary keratinocytes in culture. The results showed that propionate exerted a dose-dependent inhibitory effect on IL-33 production in keratinocytes at the mRNA level (Fig. 3 A). When keratinocytes were stimulated for 1 h with proinflammatory cytokines (IL-4 and IL-1α) and then cocultured with different concentrations of propionate for 24 h, the increase in IL-33 expression was abrogated (Fig. 3 B). The Western blotting results were in line with those of RT-qPCR (Fig. 3, C and D). Another well-studied SCFA, butyrate, also induced a dose-dependent effect on IL-33 production in keratinocytes under both steady-state and T helper cell type 2 (Th2)–mediated inflammatory conditions (Fig. S4, A–D), and the anti-inflammatory effect of butyrate was further verified in the AD mouse model induced by MC903 (Fig. S4, E–H). However, no significant changes were detected in the production of other AD-related inflammatory cytokines (IL-6, IL-8, IL-25, or TSLP) in keratinocytes following treatment with various concentrations of propionate either in the steady state or under Th2-mediated inflammatory conditions (Fig. 3, E and F). We also analyzed the effect of propionate on IL-33 production under conditions of TLR-mediated inflammation, with the results showing that propionate administration markedly abrogated the LPS (a ligand of TLR4)- or lipoteichoic acid (LTA, a ligand of TLR2)–induced increase in IL-33 expression in keratinocytes (data not shown). Collectively, these results demonstrated that propionate specifically inhibits the expression of IL-33 in human primary keratinocytes under both steady state and inflammatory conditions.
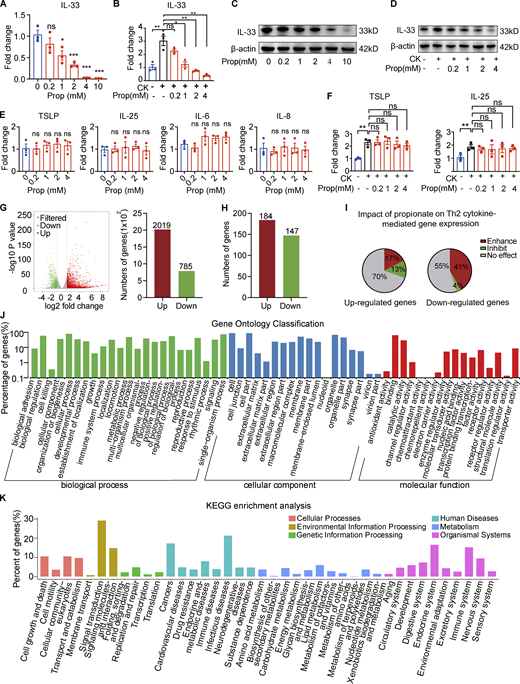
Propionate inhibits IL-33 production in keratinocytes. (A) RT-qPCR showing the expression of IL-33 mRNA in cultured human primary keratinocytes incubated with different concentrations of propionate for 24 h (n = 3 per group). (B) RT-qPCR showing the expression of IL-33 mRNA in cultured human primary keratinocytes stimulated with IL-4 (100 ng/ml) and IL-1α (10 ng/ml) for 1 h and then cocultured with different concentrations of propionate for 24 h (n = 3 per group). CK (cytokine) represents 100 ng/ml IL-4 + 10 ng/ml IL-1α. (C) Western blotting showing the protein expression of IL-33 in cultured human primary keratinocytes treated with different concentrations of propionate. (D) Western blotting showing the protein expression of IL-33 in keratinocytes cultured under Th2 inflammatory conditions and treated with different concentrations of propionate. (E) RT-qPCR showing the mRNA expression of inflammatory cytokines in human primary keratinocytes incubated with different concentrations of propionate (n = 3 per group). (F) RT-qPCR showing the mRNA expression of inflammatory cytokines in keratinocytes cultured under Th2 inflammatory conditions and treated with different concentrations of propionate (n = 3 per group). (G) Volcano plots and histogram showing the DEGs (|fold-change| >2 versus control, P < 0.05) in keratinocytes treated with propionate. (H) DEGs (|fold-change| >2 versus control, P < 0.05) in keratinocytes stimulated with Th2 cytokines. (I) The effects of propionate treatment on gene sets modulated by Th2 cytokines (enhancement: fold-change >1.5 with propionate + Th2 cytokines versus vehicle + Th2 cytokines; inhibition: fold-change <−1.5 with propionate + Th2 cytokines versus vehicle + Th2 cytokines; no effect: −1.5< fold-change <1.5). (J and K) Gene Ontology analysis and KEGG enrichment analysis of the Th2 cytokine-induced genes in keratinocytes after propionate treatment. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F3.
Propionate inhibits IL-33 production in keratinocytes. (A) RT-qPCR showing the expression of IL-33 mRNA in cultured human primary keratinocytes incubated with different concentrations of propionate for 24 h (n = 3 per group). (B) RT-qPCR showing the expression of IL-33 mRNA in cultured human primary keratinocytes stimulated with IL-4 (100 ng/ml) and IL-1α (10 ng/ml) for 1 h and then cocultured with different concentrations of propionate for 24 h (n = 3 per group). CK (cytokine) represents 100 ng/ml IL-4 + 10 ng/ml IL-1α. (C) Western blotting showing the protein expression of IL-33 in cultured human primary keratinocytes treated with different concentrations of propionate. (D) Western blotting showing the protein expression of IL-33 in keratinocytes cultured under Th2 inflammatory conditions and treated with different concentrations of propionate. (E) RT-qPCR showing the mRNA expression of inflammatory cytokines in human primary keratinocytes incubated with different concentrations of propionate (n = 3 per group). (F) RT-qPCR showing the mRNA expression of inflammatory cytokines in keratinocytes cultured under Th2 inflammatory conditions and treated with different concentrations of propionate (n = 3 per group). (G) Volcano plots and histogram showing the DEGs (|fold-change| >2 versus control, P < 0.05) in keratinocytes treated with propionate. (H) DEGs (|fold-change| >2 versus control, P < 0.05) in keratinocytes stimulated with Th2 cytokines. (I) The effects of propionate treatment on gene sets modulated by Th2 cytokines (enhancement: fold-change >1.5 with propionate + Th2 cytokines versus vehicle + Th2 cytokines; inhibition: fold-change <−1.5 with propionate + Th2 cytokines versus vehicle + Th2 cytokines; no effect: −1.5< fold-change <1.5). (J and K) Gene Ontology analysis and KEGG enrichment analysis of the Th2 cytokine-induced genes in keratinocytes after propionate treatment. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F3.

Butyrate suppresses IL-33 expression in keratinocytes and inhibits skin inflammation of AD mouse model. (A–D) IL-33 mRNA and protein expression in keratinocytes treated with different concentrations of butyrate (0.2–10 mM) in standard culture medium or supplemented with IL-4 and IL-1α for 24 h (n = 3 per group). (E–H) MC903 or MC903 plus butyrate (1 mM)/vehicle was applied topically on the ears of BALB/c mice once daily for 9 d (n = 5 per group). (E) Representative gross appearance of the ears. (F) Ear thickness of the mice in each group. (G) H&E staining of ear sections. (H) Epidermal (EM) thickness of ear sections under high-power magnification. (I–K)Cidea KO mice were topically applied with butyrate (1 mM) on the lesional skin twice daily for 21 d (n = 4 per group). (I) Gross appearance of skin lesions in Cidea KO mice before and after topical butyrate treatment. The red box indicates the skin lesions treated with butyrate. (J) Severity scores of skin lesions in Cidea KO mice before and after butyrate treatment. (K) H&E staining of skin samples from WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with butyrate. Scale bar = 1 cm (E and I); 100 μm (G and K). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test (A and B) and paired t test (J). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS4.
Butyrate suppresses IL-33 expression in keratinocytes and inhibits skin inflammation of AD mouse model. (A–D) IL-33 mRNA and protein expression in keratinocytes treated with different concentrations of butyrate (0.2–10 mM) in standard culture medium or supplemented with IL-4 and IL-1α for 24 h (n = 3 per group). (E–H) MC903 or MC903 plus butyrate (1 mM)/vehicle was applied topically on the ears of BALB/c mice once daily for 9 d (n = 5 per group). (E) Representative gross appearance of the ears. (F) Ear thickness of the mice in each group. (G) H&E staining of ear sections. (H) Epidermal (EM) thickness of ear sections under high-power magnification. (I–K)Cidea KO mice were topically applied with butyrate (1 mM) on the lesional skin twice daily for 21 d (n = 4 per group). (I) Gross appearance of skin lesions in Cidea KO mice before and after topical butyrate treatment. The red box indicates the skin lesions treated with butyrate. (J) Severity scores of skin lesions in Cidea KO mice before and after butyrate treatment. (K) H&E staining of skin samples from WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with butyrate. Scale bar = 1 cm (E and I); 100 μm (G and K). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test (A and B) and paired t test (J). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS4.
