


Recurrent and new tumors, attributed in part to lateral invasion, are frequent in squamous cell carcinomas and lead to poor survival. We identified a mechanism by which cancer subverts adjacent histologically normal epithelium to enable small clusters of cancer cells to burrow undetected under adjacent histologically normal epithelium. We show that suppression of DMBT1 within cancer promotes aggressive invasion and metastasis in vivo and is associated with metastasis in patients. Cancer cells via TGFβ1 and TNFα also suppress DMBT1 in adjacent histologically normal epithelium, thereby subverting it to promote invasion of a small population of tumor cells. The sufficiency of DMBT1 in this process is demonstrated by significantly higher satellite tumor nests in Dmbt1−/− compared with wild-type mice. Moreover, in patients, invasion of small tumor nests under adjacent histologically normal epithelium is associated with increased risk for recurrence and shorter disease-free survival. This study demonstrates a crucial role of adjacent histologically normal epithelium in invasion and its important role in the tumor microenvironment and opens new possibilities for therapeutic strategies that reduce tumor recurrence.

Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world, with ∼600,000 new cases per year (Leemans et al., 2018). Tumor recurrence and new tumors occur in up to 50% of patients with HNSCC (Leeman et al., 2017; Leemans et al., 2018). Identification of this clinical problem led to the concept of field cancerization. Slaughter et al. (1953) pioneered this concept and attributed it to discrete tumor islands lateral to the bulk of the tumor, suggesting that each served as an independent focus of carcinogenesis. Subsequent studies, using microsatellite analysis, showed that laterally invasive tumor islands have a common clonal origin (Bedi et al., 1996; Califano et al., 1996). This expansion of a mutant clone to sparsely populate an entire field is the current concept of field cancerization, which has now been described in multiple cancers (Curtius et al., 2018). The mechanism by which these laterally invasive tumor islands spread under normal (adjacent histologically normal) epithelium remains unknown; suggestions include shedding into saliva and reimplantation at a site of mucosal erosion (Bedi et al., 1996; Califano et al., 1996). Although most cancers are epithelium-derived, the adjacent histologically normal epithelium has not been investigated as a component of the tumor microenvironment. Furthermore, although developmental biology provides multiple examples of the inductive effect of the surface epithelium in epithelial–mesenchymal interactions (Kahata et al., 2018), the effect of the adjacent histologically normal epithelium on lateral invasion of tumor cells into the underlying mesenchymal tissue has not been investigated.
Invasion is a critical phenotype in development and spread of cancer. In premalignant oral lesions, genetically altered keratinocytes are restricted to the surface-stratified squamous epithelium (Scanlon et al., 2013). In HNSCC, the basement membrane, which separates surface epithelium from stroma, is disrupted, and cancer cells invade the underlying tissues. Invasion allows HNSCC cells to spread to adjacent and distant sites (Scanlon et al., 2013), which can lead to tumor recurrence and metastases.
Recurrent/new tumors and metastasis are the main causes of death in patients with HNSCC; these secondary tumors often metastasize and respond poorly to treatment (Takes et al., 2012; Ruzevick et al., 2013; Plavc et al., 2020). Thus, discovery of biological factors that regulate invasion in HNSCC is essential for identifying new treatment strategies and improving survival, with possible implications for other epithelial-derived tumors. Deleted in malignant brain tumors 1 (DMBT1) has been linked to invasion in cancer (Du et al., 2011). It is a member of the scavenger receptor cysteine-rich (SRCR) superfamily and is encoded by the DMBT1 gene on chromosome 10q26.13. The canonical DMBT1 protein exhibits 13 type B SRCR domains with intervening SRCR interspersed domains, two complement C1r/C1s, Uegf, Bmp1 domains flanking the 14th SRCR domain, and a zona pellucida domain (Reichhardt et al., 2017). Due to the 90–100% sequence identity of the first 13 SRCR repeats, alternative splicing yields different protein isoforms (Mollenhauer et al., 1999; Holmskov et al., 1999; Mollenhauer et al., 2002; Reichhardt et al., 2017). Additionally, DMBT1 may exhibit multi-allelic copy number variations in the number of repeats, potentially yielding 7–20 SRCR domains in the protein (Polley et al., 2015). DMBT1 is detected in mucus, saliva, and the extracellular matrix (Reichhardt et al., 2017).
Originally described as a bacterial agglutinating protein in saliva, subsequent studies showed that DMBT1 can serve as a tumor suppressor that is down-regulated in multiple cancers (Braidotti et al., 2004; Du et al., 2011; Imai et al., 2005). DMBT1 is also known as lung glycoprotein-340 (gp340), salivary agglutinin (SAG), salivary scavenger and agglutinin (Reichhardt and Meri, 2016), muclin (De Lisle et al., 2008), or hensin (Polley et al., 2015). In breast cancer, DMBT1 is a susceptibility gene; expression is decreased in mouse strains susceptible to breast cancer but not in strains with a low incidence of breast cancer (Blackburn et al., 2007). Consistent with this, DMBT1 is undetectable in primary human breast cancer tissue and breast cancer cell lines (Blackburn et al., 2007). Moreover, down-regulation of DMBT1 is correlated with invasion and metastasis in prostate cancer (Du et al., 2011), and is down-regulated by methylation in HNSCC (Imai et al., 2005). However, the extent to which DMBT1 regulates invasion and the mechanism by which it does so are unknown.
In the present study, we identified a DMBT1-mediated mechanism by which cancer cells induce adjacent histologically normal epithelium to release factors that attract small clusters of cancer cells to invade laterally under adjacent histologically normal epithelium. We identify the adjacent histologically normal epithelium as a significant, previously overlooked component of the tumor microenvironment with implications for field cancerization.