We also performed RNA sequencing (RNA-seq) analysis to comprehensively analyze the effects of propionate on keratinocytes. Using a fold-change cutoff of 2 compared with controls, we identified a total of 2,804 genes that were differentially expressed in keratinocytes after propionate treatment (785 downregulated and 2,019 upregulated; Fig. 3 G). Meanwhile, under Th2-mediated inflammatory conditions, we identified 184 genes that were upregulated and 147 downregulated (Fig. 3 H), almost half of which were further influenced by propionate administration (Fig. 3 I). Among the differentially expressed genes (DEGs), IL-33 was included in both the steady-state and the Th2-mediated inflammatory conditions. The Gene Ontology and KEGG enrichment analysis of these DEGs showed that propionate participated in environmental information processing, cell metabolism, and genetic information processing of keratinocytes (Fig. 3, J and K).
Propionate inhibits HDAC2 and HDAC3 in keratinocytes
As SCFAs have been shown to inhibit HDAC in immune cells such as macrophages and dendritic cells, thereby suppressing inflammatory cytokine production (Chang et al., 2014; Wang et al., 2008; Vinolo et al., 2011), we next explored whether the regulatory effects of propionate on IL-33 production in keratinocytes were mediated through HDAC inhibition. The results demonstrated that propionate exerted a dose-dependent inhibitory effect on HDAC enzyme activity (Fig. 4 A). Propionate increased the global levels of histone H3K9 and H3K27 acetylation, marks that are commonly associated with actively transcribed regions of the genome (Fig. 4 B). Furthermore, the expression level of IL-33 was decreased, and the levels of histone acetylation were increased in keratinocytes treated with trichostatin A (TSA), a commonly used broad-spectrum HDAC inhibitor (Fig. 4 C).

Propionate suppresses IL-33 expression in human primary keratinocytes through inhibiting HDAC2 and HDAC3. (A) HDAC enzyme activity in keratinocytes treated with different propionate (Prop) concentrations (n = 3 per group). (B) Western blotting showing histone acetylation in keratinocytes after treatment with different concentrations of propionate. (C) Western blotting of IL-33 expression and histone acetylation in keratinocytes treated with the HDAC inhibitor TSA. (D) The relative expression level of 11 HDACs in keratinocytes normalized to GAPDH as determined by RT-qPCR (n = 3 per group). (E–H) Expression of HDAC2/3 and IL-33 at the mRNA or protein level in keratinocytes after siRNA-mediated HDAC2/3 knockdown. (I–L) RT-qPCR results of the expression of IL-33 mRNA in keratinocytes treated with siRNA targeting HDAC1, HDAC7, HDAC8, or HDAC9. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test (A) and unpaired t tests (E, G, and I–L; n = 3 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F4.
Propionate suppresses IL-33 expression in human primary keratinocytes through inhibiting HDAC2 and HDAC3. (A) HDAC enzyme activity in keratinocytes treated with different propionate (Prop) concentrations (n = 3 per group). (B) Western blotting showing histone acetylation in keratinocytes after treatment with different concentrations of propionate. (C) Western blotting of IL-33 expression and histone acetylation in keratinocytes treated with the HDAC inhibitor TSA. (D) The relative expression level of 11 HDACs in keratinocytes normalized to GAPDH as determined by RT-qPCR (n = 3 per group). (E–H) Expression of HDAC2/3 and IL-33 at the mRNA or protein level in keratinocytes after siRNA-mediated HDAC2/3 knockdown. (I–L) RT-qPCR results of the expression of IL-33 mRNA in keratinocytes treated with siRNA targeting HDAC1, HDAC7, HDAC8, or HDAC9. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Dunnett’s test (A) and unpaired t tests (E, G, and I–L; n = 3 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F4.
We subsequently sought to identify which HDAC specifically regulates the expression of the IL33 gene in keratinocytes. For this, we first measured the relative transcript abundance of 11 HDACs in keratinocytes and found that the expression levels of HDAC1, HDAC2, HDAC3, and HDAC7 were relatively higher than those of the other seven HDACs (Fig. 4 D). When HDAC2 or HDAC3, but not HDAC1 or HDAC7, was knocked down in keratinocytes using siRNA, the IL-33 mRNA and protein levels were significantly decreased (Fig. 4, E–J). We also knocked down the expression of the genes encoding HDAC8 and HDAC9, which have been reported to promote the expression of inflammatory cytokines in keratinocytes (Sanford et al., 2016). We found that HDAC8 or HDAC9 depletion did not affect IL-33 expression (Fig. 4, K and L). Collectively, our results demonstrated that propionate inhibited IL-33 production in keratinocytes via the inhibition of HDAC2 and HDAC3.
Propionate downregulates IL-33 via the regulation of AhR
We next sought to identify the mechanism involved in the HDAC inhibition–induced downregulation of IL-33. We found that propionate treatment decreased the recruitment level of RNA polymerase II (Pol II) as well as that of its serine 5-phosphorylated (S5P) form at the IL-33 promoter in keratinocytes (Fig. 5 A). As both Pol II and S5P forms are involved in transcription initiation (Chang et al., 2014), these results indicated that propionate regulates IL-33 expression at the transcriptional level. Given that histone acetylation is associated with active gene transcription, and that chromatin immunoprecipitation (ChIP) results showed that propionate did not affect the level of H3K9 acetylation (H3K9ac) at the IL-33 promoter (Fig. 5 B), we speculated that the inhibition of HDAC activity might not directly regulate IL-33 gene transcription upon propionate treatment.

Propionate induces an increase in AhR expression and facilitates its recruitment to the IL-33 promoter. (A) A ChIP assay was used to assess the recruitment levels of polymerase II (Pol II) and its serine 5-phosphorylated form (Pol II S5P) to the IL-33 promoter in keratinocytes treated with propionate (Prop; n = 6 per group). (B) The levels of histone 3 lysine 9 acetylation (H3K9Ac) in the IL-33 promoter in keratinocytes treated with propionate as determined using a ChIP assay (n = 6 per group). (C) Western blotting of the protein expression of AhR in keratinocytes treated with propionate. (D) RT-qPCR analysis of the mRNA expression of AhR, AhRR, and CYP1A1 in keratinocytes treated with propionate or TSA (n = 3 per group). (E and F) RT-qPCR analysis of AhR mRNA expression levels in keratinocytes treated with siRNA specific for HDAC2 or HDAC3 (n = 3 per group). (G) A ChIP assay was used to measure the levels of H3K9Ac at the AhR promoter in keratinocytes treated with propionate (n = 6 per group). (H) Representative confocal images showing the localization of AhR in keratinocytes treated with propionate for different durations. (I) Western blotting analysis of the protein expression of IL-33 in keratinocytes treated with propionate or 6-formylindolo[3,2-b] carbazole (FICZ). (J) Western blotting analysis of the protein expression of IL-33 in keratinocytes treated with AhR-specific siRNA. (K) RT-qPCR analysis of IL-33 mRNA expression in keratinocytes treated with or without propionate when AhR was specifically silenced (n = 3 per group). (L–P)AhR KO mice with MC903-induced AD-like dermatitis were topically treated with propionate (n = 3 per group). Gross appearance (L), H&E staining of ear sections on day 14 (M), dynamic changes in ear thickness (N), Western blotting for IL-33 protein expression (O), and immunohistochemical staining of IL-33 in ear sections (P) are shown. (Q) The levels of AhR recruitment to the IL-33 promoter in keratinocytes following propionate treatment were assessed by ChIP assay (n = 6 per group). (R) RT-qPCR analysis of AhR and IL-33 mRNA levels in keratinocytes treated with propionate at different times (n = 3 per group). (S) A schematic figure showing the pathway through which propionate regulates IL-33. Pathway 1 (the red lines) reflects that propionate increases AhR expression by inhibiting HDAC2/3. Pathway 2 (the blue line) reflects that propionate induces AhR nuclear translocation. The black lines reflect that increased recruitment of AhR on the IL-33 promoter leads to IL-33 transcriptional repression. The schematic figure was created with BioRender.com. Scale bar = 20 μm (H); 1 cm (L); 100 μm (M and P). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed using unpaired t tests (A, B, E–G, and Q) and one-way ANOVA followed by Dunnett’s test (D, K, and R). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