Results
DMBT1 suppresses tumor progression
Previously, using a gene array (GEO accession no. GSE28501), we showed that expression of DMBT1 is down-regulated in HNSCC (Banerjee et al., 2011). A meta-analysis of HNSCC datasets available in Oncomine showed that DMBT1 is down-regulated in HNSCC (P = 0.05 level; Table S1 and Fig. S1 A). To evaluate the potential clinical relevance of this observation, we evaluated the impact of DMBT1 (mRNA) on overall and distant metastasis-free survival in an HNSCC dataset linked to clinical outcomes. The patients were stratified into groups based on the mean expression level of DMBT1; low expression of DMBT1 was strongly associated with metastasis (Fig. 1 A) and poor overall survival (Fig. S1 B). Since HNSCCs originate in keratinocytes, we evaluated DMBT1 expression in primary and immortalized human oral keratinocytes and in HNSCC. Significantly less DMBT1 mRNA (Fig. S1 C) and protein (Fig. 1 B) were observed in all six HNSCC cell lines compared with two keratinocyte cell lines (HOK5973 and HOK16B).
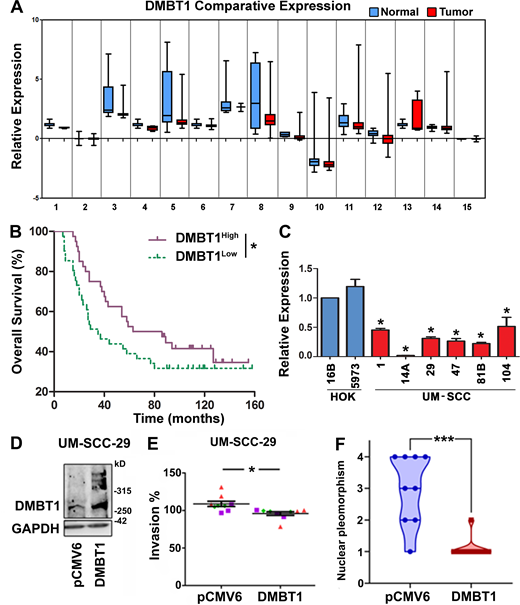
DMBT1 is down-regulated in HNSCC, and overexpression of DMBT1 suppresses invasion and tumor progression.(A) Meta-analysis showing down-regulation of DMBT1 in HNSCC relative to normal tissue. The datasets and accession numbers are given in Table S1. (B) Down-regulation of DMBT1 is correlated with poor overall survival in patients with HNSCC. The analyses were performed with the Rickman Head-Neck dataset (reporter ID, 208250_s_at; Oncomine; n = 81; *, P < 0.05; log-rank test). (C) DMBT1 gene expression in HNSCC cell lines. Total RNA from immortalized (HOK16B), primary keratinocytes (HOK5973), and HNSCC cell lines UM-SCC-(1, 14A, 29, 47, 81B, 104) was used to generate cDNAs, and Q-RT-PCR was performed. Data were analyzed by the relative quantification method with normalization to GAPDH and then relative to keratinocytes (*, P < 0.001; one-way ANOVA; error bars represent SD). Each sample was analyzed in triplicate, and the average fold-change was determined. (D) DMBT1 was stably overexpressed in UM-SCC-29 as verified by immunoblot analysis (n = 2). (E) Overexpression of DMBT1 suppresses invasion in vitro at 48 h. Each color represents an independent experiment with three replicates in each experiment (*, P < 0.05; t test). (F) Nuclear pleomorphism is significantly less in tumors with UM-SCC-29-DMBT1 cells than in control tumors with UM-SCC-29-pCMV6 cells (***, P < 0.001; t test; n = 9; error bars represent SD).
DMBT1 is down-regulated in HNSCC, and overexpression of DMBT1 suppresses invasion and tumor progression.(A) Meta-analysis showing down-regulation of DMBT1 in HNSCC relative to normal tissue. The datasets and accession numbers are given in Table S1. (B) Down-regulation of DMBT1 is correlated with poor overall survival in patients with HNSCC. The analyses were performed with the Rickman Head-Neck dataset (reporter ID, 208250_s_at; Oncomine; n = 81; *, P < 0.05; log-rank test). (C) DMBT1 gene expression in HNSCC cell lines. Total RNA from immortalized (HOK16B), primary keratinocytes (HOK5973), and HNSCC cell lines UM-SCC-(1, 14A, 29, 47, 81B, 104) was used to generate cDNAs, and Q-RT-PCR was performed. Data were analyzed by the relative quantification method with normalization to GAPDH and then relative to keratinocytes (*, P < 0.001; one-way ANOVA; error bars represent SD). Each sample was analyzed in triplicate, and the average fold-change was determined. (D) DMBT1 was stably overexpressed in UM-SCC-29 as verified by immunoblot analysis (n = 2). (E) Overexpression of DMBT1 suppresses invasion in vitro at 48 h. Each color represents an independent experiment with three replicates in each experiment (*, P < 0.05; t test). (F) Nuclear pleomorphism is significantly less in tumors with UM-SCC-29-DMBT1 cells than in control tumors with UM-SCC-29-pCMV6 cells (***, P < 0.001; t test; n = 9; error bars represent SD).

DMBT1 inhibits invasion and metastasis. (A) Down-regulation of DMBT1 is associated with distant metastasis-free survival. The analysis was performed with the Rickman Head-Neck dataset (reporter ID, 208250_s_at; Oncomine; n = 81; *, P < 0.05; log-rank test). (B) Protein lysates from immortalized (HOK16B), primary keratinocytes (HOK5973), and HNSCC cell lines UM-SCC-(1, 14A, 29, 47, 81B, 104) were immunoblotted with anti-DMBT1 antibody. Signal intensity was quantified by densitometric analysis with normalization to actin (loading control) and then expressed as percent of the signal intensity in HOK16B (n = 2). (C) Overexpression of DMBT1 inhibits tumor growth in the CAM in vivo model. UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 cells (1 × 106) were seeded on the chick CAM (*, P < 0.05; n = 6; t test). (D) Overexpression of DMBT1 suppresses invasion in the CAM assay (left scale bar = 1 mm; right scale bar = 500 µm). Tumor sections from the CAM were stained with H&E. Arrows point to invasive islands. (E) Invasive tumor islands in tumor sections were plotted as the total number of invasive islands per sample (*, P < 0.05; t test). (F) Metastases (Mets) to the lower CAM were quantified with quantitative Alu-PCR (*, P < 0.05; t test). (G) UM-SCC-29 overexpressing DMBT1 (UM-SCC-29-DMBT1) or empty vector (UM-SCC-29-pCMV6; 1 × 106) were injected subcutaneously in athymic nude mice. Days 0 and 7 denote the time of injection and the start of tumor measurements, respectively. (H) Tumor size for mice injected with UM-SCC-29-pCMV6 or UM-SCC-29-DMBT1 cells (****, P < 0.0001; n = 9/group; linear mixed model of tumor volume with random effect for mouse and fixed effects for time). (I) Quantification of mitoses from 10 high power fields (HPF) in each mouse tissue section (images at 40× magnification; *, P < 0.05; t test). (J) H&E-stained images for pCMV6 and DMBT1 groups. Representative images show the predominant pattern of invasion. Arrows denote tumor islands at the invasive front (outside scale bar = 200 µm; inside scale bar = 40 µm). (K) Predominant pattern of invasion analyzed in tissue sections from each mouse (**, P < 0.01; t test).