Propionate induces an increase in AhR expression and facilitates its recruitment to the IL-33 promoter. (A) A ChIP assay was used to assess the recruitment levels of polymerase II (Pol II) and its serine 5-phosphorylated form (Pol II S5P) to the IL-33 promoter in keratinocytes treated with propionate (Prop; n = 6 per group). (B) The levels of histone 3 lysine 9 acetylation (H3K9Ac) in the IL-33 promoter in keratinocytes treated with propionate as determined using a ChIP assay (n = 6 per group). (C) Western blotting of the protein expression of AhR in keratinocytes treated with propionate. (D) RT-qPCR analysis of the mRNA expression of AhR, AhRR, and CYP1A1 in keratinocytes treated with propionate or TSA (n = 3 per group). (E and F) RT-qPCR analysis of AhR mRNA expression levels in keratinocytes treated with siRNA specific for HDAC2 or HDAC3 (n = 3 per group). (G) A ChIP assay was used to measure the levels of H3K9Ac at the AhR promoter in keratinocytes treated with propionate (n = 6 per group). (H) Representative confocal images showing the localization of AhR in keratinocytes treated with propionate for different durations. (I) Western blotting analysis of the protein expression of IL-33 in keratinocytes treated with propionate or 6-formylindolo[3,2-b] carbazole (FICZ). (J) Western blotting analysis of the protein expression of IL-33 in keratinocytes treated with AhR-specific siRNA. (K) RT-qPCR analysis of IL-33 mRNA expression in keratinocytes treated with or without propionate when AhR was specifically silenced (n = 3 per group). (L–P)AhR KO mice with MC903-induced AD-like dermatitis were topically treated with propionate (n = 3 per group). Gross appearance (L), H&E staining of ear sections on day 14 (M), dynamic changes in ear thickness (N), Western blotting for IL-33 protein expression (O), and immunohistochemical staining of IL-33 in ear sections (P) are shown. (Q) The levels of AhR recruitment to the IL-33 promoter in keratinocytes following propionate treatment were assessed by ChIP assay (n = 6 per group). (R) RT-qPCR analysis of AhR and IL-33 mRNA levels in keratinocytes treated with propionate at different times (n = 3 per group). (S) A schematic figure showing the pathway through which propionate regulates IL-33. Pathway 1 (the red lines) reflects that propionate increases AhR expression by inhibiting HDAC2/3. Pathway 2 (the blue line) reflects that propionate induces AhR nuclear translocation. The black lines reflect that increased recruitment of AhR on the IL-33 promoter leads to IL-33 transcriptional repression. The schematic figure was created with BioRender.com. Scale bar = 20 μm (H); 1 cm (L); 100 μm (M and P). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed using unpaired t tests (A, B, E–G, and Q) and one-way ANOVA followed by Dunnett’s test (D, K, and R). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
Analysis of the intersection between transcription factors predicted to bind the IL-33 promoter and the DEGs from the RNA-seq analysis identified AhR (Fig. S5, A and B), which was selected for further study. As there are functional dioxin response elements for AhR binding located in the IL-33 promoter region (Ishihara et al., 2019) and SCFAs can reportedly enhance AhR signaling (Yang et al., 2020; Jin et al., 2017; Marinelli et al., 2019), we hypothesized that the effects on IL-33 expression resulting from propionate-mediated HDAC inhibition might be exerted through enhancing AhR responsiveness. Consistent with previously reported results, we found that propionate promoted the protein expression of AhR (Fig. 5 C). We also found that TSA promoted the expression of the AhR gene, as well as that of the AhR-regulated genes, CYP1A1 and AhRR (Fig. 5 D). Furthermore, AhR expression was significantly upregulated when either HDAC2 or HDAC3 was specifically silenced (Fig. 5, E and F). ChIP assay results further indicated that the level of H3K9ac at the AhR promoter was significantly increased after propionate treatment (Fig. 5 G). These results demonstrated that the inhibitory effect of propionate on HDAC2/3 was related to an increase in AhR expression. Fluorescence confocal microscopic analysis further showed that propionate treatment also promoted the translocation of AhR from the cytoplasm to the nucleus (Fig. 5 H), which was blocked by CH-223191, a well-characterized antagonist of the AhR-ligand binding (Fig. S5 C). Collectively, our data indicated that propionate could increase AhR expression by inhibiting HDAC2/3 and induce AhR nuclear translocation.
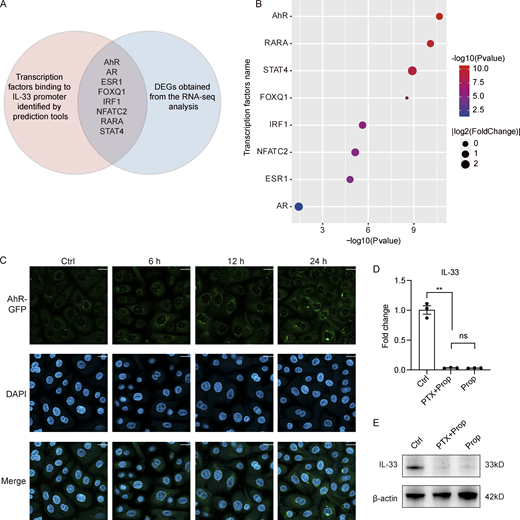
Mechanism for the inhibitory effect of propionate on IL-33 expression. (A and B) Analysis of potential transcription factors binding to IL-33 promoter. (A) The Venn diagram of intersection between transcription factors predicted to bind IL-33 promoter and DEGs obtained from the RNA-seq analysis. The red circle represents transcription factors predicted by PROMO database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) and further verified by the online software LASAGNA-search 2.0 (https://biogrid-lasagna.engr.uconn.edu/lasagna_search/). The blue circle represents DEGs (|fold-change| >1.5 versus control, P < 0.05) obtained from the RNA-seq analysis. The intersection lists transcription factors that exist in both circles. (B) The bubble chart showing P values and fold-change of the listed transcription factors. (C) Representative confocal images showing the localization of AhR in keratinocytes treated with CH-223191 for 1 h before stimulation with propionate (4 mM) for different times. Scale bar = 20 μm. (D and E) Keratinocytes were treated with PTX (0.2 μg/ml) for 1 h before stimulation with propionate (Prop; 4 mM) for 24 h. Results of RT-qPCR analysis (D) and Western blotting analysis (E) for the expression of IL-33 in keratinocytes are shown. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. **, P < 0.01. Source data are available for this figure: SourceData FS5.
Mechanism for the inhibitory effect of propionate on IL-33 expression. (A and B) Analysis of potential transcription factors binding to IL-33 promoter. (A) The Venn diagram of intersection between transcription factors predicted to bind IL-33 promoter and DEGs obtained from the RNA-seq analysis. The red circle represents transcription factors predicted by PROMO database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) and further verified by the online software LASAGNA-search 2.0 (https://biogrid-lasagna.engr.uconn.edu/lasagna_search/). The blue circle represents DEGs (|fold-change| >1.5 versus control, P < 0.05) obtained from the RNA-seq analysis. The intersection lists transcription factors that exist in both circles. (B) The bubble chart showing P values and fold-change of the listed transcription factors. (C) Representative confocal images showing the localization of AhR in keratinocytes treated with CH-223191 for 1 h before stimulation with propionate (4 mM) for different times. Scale bar = 20 μm. (D and E) Keratinocytes were treated with PTX (0.2 μg/ml) for 1 h before stimulation with propionate (Prop; 4 mM) for 24 h. Results of RT-qPCR analysis (D) and Western blotting analysis (E) for the expression of IL-33 in keratinocytes are shown. Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey’s test. **, P < 0.01. Source data are available for this figure: SourceData FS5.
We next investigated whether the inhibitory effect of propionate on IL-33 expression is dependent on AhR activation. We found that an AhR agonist, 6-formylindolo[3,2-b] carbazole, exerted an inhibitory effect similar to that of propionate on IL-33 expression in keratinocytes (Fig. 5 I), while the expression of IL-33 in keratinocytes was significantly increased when AhR was knocked down (Fig. 5 J). Moreover, the inhibitory effect of propionate on IL-33 expression in keratinocytes was abrogated when AhR was knocked down (Fig. 5 K). Additionally, no differences in gross appearance (Fig. 5 L), inflammatory cell infiltration (Fig. 5 M), and ear thickness (Fig. 5 N) were detected between AhR KO mice treated with MC903 and those treated with MC903 plus propionate. Propionate treatment did not suppress the increased expression of IL-33 in AhR KO mice treated with MC903 (Fig. 5, O and P). These results indicated that the absence of AhR abolished the propionate-mediated attenuation of skin inflammation in MC903-treated mice.