DMBT1 inhibits invasion and metastasis. (A) Down-regulation of DMBT1 is associated with distant metastasis-free survival. The analysis was performed with the Rickman Head-Neck dataset (reporter ID, 208250_s_at; Oncomine; n = 81; *, P < 0.05; log-rank test). (B) Protein lysates from immortalized (HOK16B), primary keratinocytes (HOK5973), and HNSCC cell lines UM-SCC-(1, 14A, 29, 47, 81B, 104) were immunoblotted with anti-DMBT1 antibody. Signal intensity was quantified by densitometric analysis with normalization to actin (loading control) and then expressed as percent of the signal intensity in HOK16B (n = 2). (C) Overexpression of DMBT1 inhibits tumor growth in the CAM in vivo model. UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 cells (1 × 106) were seeded on the chick CAM (*, P < 0.05; n = 6; t test). (D) Overexpression of DMBT1 suppresses invasion in the CAM assay (left scale bar = 1 mm; right scale bar = 500 µm). Tumor sections from the CAM were stained with H&E. Arrows point to invasive islands. (E) Invasive tumor islands in tumor sections were plotted as the total number of invasive islands per sample (*, P < 0.05; t test). (F) Metastases (Mets) to the lower CAM were quantified with quantitative Alu-PCR (*, P < 0.05; t test). (G) UM-SCC-29 overexpressing DMBT1 (UM-SCC-29-DMBT1) or empty vector (UM-SCC-29-pCMV6; 1 × 106) were injected subcutaneously in athymic nude mice. Days 0 and 7 denote the time of injection and the start of tumor measurements, respectively. (H) Tumor size for mice injected with UM-SCC-29-pCMV6 or UM-SCC-29-DMBT1 cells (****, P < 0.0001; n = 9/group; linear mixed model of tumor volume with random effect for mouse and fixed effects for time). (I) Quantification of mitoses from 10 high power fields (HPF) in each mouse tissue section (images at 40× magnification; *, P < 0.05; t test). (J) H&E-stained images for pCMV6 and DMBT1 groups. Representative images show the predominant pattern of invasion. Arrows denote tumor islands at the invasive front (outside scale bar = 200 µm; inside scale bar = 40 µm). (K) Predominant pattern of invasion analyzed in tissue sections from each mouse (**, P < 0.01; t test).
To identify the role of DMBT1 in regulating invasion, we stably overexpressed DMBT1 in HNSCC cell lines (UM-SCC-29 and UM-SCC-1). In a Matrigel invasion assay, overexpression of DMBT1 reduced invasion of cancer cells (Fig. S1, D and E). To investigate the impact of DMBT1 on tumor growth and invasion in vivo, stably transfected cells were implanted on the chick chorioallantoic membrane (CAM; Liu et al., 2013) and in mice. In the CAM assay, overexpression of DMBT1 significantly reduced tumor growth (Fig. 1 C) and invasion (Fig. 1 D) compared with the isogenic control cell line. Control tumors with low DMBT1 had almost six times the number of invasive tumor islands observed in tumors overexpressing DMBT1 (Fig. 1 E). Since metastasis is distant invasion, we evaluated the impact of DMBT1 on metastases to the lower CAM and liver of the chick embryo. 0 of 9 tumors that overexpressed DMBT1 metastasized, whereas 9 of 10 of control tumors, with low DMBT1, metastasized (Fig. 1 F).
The findings from the CAM assay were corroborated in mice, which allow for longer observation of tumor growth. In mice, cells were injected subcutaneously and allowed to progress for ∼7 wk (Fig. 1 G); overexpression of DMBT1 significantly reduced tumor progression compared with control tumors (Fig. 1 H; P < 0.0001). Consistent with the slower growth of tumors overexpressing DMBT1, the average number of mitoses per high-power field was less than half that of control tumors (Fig. 1 I). Xenografts generated from control HNSCC cells expressing low DMBT1 exhibited cords of tumor cells and small invasive tumor islands at the invasive front (Fig. 1 J). In contrast, tumors overexpressing DMBT1 mostly exhibited broad, pushing tumor islands at the invasive front, consistent with less aggressive tumors (Brandwein-Gensler et al., 2010; Fig. 1 J). The quantification of predominant patterns of invasion is shown in Fig. 1 K. The total malignancy score, generated from morphological features including degree of keratinization, predominant pattern of invasion, and nuclear pleomorphism (Fig. S1 F), was higher in tumors with low DMBT1, consistent with more aggressive tumors (Table S2). Taken together, these data show that overexpression of DMBT1 suppresses aggressive tumor progression, including growth, invasion, and metastasis.