Furthermore, ChIP assay results showed that the binding of AhR to the IL-33 promoter in keratinocytes was significantly increased with propionate treatment (Fig. 5 Q), while changes in AhR expression preceded that of IL-33 in propionate-treated keratinocytes (Fig. 5 R). Collectively, these data revealed that the effect of propionate on IL-33 production was achieved via two pathways of regulation on AhR. A schematic figure showing the regulation of IL-33 by propionate was shown in Fig. 5 S.
Previous studies have shown that several membrane-bound G protein–coupled receptors (GPCRs) in the gut act as receptors for SCFAs and have attributed some of SCFAs’ effects to the activation of these GPCRs (Brown et al., 2003; Kim et al., 2013; Singh et al., 2014). We incubated keratinocytes with the Gαi-subunit inhibitor, Pertussis toxin (PTX, 0.2 μg/ml), for 1 h prior to the stimulation with propionate for 24 h. Both the results of qRT-PCR and Western blotting showed that PTX failed to abrogate the inhibitory effect of propionate on the expression of IL-33 in keratinocytes (Fig. S5, D and E). Additionally, Sanford et al. have shown that the expression levels of GPR41, GPR43, and GPR109a were extremely low in cultured epidermal keratinocytes (Sanford et al., 2016). Collectively, these data suggested that GPCRs might not be involved in the inhibition of IL-33 expression by propionate.
Sebum-deficient mice spontaneously develop AD-like dermatitis
Cidea is highly expressed in sebaceous glands (Wang et al., 2012) and is a crucial regulator of lipid storage and sebum lipid secretion in mammals, including humans (Zhang et al., 2014). We have previously shown that sebum levels on the skin surface of Cidea KO mice are markedly lower than those of WT controls, as are the contents of the constituents of sebum lipids, including triglycerides, wax diesters, diglycerides, phospholipids, and phosphatidylcholine (Zhang et al., 2014). In this study, we found that Cidea KO mice spontaneously developed AD-like dermatitis characterized by dry skin, hair loss, skin redness, scales, and scratches (Fig. 6 A). These mice also demonstrated significantly higher TEWL, lower levels of epidermal hydration, higher serum concentrations of total IgE, and higher scratching frequency than WT controls (Fig. 6, B–E).

Propionate attenuates spontaneous AD-like dermatitis in sebum-deficient mice. (A) Gross appearance of skin lesions in Cidea KO and WT mice. The line down the middle of this panel is used to divide the two types of mice. (B–E) TEWL (B), epidermal hydration (C), total serum IgE (D), and scratching frequency (E) in Cidea KO and WT mice (n = 8 per group). (F) SCFA levels on the skin surface of Cidea KO and WT mice (n = 6 per group). (G) Gross appearance of skin lesions in Cidea KO mice before and after topical propionate treatment. The red box indicates the skin lesions treated with propionate. (H) The severity scores of skin lesions in Cidea KO mice before and after propionate treatment (n = 4). (I) H&E staining of skin samples from WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with propionate. (J) Western blotting showing the protein expression of IL-33 in skin samples of WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with propionate (Prop). Scale bar = 1 cm (A and G); 100 μm (I). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by unpaired t test with Welch’s correction (B–F) and paired t test (H). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F6.
Propionate attenuates spontaneous AD-like dermatitis in sebum-deficient mice. (A) Gross appearance of skin lesions in Cidea KO and WT mice. The line down the middle of this panel is used to divide the two types of mice. (B–E) TEWL (B), epidermal hydration (C), total serum IgE (D), and scratching frequency (E) in Cidea KO and WT mice (n = 8 per group). (F) SCFA levels on the skin surface of Cidea KO and WT mice (n = 6 per group). (G) Gross appearance of skin lesions in Cidea KO mice before and after topical propionate treatment. The red box indicates the skin lesions treated with propionate. (H) The severity scores of skin lesions in Cidea KO mice before and after propionate treatment (n = 4). (I) H&E staining of skin samples from WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with propionate. (J) Western blotting showing the protein expression of IL-33 in skin samples of WT mice, lesional skin of Cidea KO mice, and lesional skin of Cidea KO mice treated with propionate (Prop). Scale bar = 1 cm (A and G); 100 μm (I). Data are representative of three independent experiments and are expressed as means ± SEM. Statistical significance was analyzed by unpaired t test with Welch’s correction (B–F) and paired t test (H). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F6.
We further assessed SCFA levels on the skin surface of Cidea KO mice and found that the acetate, propionate, butyrate, isovalerate, and valerate contents were significantly lower in Cidea KO mice than in WT controls (Fig. 6 F). However, when we applied propionate topically on the lesional skin of Cidea KO mice twice daily, we found that dermatitis symptoms in the treated areas were alleviated after 21 d of application (Fig. 6, G and H). The curative effects of propionate were further confirmed by H&E staining of sections of propionate-treated lesional skin obtained from Cidea KO mice, as evidenced by a reduction in inflammatory cell infiltration (Fig. 6 I). Propionate ameliorated the increased level of IL-33 expression in the lesional skin of Cidea KO mice (Fig. 6 J). Butyrate also exhibited an inhibitory effect on the spontaneous skin inflammation of Cidea KO mice (Fig. S4, I–K). Combined, these results demonstrated that sebum from sebaceous glands might play a key role in the pathogenesis of AD through the microbial metabolites.
Topical propionate application ameliorates skin lesions in AD patients
To explore the effect of propionate on patients with AD, we enrolled 11 patients with mild to moderate AD and topically applied a cream containing 0.05% propionate or vehicle onto their skin lesions of symmetrical parts twice daily. After 2 wk of the treatment, dermatitis of the propionate-treated regions in the AD patients exhibited a significant improvement compared with the vehicle-treated regions (Fig. 7 A). The enrolled AD patients presented decreased skin symptom intensity and less subjective pruritus of the propionate-treated regions measured by SCORAD criteria (Fig. 7, B and C). The regional SCORAD (sum of local intensity and pruritus scores) after topical propionate treatment was also decreased (Fig. 7 D). The regional skin of AD patients with topical propionate exhibited decreased TEWL and increased skin hydration after 2 wk of treatment (Fig. 7, E and F). These results demonstrated that topical propionate application could alleviate AD symptoms in patients, suggesting a new strategy for AD treatment.

Topical propionate application ameliorates the symptoms of AD patients. (A) Representative photographs of symmetrical lesions in AD patients at enrollment (week 0) and week 2 after topical propionate or vehicle application. Scale bar = 1 cm. (B–D) Before- and after-treatment scores for skin symptom intensity (B), subjective pruritus (C), and treated region-specific SCORAD (D) of the propionate- or vehicle-treated region as measured by SCORAD protocol (n = 11). Treated region-specific SCORAD score is the sum of skin symptom intensity and pruritus scores. (E and F) Before- and after-treatment levels of TEWL (E) and skin hydration (F) of the propionate- or vehicle-treated region in enrolled AD patients (n = 6). Statistical significance was analyzed using paired t test. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Topical propionate application ameliorates the symptoms of AD patients. (A) Representative photographs of symmetrical lesions in AD patients at enrollment (week 0) and week 2 after topical propionate or vehicle application. Scale bar = 1 cm. (B–D) Before- and after-treatment scores for skin symptom intensity (B), subjective pruritus (C), and treated region-specific SCORAD (D) of the propionate- or vehicle-treated region as measured by SCORAD protocol (n = 11). Treated region-specific SCORAD score is the sum of skin symptom intensity and pruritus scores. (E and F) Before- and after-treatment levels of TEWL (E) and skin hydration (F) of the propionate- or vehicle-treated region in enrolled AD patients (n = 6). Statistical significance was analyzed using paired t test. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Discussion
The interaction between the skin microbiota and the host is crucial for the maintenance of skin homeostasis (Rooks and Garrett, 2016; Shapiro et al., 2014; Flowers and Grice, 2020). Various studies have reported that skin dysbiosis is involved in the pathogenesis of AD (Kennedy et al., 2018; Brunner et al., 2018; Weidinger and Novak, 2016); however, whether and how changes in the skin microbiome initiate skin inflammation, or whether it is skin inflammation that promotes changes in the composition of the skin microbiome, remains poorly understood. In the present study, we focused on the significance of decreased sebum production and its microbial metabolite, propionate, in AD patients to elucidate whether the skin microbiome has an initiating role in AD pathogenesis. Using LC-MS/MS, we detected a substantial amount of propionate on the skin surface and found that the propionate content was significantly lower in AD patients than in healthy individuals. Topical propionate application attenuated skin inflammation in mice with MC903-induced AD by inhibiting IL-33 production, an effect that was mediated through HDAC inhibition and AhR activation. This is the first study to report on the role of SCFAs in skin homeostasis based on the determination of SCFA levels on the skin surface, with the results showing that the sebum–microbial metabolite–IL-33 axis might play an essential role in the initiation of AD-related skin inflammation.