The low expression of endogenous DMBT1 in HNSCC cell lines (Fig. 1 B), due to methylation (Imai et al., 2005), precluded their use for loss-of-function studies. In a previous study, DMBT1 was one of the top 100 genes suppressed by EZH2, a histone methyltransferase that is overexpressed in HNSCC (GEO accession no. GSE28501; Banerjee et al., 2011). Therefore, we reasoned that by blocking methylation, endogenous DMBT1 would be increased, and these cells could then be transfected with siDMBT1 (siRNA targeting DMBT1) to investigate the impact of loss of DMBT1. shEZH2 was used as a tool to generate HNSCC cell lines with increased endogenous DMBT1 expression. Significant and sustained up-regulation of DMBT1 was observed after stable down-regulation of EZH2 in multiple cancer cell lines including UM-SCC-29, UM-SCC-47 (Fig. 2 A), and UM-SCC-1 (Fig. S2 A). The impact of loss of endogenous DMBT1 expression on invasion was investigated in UM-SCC-29-shEZH2. Down-regulation of EZH2 in HNSCC suppressed invasion to almost half that of control (shVSVG [scramble short hairpin RNA]) cells (Fig. 2 B), consistent with earlier findings (Banerjee et al., 2012). Importantly, siRNA-mediated suppression of DMBT1 in cells with low EZH2 rescued the EZH2-mediated invasive phenotype (Fig. 2 B and Fig. S2 E). Down-regulation of DMBT1 was verified by immunoblot analysis (Fig. 2 C). Similar results were obtained with UM-SCC-1, an independent cell line (Fig. S2, A and D).

Suppression of DMBT1 promotes invasion and metastasis. (A) Lysates from UM-SCC-29 and UM-SCC-47 stably transduced with shEZH2 were immunoblotted with DMBT1, EZH2, and actin (loading control) antibodies (n = 2). (B) UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 were transfected with siDMBT1 or siNT. Invasion was quantified at 48 h after seeding. Scatter plots show three independent experiments; each color represents one experiment with three replicates in each (*, P < 0.05; ****, P < 0.0001; one-way ANOVA; error bars represent SD). (C) Lysates were immunoblotted with DMBT1, E-cadherin (E-Cad), MMP9, MMP2, EZH2, and actin antibodies. Quantification represents densitometric unit (DU) normalized to actin and expressed as percent of corresponding control (n = 2). (D) GFP-labeled UM-SCC-29-shEZH2 cells transfected with siNT or siDMBT1 (1 × 106) were seeded on the CAM. After 4 d, the upper CAM was photographed (brightfield and GFP) and merged (scale bar = 2 mm). Representative images shown. (E) Tumor area (*, P < 0.05; siNT: n = 6; siDMBT1: n = 4; t test; error bars represent SD). (F) Tumor islands in H&E-stained sections were quantified (top scale bar = 1 mm; bottom scale bar = 500 µm). Sections stained with anti-collagen IV and DAPI to highlight basement membrane and nuclei, respectively (bottom). Tumor cells have GFP label (scale bar = 100 µm). Arrows show invasive islands. (G) Quantification of invasive islands (*, P < 0.05; siNT: n = 5; siDMBT1: n = 4; t test; error bars represent SD). (H) Metastases (Mets) to the lower CAM (n = 6) and liver (n = 6) were quantified. y axis = metastatic cells/µg of DNA from lower CAM (*, P < 0.05; t test; error bars represent SD).
Suppression of DMBT1 promotes invasion and metastasis. (A) Lysates from UM-SCC-29 and UM-SCC-47 stably transduced with shEZH2 were immunoblotted with DMBT1, EZH2, and actin (loading control) antibodies (n = 2). (B) UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 were transfected with siDMBT1 or siNT. Invasion was quantified at 48 h after seeding. Scatter plots show three independent experiments; each color represents one experiment with three replicates in each (*, P < 0.05; ****, P < 0.0001; one-way ANOVA; error bars represent SD). (C) Lysates were immunoblotted with DMBT1, E-cadherin (E-Cad), MMP9, MMP2, EZH2, and actin antibodies. Quantification represents densitometric unit (DU) normalized to actin and expressed as percent of corresponding control (n = 2). (D) GFP-labeled UM-SCC-29-shEZH2 cells transfected with siNT or siDMBT1 (1 × 106) were seeded on the CAM. After 4 d, the upper CAM was photographed (brightfield and GFP) and merged (scale bar = 2 mm). Representative images shown. (E) Tumor area (*, P < 0.05; siNT: n = 6; siDMBT1: n = 4; t test; error bars represent SD). (F) Tumor islands in H&E-stained sections were quantified (top scale bar = 1 mm; bottom scale bar = 500 µm). Sections stained with anti-collagen IV and DAPI to highlight basement membrane and nuclei, respectively (bottom). Tumor cells have GFP label (scale bar = 100 µm). Arrows show invasive islands. (G) Quantification of invasive islands (*, P < 0.05; siNT: n = 5; siDMBT1: n = 4; t test; error bars represent SD). (H) Metastases (Mets) to the lower CAM (n = 6) and liver (n = 6) were quantified. y axis = metastatic cells/µg of DNA from lower CAM (*, P < 0.05; t test; error bars represent SD).

Suppression of DMBT1 down-regulates E-cadherin (E-cad), up-regulates MMPs, and promotes invasion. (A) Lysates from UM-SCC-1 were immunoblotted with DMBT1, EZH2, E-cad, MMP9, MMP2, and actin (loading control) antibodies. Quantification represents DUs normalized to actin and expressed as percent of control (n = 2). (B and C) UM-SCC-1-shEZH2-DOX (B) and UM-SCC-29-shEZH2 (C) were transfected with siDMBT1 or control siRNA (siNT). MMP9 and MMP2 in CM were detected by gelatin zymography. Data represent three independent experiments. (D) UM-SCC1-shEZH2-Dox was transfected with siDMBT1 or siNT. Invasion was quantified at 48 h after seeding (*, P < 0.05; t test; error bars represent SD). (E) UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 were transfected with siDMBT1-2 or siNT. Invasion was quantified at 24 h after seeding (****, P < 0.0001; one-way ANOVA; error bars represent SD).
Suppression of DMBT1 down-regulates E-cadherin (E-cad), up-regulates MMPs, and promotes invasion. (A) Lysates from UM-SCC-1 were immunoblotted with DMBT1, EZH2, E-cad, MMP9, MMP2, and actin (loading control) antibodies. Quantification represents DUs normalized to actin and expressed as percent of control (n = 2). (B and C) UM-SCC-1-shEZH2-DOX (B) and UM-SCC-29-shEZH2 (C) were transfected with siDMBT1 or control siRNA (siNT). MMP9 and MMP2 in CM were detected by gelatin zymography. Data represent three independent experiments. (D) UM-SCC1-shEZH2-Dox was transfected with siDMBT1 or siNT. Invasion was quantified at 48 h after seeding (*, P < 0.05; t test; error bars represent SD). (E) UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 were transfected with siDMBT1-2 or siNT. Invasion was quantified at 24 h after seeding (****, P < 0.0001; one-way ANOVA; error bars represent SD).
Loss of EZH2 also inhibits tumor growth and invasion in vivo (Banerjee et al., 2012; Liu et al., 2013). To investigate whether suppression of DMBT1 enhances tumor growth and invasion in vivo, studies were performed in the CAM with UM-SCC-29-shEZH2, which has increased endogenous DMBT1. 4 d after grafting on the CAM, GFP-expressing UM-SCC-29-shEZH2-siDMBT1 formed tumors approximately threefold the size of tumors with the same cells transfected with nontarget siRNA (Fig. 2, D and E). This disparity was also observed with invasion; tumors induced by UM-SCC-29-shEZH2-siDMBT1 exhibited approximately six times as many invasive islands as control tumors (Fig. 2, F and G). To investigate the impact of DMBT1 on invasion-mediated destruction of the basement membrane (Liu et al., 2013), CAM sections were stained with anti-collagen IV. Cells with suppressed DMBT1 expression exhibited more extensive destruction of the basement membrane than corresponding control cells, which showed a relatively intact basement membrane (Fig. 2 F, bottom). Tumors with siDMBT1 exhibited significantly more metastatic tumor cells to the lower CAM and liver than corresponding control tumors (non-target siRNA [siNT]; Fig. 2 H). Taken together, the gain-of-function (Fig. 1) and down-regulation of DMBT1 expression (Fig. 2) studies showed that DMBT1 suppresses tumor growth, invasion, and metastasis.
DMBT1 inhibits invasion via ZEB1-mediated up-regulation of E-cadherin and suppression of MMP9
Given its role in invasion, we investigated the impact of DMBT1 on E-cadherin expression, which is associated with an epithelial phenotype, and gelatinases, which promote invasion (Scanlon et al., 2013; Mitra et al., 2008). Consistent with an invasive phenotype, down-regulation of DMBT1 decreased E-cadherin and increased MMP9 and MMP2 expression compared with nontarget siRNA control in two cell lines (Fig. 2 C; Fig. S2 A, compare lane 4 with lane 3). Conversely, down-regulation of EZH2 alone, which increased DMBT1, increased E-cadherin and decreased MMP9 and MMP2 expression in UM-SCC-29 and UM-SCC-1 (Fig. 2 C; Fig. S2 A, compare lane 2 with lane 1). To confirm whether the increase in expression of gelatinases MMP2 and MMP9 leads to enhanced secretion of gelatinases that are proteolytically active, gelatin zymography was performed on conditioned medium (CM) from these cells. Down-regulation of DMBT1 increases the activity of MMP9 and MMP2 in both cell lines (Fig. S2, B and C). Together, these studies show that loss of DMBT1 is correlated with an invasive phenotype.
In complementary studies, we observed that E-cadherin expression was increased in HNSCC cells overexpressing DMBT1, whereas MMP9 was down-regulated (Fig. 3 A and Fig. S3 A); ZEB1, a transcription factor that promotes epithelial to mesenchymal transition, was also down-regulated (Fig. 3 A). To identify the extent to which DMBT1-mediated up-regulation of E-cadherin inhibits invasion, UM-SCC-29 overexpressing DMBT1 was transiently transfected with two different siRNAs targeting E-cadherin, siE-cad#1 and siE-cad#2. Reduction of E-cadherin rescued the invasive phenotype, comparable to control cells expressing low DMBT1 (pCMV6-siNT; Fig. 3, B–E). Similarly, overexpression of MMP9 and ZEB1 in cells with high DMBT1 rescued the invasive phenotype, comparable to cells with low DMBT1 (Fig. 3, F and G).

DMBT1 regulates mRNA stability and transcription of MMP9 and E-cadherin via ZEB1. (A) Immunoblot for EMT markers in UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 cells (n = 2). (B–E) DMBT1 inhibits invasion via up-regulation of E-cadherin (E-cad). UM-SCC-29-DMBT1 cells were transiently transfected with siNT or siE-cad-1 (B and C) or siE-cad-2 (D and E). (B and D) Immunoblots verify suppression of E-cadherin. (C and E) Invasion assays. Scatter plots show three independent experiments; each color represents one experiment with at least duplicates in each (**, P < 0.001; ***, P < 0.0001; one-way ANOVA; error bars represent SD). (F and G) Overexpression of MMP9 and ZEB1 rescue invasion. UM-SCC-29-DMBT1 cells were transduced with empty vector (pLX304) or MMP9 or ZEB1. UM-SCC-29-pCMV was transduced with pLX304 as a control for UM-SCC-29-DMBT1-pLX304. Lysates were immunoblotted with anti-MMP9, anti-ZEB1, and anti-GAPDH (F). Invasion was quantified at 48 h after seeding (G). Scatter plots show two independent experiments; each color represents an independent experiment with three replicates in each (****, P < 0.0001; one-way ANOVA; error bars represent SD). (H and I) UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 were incubated with actinomycin D (1 µg/ml), and RNA was isolated at the indicated time. MMP-9 (H) and E-cadherin (I) transcripts, quantified by Q-RT-PCR, were normalized to GAPDH. Results are presented as percent of corresponding transcript at time 0. Values are mean ± SEM (n = 3). (J and K) Normalized luciferase activity of MMP9 (J) and CDH1 (K) promoters in UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1. Scatter plots show three independent experiments; each color represents an independent experiment (***, P < 0.001; t test; error bars represent SD).