SCFAs, produced by the fermentation of nondigestible polysaccharides in the diet, are the most studied microbial metabolites in the gut (Rooks and Garrett, 2016; Levy et al., 2017). In contrast, the importance of SCFAs in skin homeostasis has long been overlooked, as the skin surface is almost devoid of polysaccharides (Chen et al., 2018); however, the pilosebaceous unit provides an abundance of sebum that can serve as a substrate for skin microbiota-mediated lipid metabolism, which results in the production of SCFAs (Chen et al., 2018; Byrd et al., 2018; James et al., 2004). This is the first study to properly quantify the levels of SCFAs on the skin surface. Subsequently, we uncovered that the propionate level is significantly lower in the skin of AD patients compared with healthy individuals, which is consistent with reduced sebum production, a decreased abundance of C. acnes, and alterations in microbial lipid metabolism in the skin of AD patients (Li et al., 2019; Shi et al., 2015; Chng et al., 2016). Cutibacterium is the most abundant genus in the skin, and the pilosebaceous unit contains a substantially higher level of sebum and Cutibacterium than the skin surface (Byrd et al., 2018); thus, the actual level of SCFAs in the skin should be higher than reported in this study.
In our study, the importance of sebum and propionate in the development of AD was first demonstrated by the propionate-mediated amelioration of AD-like dermatitis in mice with MC903, an effect that was further confirmed by topical propionate application in sebum-deficient Cidea KO mice which spontaneously developed AD-like dermatitis. Previous studies have shown that propionate suppresses the growth of Staphylococcus aureus both in vitro and in vivo (Shu et al., 2013; Wang et al., 2014). Consequently, the decreased levels of C. acnes and its metabolite propionate in AD patients may lead to an increase in the abundance of S. aureus in the skin of these patients. We also demonstrated that topical C. acnes application effectively alleviated AD-like dermatitis in mouse models. Considering that C. acnes helps maintain the pH of the skin (Youn et al., 2013) and promotes the expression of filaggrin and antimicrobial peptides in keratinocytes (Lee et al., 2019; Ramasamy et al., 2019), we propose that C. acnes plays an essential protective role in AD and has potential as a topical probiotic for the prevention and treatment of AD. Conversely, C. acnes is traditionally considered as pathogen in acne (Dréno et al., 2018; Ramasamy et al., 2019). The paradoxical regulatory role of C. acnes between AD and acne implies that C. acnes might be one of the key skin commensals regulating different types of skin inflammation, which is worthy of further study. Another SCFA, butyrate, also exhibited an inhibitory effect on IL-33 production in keratinocytes. The antiinflammatory effect of butyrate was further verified in the AD mouse model and was even stronger than that of propionate, which might be due to their differential effects on HDAC activity and AhR signaling (Silva et al., 2018; Denison and Faber, 2017). Although the level of butyrate on the skin surface was extremely low and was similar between AD patients and healthy people, butyrate’s strong inhibitory effect on IL-33 expression suggests that it may also have a potential application in AD treatment. Notably, butyrate has a strong and conspicuous odor, which may hinder its use; therefore, more efforts on butyrate are needed in the future to optimize its clinical application. Besides, SCFAs have been well described to be able to modulate the number and function of colonic Tregs (Levy et al., 2017; Shapiro et al., 2014); Schwarz et al. (2017) have reported that butyrate promotes the development of Tregs in the skin of a contact hypersensitivity mouse model. However, our results suggest that Tregs might not be involved in the propionate-induced suppression of skin inflammation. The changes of Tregs in the skin of MC903-treated mice might be a downstream response to the inflammation, as previous studies have shown that the development of Tregs is affected by IL-33 (Schiering et al., 2014; Toyama et al., 2021). The discrepancy between the study of Schwarz et al. (2017) and our work may be due to the differences in inflammation types of different mouse models, observation time points, or the activity of butyrate and propionate in the two studies.
IL-33, a proinflammatory cytokine highly expressed in epithelial and endothelial cells, is often considered an upstream actor in type 2 immune responses (Molofsky et al., 2015; Cayrol and Girard, 2018) and plays a crucial role in allergic diseases via the receptor ST2, which is abundantly expressed in Th2 cells and group 2 innate lymphoid cells (Molofsky et al., 2015; Cayrol and Girard, 2018; Chan et al., 2019). The expression level of IL-33 is significantly increased in keratinocytes of the lesional skin of AD patients, and IL-33 not only acts as an initiator of the regional Th2 response in the skin (Li et al., 2017; Imai et al., 2013; Savinko et al., 2012) but also promotes gastrointestinal food allergies (Leyva-Castillo et al., 2019). Previous studies have mainly focused on the downstream effects mediated by IL-33, while the upstream regulation of IL-33 expression has been largely ignored. To our knowledge, our demonstration of the regulatory effect of SCFAs on IL-33 expression has not been previously reported. Our findings extend the range of immunomodulatory effects associated with SCFAs and hint that they may participate in diseases other than AD in which IL-33 is involved. IL-33 is constitutively expressed in cells of epithelial surfaces such as the skin, intestine, and lung and is extensively involved in the homeostasis of adipose tissue, intestine, and nerves (Cayrol and Girard, 2018; Molofsky et al., 2015; Chan et al., 2019; Leyva-Castillo et al., 2019; Savinko et al., 2012; Du et al., 2018). Additionally, as a proinflammatory factor, IL-33 is involved in the pathogenesis of various diseases such as asthma, inflammatory bowel disease, and neurodegenerative diseases, among others (Cayrol and Girard, 2018; Molofsky et al., 2015; Chan et al., 2019; Leyva-Castillo et al., 2019; Savinko et al., 2012; Du et al., 2018). Thus, the regulatory effect of SCFAs on IL-33 may be important for maintaining the homeostasis of multiple human tissues, and IL-33 inhibition by SCFAs could be developed as a potential therapeutic strategy for the treatment of many conditions. Drugs targeting IL-33 have already been used to treat several diseases and have demonstrated high efficacy (Chen et al., 2019). Our work opens a new avenue for future study on the treatment of IL-33–related diseases. IL-25 (also known as IL-17E) not only is produced by keratinocytes, but also is derived from various immune cells, such as Th2 cells, eosinophils, and dendritic cells (Fort et al., 2001; Tang et al., 2018; Hvid et al., 2011). To our knowledge, few studies have explored the direct interactions between IL-25 and IL-33, whereas there are clues indicating that IL-33 may influence the expression of IL-25. Previous studies have shown that binding of IL-33 to the cells that express ST2 (including some immune cells and keratinocytes; Zeng et al., 2021) results in the activation of NF-κB and MAPKs (Schmitz et al., 2005), and activation of the MAPK pathway is involved in production of IL-25 (Liu et al., 2018; Zhou et al., 2017), suggesting that the expression of IL-25 might be regulated by IL-33. Besides, RNA-seq data of mouse keratinocytes show that the genes involved in IL-17 signaling pathway are upregulated by IL-33 stimulation (Zeng et al., 2021). These studies suggest that IL-33 may induce the expression of IL-25, and the decrease of IL-25 expression in the MC903 plus propionate group may be due to the decreased expression of IL-33 caused by propionate.
The mechanism underlying how IL-33 expression is regulated is poorly understood. As SCFAs are well established as HDAC inhibitors (Chang et al., 2014; Wang et al., 2008; Vinolo et al., 2011), and the inhibitory effect of propionate on HDAC enzyme activity increased in a concentration-dependent manner in our study (Fig. 5 A), we first assessed whether propionate exerted its suppressive effects on IL-33 expression in keratinocytes through the inhibition of HDACs. As SCFAs and TSA have broad-spectrum activity toward 11 HDACs, we next sought to determine which HDACs regulate IL-33 expression in keratinocytes. HDAC2 and HDAC3 were then sorted out, which is consistent with previous studies that have implicated these two HDACs in the regulation of inflammation-related gene expression in macrophages and intestinal epithelial cells (Jeong et al., 2014; Alenghat et al., 2013). Meanwhile, James et al. (2004) showed that, in keratinocytes, the inhibition of HDAC8 and HDAC9 by sodium butyrate promoted the expression of inflammatory cytokines such as IL-6, IL-8, and TNFα when stimulated by TLR ligands (Sanford et al., 2016); however, when we silenced the gene expression of HDAC8 or HDAC9, no effect on IL-33 expression was detected. Nevertheless, we observed that propionate also inhibited IL-33 expression in keratinocytes when stimulated with TLR ligands. The mechanism involved in the differential effects of propionate on the production of different inflammatory cytokines in keratinocytes requires further investigation.