DMBT1 regulates mRNA stability and transcription of MMP9 and E-cadherin via ZEB1. (A) Immunoblot for EMT markers in UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 cells (n = 2). (B–E) DMBT1 inhibits invasion via up-regulation of E-cadherin (E-cad). UM-SCC-29-DMBT1 cells were transiently transfected with siNT or siE-cad-1 (B and C) or siE-cad-2 (D and E). (B and D) Immunoblots verify suppression of E-cadherin. (C and E) Invasion assays. Scatter plots show three independent experiments; each color represents one experiment with at least duplicates in each (**, P < 0.001; ***, P < 0.0001; one-way ANOVA; error bars represent SD). (F and G) Overexpression of MMP9 and ZEB1 rescue invasion. UM-SCC-29-DMBT1 cells were transduced with empty vector (pLX304) or MMP9 or ZEB1. UM-SCC-29-pCMV was transduced with pLX304 as a control for UM-SCC-29-DMBT1-pLX304. Lysates were immunoblotted with anti-MMP9, anti-ZEB1, and anti-GAPDH (F). Invasion was quantified at 48 h after seeding (G). Scatter plots show two independent experiments; each color represents an independent experiment with three replicates in each (****, P < 0.0001; one-way ANOVA; error bars represent SD). (H and I) UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 were incubated with actinomycin D (1 µg/ml), and RNA was isolated at the indicated time. MMP-9 (H) and E-cadherin (I) transcripts, quantified by Q-RT-PCR, were normalized to GAPDH. Results are presented as percent of corresponding transcript at time 0. Values are mean ± SEM (n = 3). (J and K) Normalized luciferase activity of MMP9 (J) and CDH1 (K) promoters in UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1. Scatter plots show three independent experiments; each color represents an independent experiment (***, P < 0.001; t test; error bars represent SD).
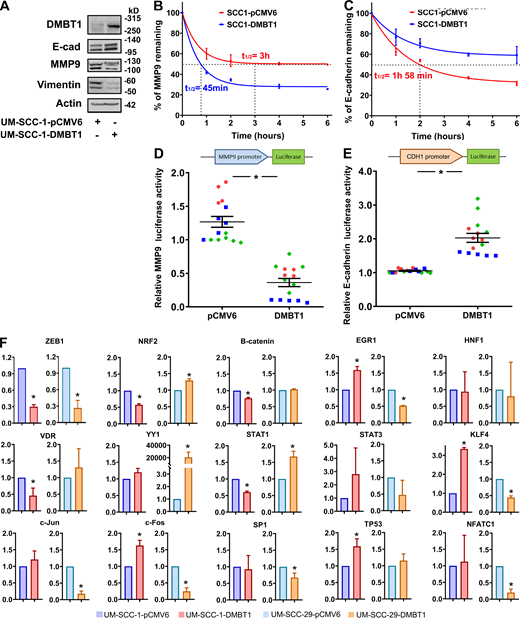
Overexpression of DMBT1 regulates mRNA stability and transcription of MMP9 and E-cadherin (E-cad).(A) Expression of E-cad and MMP9 in UM-SCC-1-pCMV6 and UM-SCC-1-DMBT1 cells (n = 2). (B and C) UM-SCC-1-pCMV6 and UM-SCC-1-DMBT1 were incubated with actinomycin D (1 µg/ml), and RNA was isolated at different time points. MMP9 (B) and E-cad (C) transcripts were quantified by Q-RT-PCR. MMP9 and E-cad mRNA expression was normalized to GAPDH. Results are presented as percent of corresponding transcript at time 0. Values are mean ± SEM. (D and E) Normalized luciferase activity of MMP9 promoter (D) is reduced whereas activity of CDH1 promoter (E) is significantly higher in UM-SCC-1-DMBT1 compared with control UM-SCC-1-pCMV6 cells (*, P < 0.001; n = 2; t test; error bars represent SD). (F) Q-RT-PCR of UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1. Primers for ZEB1, NRF2 (NFE2), β-catenin, VDR, YY1, c-Jun (AP1), c-Fos (AP1), SP1, EGR1, KLF4, NFATC1 (NFAT), HNF1 (HNF1A), STAT1, STAT3, and TP53 are listed in Table S3. Data are normalized to GAPDH. Relative expression of each transcription factor in cells overexpressing DMBT1 is shown as fold change with respect to control (pCMV6) cells (*, P < 0.05; t test; error bars represent SD).
Overexpression of DMBT1 regulates mRNA stability and transcription of MMP9 and E-cadherin (E-cad).(A) Expression of E-cad and MMP9 in UM-SCC-1-pCMV6 and UM-SCC-1-DMBT1 cells (n = 2). (B and C) UM-SCC-1-pCMV6 and UM-SCC-1-DMBT1 were incubated with actinomycin D (1 µg/ml), and RNA was isolated at different time points. MMP9 (B) and E-cad (C) transcripts were quantified by Q-RT-PCR. MMP9 and E-cad mRNA expression was normalized to GAPDH. Results are presented as percent of corresponding transcript at time 0. Values are mean ± SEM. (D and E) Normalized luciferase activity of MMP9 promoter (D) is reduced whereas activity of CDH1 promoter (E) is significantly higher in UM-SCC-1-DMBT1 compared with control UM-SCC-1-pCMV6 cells (*, P < 0.001; n = 2; t test; error bars represent SD). (F) Q-RT-PCR of UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1. Primers for ZEB1, NRF2 (NFE2), β-catenin, VDR, YY1, c-Jun (AP1), c-Fos (AP1), SP1, EGR1, KLF4, NFATC1 (NFAT), HNF1 (HNF1A), STAT1, STAT3, and TP53 are listed in Table S3. Data are normalized to GAPDH. Relative expression of each transcription factor in cells overexpressing DMBT1 is shown as fold change with respect to control (pCMV6) cells (*, P < 0.05; t test; error bars represent SD).
To further understand the major regulatory mechanism by which DMBT1 down-regulates MMP9 and up-regulates E-cadherin, we focused on transcript stability and transcription. For transcript stability experiments, HNSCC cell lines were incubated with actinomycin D (transcriptional inhibitor). Increased DMBT1 expression significantly reduced the stability of MMP9 (Fig. 3 H and Fig. S3 B) and increased the stability of E-cadherin (Fig. 3 I and Fig. S3 C) transcripts in both cell lines. Luciferase assays were performed with reporter constructs of the promoter regions of MMP9 and CDH1 and showed that overexpression of DMBT1 reduced transcription of MMP9 (Fig. 3 J and Fig. S3 D) and enhanced transcription of CDH1 (Fig. 3 K and Fig. S3 E). Together, these data indicate that loss of DMBT1 expression increases MMP9 and suppresses E-cadherin via translational and transcriptional effects.