The involvement of HDACs in the regulatory effects of SCFAs suggests that the skin microbiota can influence skin homeostasis through epigenetic modifications. However, the inhibitory effect of propionate on HDACs is inconsistent with the observed inhibition of IL-33 transcription following HDAC inhibition. HDACs are enzymes that remove acetyl groups from histones, thereby allowing the latter to bind DNA more tightly (Milazzo et al., 2020), and inhibition of HDAC activity leads to histone acetylation, resulting in transcriptional activation (Milazzo et al., 2020). Following the analysis of transcription factors predicted to bind the IL-33 promoter and DEGs identified from our RNA-seq data, AhR was screened out. We found that propionate increased AhR expression by inhibiting HDAC2/3 and induced AhR nuclear translocation acting as a potential AhR ligand, in line with that previously reported (Marinelli et al., 2019). Additionally, we demonstrated that AhR exerts a negative regulatory effect on IL-33 gene transcription in keratinocytes. Our previous study showed that activation of AhR by a tryptophan metabolite suppressed the expression of TSLP in MC903-mediated inflamed skin of mice, but in this study activation of AhR by propionate had no effect on the expression of TSLP. This discrepancy may be due to the nature and manner of function of AhR. Generally, AhR exists in the cytosol in an inactive form. After ligand binding, AhR translocates into the nucleus and forms a dimer with a nuclear protein called ARNT (AhR nuclear translocator). Then the AhR: ARNT heterodimer binds to a specific DNA recognition site called DRE (the dioxin responsive element) and influences the transcription of target genes (Shinde and McGaha, 2018; Denison et al., 2002; Stockinger et al., 2014). Previous studies have demonstrated that different AhR ligands may exert different effects on the same genes, which may result from ligand-specific changes in the structure of AhR, AhR: ARNT, and/or other AhR: protein complexes (Denison and Faber, 2017; Denison et al., 2011; Nault et al., 2013; Goodale et al., 2013). These complexes could interact with different subsets of transcriptional modulators, thereby inducing different responses of gene expression (Soshilov and Denison, 2008; Zhang et al., 2008). The differences in ligand-specific effects on target gene expression may account for the discrepancy in effects of propionate and the tryptophan metabolite on TSLP gene. Detailed mechanisms for the differential ligand responses on TSLP gene in human primary keratinocytes remain to be elucidated.
A limitation of our study is that we did not enroll people >45 yr of age for the SER study. Therefore, caution should be used when extending conclusions to people >45 yr of age. In addition, because of the volatile nature of SCFAs, we cannot accurately assess the actual amounts of SCFAs in the skin, which might be higher than the value we measured in this study.
In conclusion, our investigations have uncovered a sebum–microbial metabolite–IL-33 axis that initiates the skin inflammation in AD. The results of the proof-of-concept clinical study further confirmed the inhibitory effects of propionate on skin inflammation. Formally designed clinical trials should be carried out in the future. SCFAs have advantages as natural metabolites fermented by commensal microbiota, indicating a safer, more economical, and efficient strategy for the treatment of AD and other IL-33–related diseases.
Materials and methods
Subjects and SER assessments
Two cohorts applied by two methods were included for the evaluation of SER. The UK refinement of Hanifin and Rajka’s criteria was applied to diagnose AD (Williams et al., 1994). Healthy subjects were defined as those without any history of allergic skin disease or systemic disease. The subjects were asked not to bathe for ≥8 h before the sampling. This study was approved by the institutional review boards (IRBs) of Xijing Hospital and Huashan Hospital, and all participants provided written informed consent. Sample size was determined on the basis of our previous study of tryptophan metabolite levels on human skin surface (Yu et al., 2019).
One cohort included 95 AD patients and 64 age- and gender-matched healthy individuals aged 1–45 yr. In this cohort, Sebutape patches (CuDerm) were applied to assess the SER as previously described (Bissonnette et al., 2015). Briefly, the SER was measured at four body sites: the forehead (0.5 cm from the upper edge of the eyebrow arch), chest (2 cm below the upper edge of the sternum), abdomen (1 cm above the umbilicus), and flexion side of the forearm (1 cm below the Af transverse lines). These test sites were first washed three times with medical gauze soaked in soapy water and then three times with 75% ethanol solution to ensure the removal of sebum remaining on the skin surface. Sebutape patches (3 × 1.5 cm) were then applied to the skin surface at each site, and the subjects were asked to remain in an environment with controlled temperature (23 ± 1°C) and relative humidity (50 ± 10%). After 1 h, the Sebutape patches were peeled off and applied onto a black plastic card, scanned with a CanoScan LiDE300 scanner (Canon), and analyzed using Image-Pro Plus software. The SER was expressed as areas occupied by sebum per square centimeter of a Sebutape in 1 h (pixels/cm2/h). The association between SER and age was visualized as fitted smooth curves with confidence regions using the R function LOESS.
The other cohort included 40 AD patients and 40 age- and gender-matched healthy individuals. Here, SERs were measured using the Delfin SebumScale (Delfin Technologies) at two body sites (antecubital fossa and forehead), 1 h after the washing of skin as mentioned above. The SER was expressed as the amount of sebum secreted in 1 h (AU/h). TEWL was measured with a VapoMeter (Delfin Technologies), and skin hydration was measured with a MoistureMeter SC (Delfin Technologies). SCORAD values were determined under standard approaches (Kunz et al., 1997). All the tests were carried out in a room with controlled temperature (23 ± 1°C) and relative humidity (50 ± 10%).
SCFA collection and quantification
Samples for the assessment of skin SCFAs were collected from 30 AD patients and 30 age- and gender-matched healthy individuals by tape stripping. This study was approved by the Huashan Hospital IRB, and all participants provided written informed consent. The subjects were enrolled as previously described and were asked not to bathe for ≥8 h before the sampling. The patients provided subjective values (score 0–10) for pruritus scores. A 2 × 4-cm tape (3M 810 Scotch Tape, S-9783) was stuck on the Af and the back of subjects with the same pressure and then peeled off. The sticking and peeling cycle was repeated 20 times with the same tape on a delimited area (16 cm2) of the Af and back. For Cidea KO mice, tape stripping was performed as mentioned above on a shaved area (4 cm2) on the back of the mice.
The tapes were cut into small pieces with sterile scissors and collected in a 1.5-ml EP tube containing 250 μl of 50% aqueous acetonitrile, following which the tubes were vortexed (1,200 rpm) for 1 min to extract the SCFAs. The samples were then centrifuged at 12,000 rpm for 5 min at 4°C (Eppendorf), and the clear supernatants were collected. Aliquots (40 μl) of the supernatants were mixed with a solution containing 20 μl 3-nitrophenylhydrazine · HCl (200 mM), 20 μl N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide · HCl (120 mM), and 6% pyridine and incubated at 40°C for 30 min for derivatization. After the reaction, the mixtures were cooled on ice for 1 min before dilution with 320 μl of 10% aqueous acetonitrile. A 50-μl volume of this mixture was mixed with 50 μl of isotope-labeled solutions as previously described (Han et al., 2015), and a 20-μl aliquot of the final mixture was subjected to LC–MS/MS analysis. The quantification of SCFAs was performed on an Agilent 1290 LC system coupled to an Agilent 6545 quadrupole time-of-flight mass spectrometer. Chromatographic separation was performed on an Agilent ZORBAX Eclipse Plus C18 (2.1 × 100 mm, 1.8 µm) column, using water (containing 0.1% formic acid, vol/vol) and acetonitrile (containing 0.1% formic acid, vol/vol) as the mobile phase. The flow rate was 0.35 ml/min, and samples were kept at 5°C. MS data were collected as previously described (Han et al., 2015). The raw MS data were analyzed using Agilent Technologies MassHunter Workstation B.09.00 Software Quantitative Analysis. The samples were quantified in relation to SCFA standards (Dr. Ehrenstorfer) measured in parallel.