To identify potential transcription factors that regulate both CDH1 and MMP9, the promoter region of each gene was interrogated with TRANSFEC software, and 10 transcription factors with binding sites in both promoter regions were selected. Transcription factors ZEB1, NRF2, VDR, YY1, AP1, EGR1, KLF4, NFAT, HNF1A, and SP1 have binding sites in the promoter regions of both MMP9 and CDH1 (Fig. 4 A). To investigate the expression of these transcription factors in HNSCC cells overexpressing DMBT1, quantitative RT-PCR (Q-RT-PCR) was performed. The fold-change for each transcription factor in two independent cell lines is shown in Fig. S3 F. Only ZEB1 transcript was reduced in both HNSCC cell lines stably overexpressing DMBT1 compared with control cells (Fig. 4, B and C). Since ZEB1 induces matrix metalloproteinases (Guo et al., 2017) but is a negative regulator of E-cadherin (Maturi et al., 2018; Caramel et al., 2018), using chromatin immunoprecipitation (ChIP), changes in binding of ZEB1 to the promoter regions of MMP9 and CDH1 were investigated. Binding of ZEB1 to MMP9 (Fig. 4 D) and CDH1 (Fig. 4 E) is reduced in cells overexpressing DMBT1. In complementary experiments, binding of ZEB1 to the promoter regions of MMP9 and CDH1 was investigated after suppression of DMBT1, which was verified by immunoblot analysis (Fig. 4 F). Binding of ZEB1 to the promoter region of MMP9 (Fig. 4 G) and CDH1 (Fig. 4 H) was significantly increased after down-regulation of DMBT1 compared with nontarget siRNA (shEZH2-siDMBT1 compared with shEZH2-NT). This increase was more prominent with siDMBT1-1 than with siDMBT1-2, which corresponded to greater suppression of DMBT1 expression with siDMBT-1 than siDMBT1-2 (Fig. 4 F).

ZEB1 down-regulates DMBT1 by binding promoter regions of MMP9 and CDH1. (A) Venn diagram showing transcription factors with binding sites in the promoter regions of MMP9 and CDH1, including ZEB1, NRF2 (NFE2), VDR, YY1, c-Jun (AP1), SP1, EGR1, KLF4, NFATC1 (NFAT), HNF1 (HNF1A), STAT1, STAT3, and TP53. (B and C) Q-RT-PCR of UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1. Data are normalized to GAPDH. Relative expression of ZEB1 transcription factor in cells overexpressing DMBT1 is shown as fold change with respect to control (pCMV6) cells (***, P < 0.001; ****, P < 0.0001; t test; n = 2; error bars represent SD). (D and E) Q-RT-PCR was performed with ChIP-eluted DNA from UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1 with validated primers specific to the ZEB1-binding site in the promoter region of MMP9 (D) and CDH1 (E; *, P < 0.05; t test; n = 2; error bars represent SD). (F) Immunoblot validation of DMBT1 down-regulation in lysates from UM-SCC-29-shVSVG and -shEZH2 transfected with nontarget siRNA (NT) or siDMBT1 (-1 and -2), as indicated. (G and H) Q-RT-PCR with ChIP-eluted DNA from UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 (NT, siDMBT1-1, and siDMBT1-2) for standardized primers targeting the ZEB1-binding site in the promoter region of MMP9 (G) and CDH1 (H). Scatter plots show two independent experiments; each color represents an independent experiment (**, P < 0.01; ****, P < 0.0001; one-way ANOVA; error bars represent SD).
ZEB1 down-regulates DMBT1 by binding promoter regions of MMP9 and CDH1. (A) Venn diagram showing transcription factors with binding sites in the promoter regions of MMP9 and CDH1, including ZEB1, NRF2 (NFE2), VDR, YY1, c-Jun (AP1), SP1, EGR1, KLF4, NFATC1 (NFAT), HNF1 (HNF1A), STAT1, STAT3, and TP53. (B and C) Q-RT-PCR of UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1. Data are normalized to GAPDH. Relative expression of ZEB1 transcription factor in cells overexpressing DMBT1 is shown as fold change with respect to control (pCMV6) cells (***, P < 0.001; ****, P < 0.0001; t test; n = 2; error bars represent SD). (D and E) Q-RT-PCR was performed with ChIP-eluted DNA from UM-SCC-1-pCMV6, UM-SCC-1-DMBT1, UM-SCC-29-pCMV6, and UM-SCC-29-DMBT1 with validated primers specific to the ZEB1-binding site in the promoter region of MMP9 (D) and CDH1 (E; *, P < 0.05; t test; n = 2; error bars represent SD). (F) Immunoblot validation of DMBT1 down-regulation in lysates from UM-SCC-29-shVSVG and -shEZH2 transfected with nontarget siRNA (NT) or siDMBT1 (-1 and -2), as indicated. (G and H) Q-RT-PCR with ChIP-eluted DNA from UM-SCC-29-shVSVG and UM-SCC-29-shEZH2 (NT, siDMBT1-1, and siDMBT1-2) for standardized primers targeting the ZEB1-binding site in the promoter region of MMP9 (G) and CDH1 (H). Scatter plots show two independent experiments; each color represents an independent experiment (**, P < 0.01; ****, P < 0.0001; one-way ANOVA; error bars represent SD).
Microinvasion is associated with tumor progression
Notably, in vivo effects of DMBT1 on invasion in both the CAM assay and mice were more profound than in vitro effects, likely due to the tumor microenvironment (Figs. 1 and 2). To investigate a role for DMBT1 in the tumor microenvironment, we generated Dmbt1+/+ (wild-type) and Dmbt1−/− mice and injected them with MOC-1, a syngeneic mouse HNSCC cell line (Fig. 5 A). Notably, Dmbt1−/− mice showed higher numbers of small tumor islands, i.e., “satellite lesions” (Fig. 5 B), at a distance from the bulk of the tumor (Fig. 5 C). Tumor volumes in Dmbt1−/− mice exceeded 1,000 µm3 faster than in wild-type mice, showing that tumors in Dmbt1−/− were more aggressive (Fig. 5 D). Consistent with this faster growth, the average number of mitoses in tumors from Dmbt1−/− mice was more than twice that in wild-type mice (Fig. 5 E).