Mouse studies
WT C57BL/6 and BALB/c mice were purchased from Shanghai Slac Laboratory Animal Company. AhR KO mice (#002831; JAX stock) in the background of C57BL/6 were purchased from The Jackson Laboratory. Cidea KO mice in the background of C57BL/6 were a gift from Peng Li’s laboratory (School of Life Sciences, Tsinghua University, Beijing, China; Wang et al., 2012). IL33 KO mice in the background of C57BL/6 were provided by Dr. Hiroshi Kiyonari at the Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology, Institute of Physical and Chemical Research, Kobe, Japan (Oboki et al., 2010). ST2 KO mice in the background of BALB/c were provided by Dr. Andrew McKenzie at the MRC Laboratory of Molecular Biology (Townsend et al., 2000). All KO mice had been backcrossed ≥10 times to their respective background. Corresponding WT littermates were used as controls for KO mice. All the mice used in this study were kept in sterile specific pathogen–free environments. Age- and sex-matched 6–8-wk-old mice were randomly assigned to animal studies. All animal experiments were approved by the Department of Laboratory Animal Science, Fudan University.
The MC903-induced AD-like dermatitis was performed by applying 2 nmol MC903 (calcipotriol; Leo Pharma) on mice ears for 9–14 d (9 d for WT BALB/c mice and ST2 KO mice; 14 d for WT C57BL/6 mice, IL33 KO mice, and AhR KO mice). IMQ-induced psoriatic dermatitis was produced by painting 5 mg IMQ cream (containing 5% IMQ; MED-SHINE) on mouse ears once daily for 7 consecutive days. The OXA–induced contact hypersensitivity was prepared by sensitizing the mice with 100 μl of 3.0% OXA (dissolved in anhydrous ethanol; Sigma-Aldrich) on the shaved abdomen and challenging them with 10 μl of 1% OXA on ears 5 d later. Three groups were set up for each mouse model: NC group, model control group, and propionate-treated group, with 3–5 mice per group. Mice in the propionate-treated group were applied with 30 μl of 1 mM sodium propionate (dissolved in a 7:3 mix solvent of 1,2-propanediol: isopropyl alcohol) topically onto ears 30 min after MC903, IMQ, or OXA application once a day; in the model control group, 30 μl of the solvent was used as the vehicle. Mice without any treatment served as the NC group. Mice in the butyrate-treated group were applied with 30 μl of 1 mM sodium butyrate (dissolved in a 7:3 mix solvent of 1,2-propanediol: isopropyl alcohol) topically onto ears 30 min after MC903 application once daily. The full thickness of mouse ears was assessed with a micrometer. Formalin-fixed and paraffin-embedded ear sections were stained with H&E or anti-mouse IL-33 antibody (Clone# EPR17831, Cat# ab187060; Abcam) for immunohistochemical staining. The fresh ear tissues were collected for RNA or protein extraction.
For testing the effect of external use of C. acnes on AD mouse model, C. acnes strain ATCC6919 was purchased from ATCC and cultured on blood agar plates (37°C, anaerobically). Age-matched BALB/c mice were randomly divided into four groups: 30 μl of 2% glycerol (glycerol control group), C. acnes (1 × 105 CFU/ml; C. acnes control group), or 2% glycerol plus C. acnes (1 × 105 CFU/ml; experimental group). Treatments were topically painted onto the ears of mice 30 min after the application with MC903, once a day for 9 consecutive days, and mice without any treatment served as the NC group.
For Cidea KO mice, TEWL and skin hydration were performed in compliance with that of humans as described above. The number of scratching movements recorded by video in 30 min was evaluated. Blood samples were collected for ELISA of IgE. 30 μl of 1 mM sodium propionate/butyrate (dissolved in a 7:3 mix solvent of 1,2-propanediol:isopropyl alcohol; Schwarz et al., 2017) was applied topically on lesions of Cidea KO mice twice a day for 3 wk. The severity scores of AD-like lesions of mice were assessed before and after the sodium propionate treatment according to four symptoms: (1) erythema, (2) scarring/dryness, (3) edema, and (4) erosion/excoriation as described previously (Sung et al., 2018). Each symptom was scored depending on its severity: 0 = none, 1 = mild, 2 = moderate, and 3 = severe. The dermatitis severity score was the sum of these four values. The lesional tissues before and after the sodium propionate treatment were collected for histopathology assessment as described previously (Yu et al., 2019). Skin tissues were fixed in 4% paraformaldehyde, paraffin-embedded, sectioned, and stained with H&E.
Cell isolation and flow cytometry
Mice ear tissues were cut into pieces and placed in 2.5 mg/ml Dispase II (Roche) solution at 4°C overnight. Then the epidermis was torn off and digested with 0.025% trypsin at 37°C for 15 min, and an equal volume of FBS was added. After being filtered through a 70-μm cell strainer, the cells were resuspended in PBS with 1% FBS. For detecting Tregs, mice ear tissues were cut into pieces and placed in the digestion cocktail (a solution consisting of 2 mg/ml collagenase XI [Cat# C9407; Sigma-Aldrich], 0.5 mg/ml hyaluronidase [Cat# H3506; Sigma-Aldrich], and 0.1 mg/ml DNase [Cat# D5025; Sigma-Aldrich]) with shaking at 255 rpm in 37°C for 40 min (Moreau et al., 2021). Then an equal volume of FBS was added. After being filtered through a 70-μm cell strainer, the cells were resuspended in PBS with 1% FBS. Dead cells were excluded using Fixable Viability Dye eFluor 780 (eBioscience). The single-cell suspensions were stained per the manufacturer’s recommendations with anti-CD207 (Cat# 144206; BioLegend), anti-MHC II (Cat# 107605; BioLegend), anti-CD3ε (Cat# 152311; BioLegend), anti-γδ TCR (Cat# 118107; BioLegend), anti-CD3 (Cat# 152311; BioLegend), anti-CD4 (Cat# 100406; BioLegend), and anti-Foxp3 (Cat# 563101; BD Pharmingen). After washing with PBS, the cells were subjected to flow cytometry analysis (BD Fortessa II), and the data were analyzed using FlowJo v10.4.
Cell culture and stimulation
Human primary keratinocytes were isolated from normal foreskin tissues as described above (Peng et al., 2017). The normal foreskin tissues obtained from circumcisions were cut into pieces and placed in 2.5 mg/ml Dispase II (Roche) solution at 4°C overnight. Then the epidermis was torn off and digested with 0.025% trypsin for 10 min, and an equal volume of FBS was added. After being filtered through a 70-μm cell strainer, the cells were cultured with Keratinocyte Growth Medium 2 (PromoCell) in a 25-cm2 culture flask at 37°C with 5% CO2. The primary keratinocytes were incubated with different concentrations (0.2–10 mM) of sodium propionate/butyrate for 24 h or stimulated with 100 ng/ml IL-4 (R&D Systems) and 10 ng/ml IL-1α (R&D Systems) and then cocultured with sodium propionate/butyrate (0.2–4 mM) for 24 h. For siRNA-mediated gene silencing, HDACs and AhR genes were knocked down in keratinocytes using siRNA purchased from RiboBio. siRNAs were transfected into keratinocytes using RNAiMax (Invitrogen) following the manufacturer’s protocol. The cells were then collected for RNA or protein extraction. The sequence for siRNAs were as follows: HDAC1 siRNA (target sequence: 5′-GCGACTGTTTGAGAACCTT-3′), HDAC2 siRNA (target sequence: 5′-GACCCATAACTTGCTGTTA-3′), HDAC3 siRNA (target sequence: 5′-GTGGTTATACTGTCCGAAA-3′), HDAC7 siRNA (target sequence: 5′-CTCACGTCCAGGTGATCAA-3′), HDAC8 siRNA (target sequence: 5′-GAAGCATATGCACTGCATA-3′), HDAC9 siRNA (target sequence: 5′-AGCCACCCTCATGTTACTT-3′), AhR siRNA (target sequence: 5′-GCACCGATGGGAAATGATA-3′), and control siRNA (5′-TTCTCCGAACGTGTCACGT-3′). JB6 cells (#CRL-2010; ATTC) cultured in DMEM containing 10% FBS were stimulated with MC903 (100 ng/ml; Sigma-Aldrich) for 1 h and then treated with or without 4 mM sodium propionate for 24 h. The cells were collected for protein extraction.
Western blotting
Western blotting was performed as described previously (Yu et al., 2019). The cells were harvested and lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime) for protein extraction. The tissues were ground with liquid nitrogen and then lysed in RIPA buffer. The BCA Protein Assay Kit (Beyotime) was used to measure the concentrations of proteins in lysates. The protein lysates were added with 1× SDS loading buffer and denatured at 100°C for 10 min. Equal total proteins were run on 10% SDS-PAGE gel and transferred to 0.45 μm nitrocellulose membranes (Millipore Immobilon). The membranes were blocked with 5% BSA and incubated with primary antibodies against indicated targets at 4°C overnight. After being incubated with goat anti-rabbit IgG secondary antibody (cat# 7074; Cell Signaling Technology) for 1 h at room temperature, the protein blots were subjected to ECL reaction and were imaged by a TANON 5200 imaging system or a LAS 3000 imager (FujiFilm). Primary antibodies in this study included anti–IL-33 antibody (Clone# EPR20417, Cat# ab207737; Abcam; Clone# EPR17831, Cat# ab187060; Abcam), anti-TSLP antibody (rabbit polyclonal, ab188766; Abcam), anti–β-actin antibody (#4967; Cell Signaling Technology), anti-histone H3 acetyl K9 antibody (Clone# Y28, Cat# ab32129; Abcam), anti-histone H3 acetyl K27 antibody (Clone# EPR21218, Cat# ab213279; Abcam), anti-HDAC2 antibody (Clone# Y461, Cat# ab32117; Abcam), anti-HDAC3 antibody (Clone# EP462Y, Cat# ab76295; Abcam), and anti-AhR antibody (Clone# EPR7119, Cat# ab190797; Abcam).
RT-qPCR
Total RNA was extracted from cells or mouse tissues using Trizol following the manufacturer’s protocol (Invitrogen, Life Technologies). The RNA was measured for concentration with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and reverse-transcribed to cDNA using the PrimerScript RT Reagent Kit. RT-qPCR was performed in a QuantStudio 6Flex Real-time PCR System (Thermo Fisher Scientific) using T.B. Green Premix Ex Taq II according to the manufacturer’s instructions. The analysis of relative gene expression (fold-change) was determined by ΔΔCt normalized to β-actin. The primers were listed in Table S1.
RNA-seq
Purified RNA was extracted with the mirVana miRNA Isolation Kit (Ambion). The libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina) and sequenced on the Illumina sequencing platform (HiSeqTM 2500). Raw data were processed using Trimmomatic (Bolger et al., 2014). DEGs were identified using the DESeq R package. Gene Ontology enrichment (Huang et al., 2009) and KEGG (Kanehisa et al., 2008) pathway enrichment analysis of DEGs was performed using R based on the hypergeometric distribution.
HDAC activity assay
HDAC activity was measured with an HDAC activity colorimetric assay kit (cat# K331; Biovision) according to the manufacturer’s protocol. Briefly, 100 μg of cell lysates were added with 10 μl of 10× HDAC assay buffer and 5 μg of HDAC colorimetric substrates and incubated at 37°C for 1 h. The mixture was then added with 10 μl of Lysine Developer and set at 37°C for another 30 min. The sample signals (OD values) were read in an ELISA plate reader at 400 nm. The HDAC activity was quantified according to the standard curve measured in parallel.
ChIP
ChIP assay was determined with a SimpleChIP Enzymatic Chromatin IP Kit (cat# 9003; Cell Signaling Technology) according to the manufacturer’s protocol. Briefly, cells were fixed with 1% formaldehyde and disrupted by sonication. Fragmented chromatin was incubated with anti-RNA polymerase II (phospho S5) antibody (clone# 4H8, cat# ab5408; Abcam), anti-RNA polymerase II antibody (cat# 664911; BioLegend), anti-histone H3 acetyl K9 antibody (clone# Y28, cat# ab32129; Abcam), and anti-AhR antibody (clone# EPR7119, cat# ab190797; Abcam), together with protein G magnetic beads at 4°C overnight. After magnetic beads washing, cross-links reversing, and DNA purification, qPCR was used to assess the amount of immunoprecipitated DNA by using primers listed in Table S1. The fold enrichment was calculated as 2−ΔΔCt and expressed as the percentage of the total input chromatin (input%).
AhR localization experiments
To determine the localization of AhR driven by propionate, keratinocytes were transfected with a pEGFP-C3-AhR plasmid (RiboBio) using Lipofectamine 2000 Reagent (Invitrogen) following the manufacturer’s protocol. Briefly, the AhR gene was cloned into a pEGFP-C3 vector, and GFP-fusion proteins were constructed. Keratinocytes were cultured in KGM containing 1 μg/ml of pEGFP-C3-AhR plasmid and 1.5 μl/ml lipofectamine 2000 for 6 h, followed by culturing in fresh KGM for 24 h. Then the keratinocytes were stimulated with 4 mM sodium propionate for a different period (0, 6, 12, and 24 h). For AhR translocation assay using CH-223191, keratinocytes that had been transfected with plasmids were pre-treated with CH-223191 (10 μM, #C8124; Sigma-Aldrich) for 1 h, and then treated with sodium propionate (4 mM) for a different period (0, 6, 12, and 24 h), After nuclear staining with DAPI (Beyotime), confocal images of the localization of AhR in keratinocytes at different time points were taken with LSM 800 confocal microscope (Zeiss). Image processing was performed with the Zeiss Zen software (v2.6) and Adobe Photoshop software.
Clinical application of propionate
For evaluating the clinical effect of propionate application on AD patients, 11 patients with mild to moderate AD were recruited and enrolled at Huashan Hospital, Fudan University, under clinical trial no. ChiCTR2100043963 registered in Chinese Clinical Trials Registry Platform with approval from Huashan Hospital IRB. Before enrollment, all patients signed informed consent. The enrolled patients were given a cream containing 0.05% propionate or vehicle onto their skin lesions of symmetrical parts. The creams were used twice daily. Clinical characteristics of patients were assessed as described previously (Myles et al., 2018) by a blinded investigator. SCORAD values were determined under standard approaches (Kunz et al., 1997). The skin symptom intensity of the treated region was valued by dryness, erythema, edema, oozing, excoriation, and lichenification (score 0–3, respectively). The patients provided subjective values (score 0–10) for regional pruritus. The treated region-specific SCORAD score is the sum of skin symptom intensity values and pruritus scores. TEWL was measured with a VapoMeter (Delfin Technologies), and skin hydration was measured with a MoistureMeter SC (Delfin Technologies). All the tests were carried out in a room with controlled temperature (23 ± 1°C) and relative humidity (50 ± 10%).
Statistical analysis
Student’s t test or Wilcoxon rank-sum test was performed to compare two groups depending on whether the data were normally distributed. Comparisons among multiple groups in this study were performed with one-way ANOVA followed by Tukey’s test or Dunnett’s test, which were used as described in the figure legends. The correlation between the two groups was assessed by the Spearman correlation test. All the data analysis was performed with GraphPad Prism 8.0 software. Data are presented as mean ± SEM. The following symbols were used in figures to indicate statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
Online supplemental material
Fig. S1 shows that the absence of IL-33 or ST2 abolishes the propionate-mediated attenuation in skin inflammation of the MC903-treated mice. Fig. S2 shows that topical propionate treatment attenuates IMQ-, OXA-, or MC903-induced skin inflammation in mice. Fig. S3 shows that lipophilic microbes cultured in lipid-rich conditions attenuate MC903-induced AD-like dermatitis in mice. Fig. S4 shows that butyrate suppresses IL-33 expression in keratinocytes and inhibits skin inflammation of AD mouse models. Fig. S5 shows the mechanism for the inhibitory effect of propionate on IL-33 expression. Table S1 lists primers for RT-qPCR analysis and ChIP assay.
Data availability
The RNA-seq data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, and are publicly available as of the date of publication; accession no. GSA HRA001005 (http://bigd.big.ac.cn/gsa-human/). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Prof. Peng Li for providing the Cidea KO mice. We also thank Prof. Shau-Ku Huang and Prof. Yuping Lai for their helpful discussions and suggestions.
This work was funded by the Key Project of the Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-07-E00078), National Natural Science Foundation of China (81972939, 82073446, 82003349, 82003357), the Milstein Medical Asian American Partnership Foundation (W. Li), Nanjing Incubation Program for National Clinical Research Centre (2019060001), Key Project of Social Development in Jiangsu Province (BE2020632), and the CAMS Innovation Fund for Medical Sciences (no. 2021-I2M-1-059). The graphical abstract and Fig. 5 S were created with BioRender.com.
Author contributions: W. Li and X. Yao designed the experiments, analyzed the data, and wrote the manuscript. Z. Qiu performed experiments, analyzed the data, and wrote the manuscript. Z. Zhu, X. Liu, B. Chen, H. Yin, C. Gu, X. Fang, R. Zhu, and T. Yu performed experiments. W. Mi, H. Zhou, and Y. Zhou coordinated the research and analyzed the data.