HNSCCs in Dmbt1−/− mice exhibit satellite lesions; HNSCCs with lateral (sub-epithelial) invasion are associated with poor clinical outcome. (A) Schematic for Dmbt1−/− mouse model (left) and method of sectioning tumors (right; distance between arrowheads = 1 cm). (B) Tumor tissue sections from Dmbt1+/+ and Dmbt1−/− mice. Black box shows main tumor, and red box indicates satellite lesions, when present. Middle scale bar = 2,000 µm; side scale bar = 200 µm. (C) Percentage of mouse tumors with satellite lesions, i.e., Y in green, and samples that do not, i.e., N in gray (Dmbt1+/+, n = 16; and Dmbt1−/−, n = 17). (D) Kaplan–Meier curve showing time to tumors reaching 1,000 µm3 in male Dmbt1+/+ and Dmbt1−/− mice (*, P = 0.01; log-rank test). (E) Mitoses were counted at 40× magnification in five fields of tumors from each mouse (***, P < 0.001; t test). (F) Human HNSCC stained with H&E or cytokeratin to identify/verify lateral (sub-epithelial) invasion. Left scale bar = 1 mm; right scale bar = 200 µm. Black arrows show tumor nests invading laterally under adjacent epithelium. Red arrows highlight bulk of tumor. (G and H) Disease-specific (G; *, P = 0.03; log-rank test) and recurrence-free (H; *, P = 0.02; log-rank test) survival in patients with (Y) and without (N) laterally invasive tumor islands. HPF, high power field.
HNSCCs in Dmbt1−/− mice exhibit satellite lesions; HNSCCs with lateral (sub-epithelial) invasion are associated with poor clinical outcome. (A) Schematic for Dmbt1−/− mouse model (left) and method of sectioning tumors (right; distance between arrowheads = 1 cm). (B) Tumor tissue sections from Dmbt1+/+ and Dmbt1−/− mice. Black box shows main tumor, and red box indicates satellite lesions, when present. Middle scale bar = 2,000 µm; side scale bar = 200 µm. (C) Percentage of mouse tumors with satellite lesions, i.e., Y in green, and samples that do not, i.e., N in gray (Dmbt1+/+, n = 16; and Dmbt1−/−, n = 17). (D) Kaplan–Meier curve showing time to tumors reaching 1,000 µm3 in male Dmbt1+/+ and Dmbt1−/− mice (*, P = 0.01; log-rank test). (E) Mitoses were counted at 40× magnification in five fields of tumors from each mouse (***, P < 0.001; t test). (F) Human HNSCC stained with H&E or cytokeratin to identify/verify lateral (sub-epithelial) invasion. Left scale bar = 1 mm; right scale bar = 200 µm. Black arrows show tumor nests invading laterally under adjacent epithelium. Red arrows highlight bulk of tumor. (G and H) Disease-specific (G; *, P = 0.03; log-rank test) and recurrence-free (H; *, P = 0.02; log-rank test) survival in patients with (Y) and without (N) laterally invasive tumor islands. HPF, high power field.
In human HNSCC, small tumor islands, i.e., “microinvasion” or lateral spread, is sometimes observed under adjacent histologically normal epithelium distal from the bulk of the tumor (Fig. 5 F). We investigated the clinical relevance of microinvasion beneath adjacent histologically normal epithelium in HNSCC by examining tissue sections from 57 patients previously stained with H&E and cytokeratin antibody (Schmitd et al., 2018a) to detect lateral invasion (Fig. 5 F; and Fig. S4, A and B), and investigated its association with clinical outcome. 54% of patients had lateral invasion. Importantly, this was associated with poor patient outcomes by log-rank test, including poor overall survival (Fig. S4 C), poor disease-specific survival (Fig. 5 G), and increased recurrences (Fig. 5 H).

Lateral (sub-epithelial) invasion in patients with HNSCC.(A and B) HNSCC tissue sections from two patients stained with H&E (top) or cytokeratin. Scale bar = 1 mm in left and 200 µm in right . (C) Kaplan–Meier plot of overall survival for patients with (Y) and without (N) sub-epithelial invasion (P = 0.13; log-rank test).
Lateral (sub-epithelial) invasion in patients with HNSCC.(A and B) HNSCC tissue sections from two patients stained with H&E (top) or cytokeratin. Scale bar = 1 mm in left and 200 µm in right . (C) Kaplan–Meier plot of overall survival for patients with (Y) and without (N) sub-epithelial invasion (P = 0.13; log-rank test).
HNSCC suppresses DMBT1 in adjacent histologically normal epithelium to promote invasion of HNSCC
It is known that cancer cells interact with components of the microenvironment to promote invasion. For example, in HNSCC, perineural invasion occurs due to the dynamic interaction between cancer and nerves (Schmitd et al., 2018b; Scanlon et al., 2015). Since microinvasion is observed beneath adjacent histologically normal surface epithelium in human HNSCC, we investigated the extent to which adjacent histologically normal epithelium might play a role in promoting tumor cell invasion. In initial studies, we investigated DMBT1 expression in human HNSCC and normal oral epithelium. Using laser capture, normal oral epithelium was isolated from tissue specimens without cancer (Fig. 6 A). Additionally, tumor islands from HNSCC and adjacent histologically normal epithelium to HNSCC were isolated. Salivary glands, which normally express high DMBT1 (Reichhardt et al., 2017), were isolated as a positive control. Q-RT-PCR detected no significant difference in DMBT1 in salivary glands from nontumor or tumor specimens or between salivary glands and normal epithelium (Fig. 6 B). In contrast, adjacent histologically normal epithelium to HNSCC and tumor islands from HNSCC expressed significantly less DMBT1 than normal epithelium (Fig. 6 B). To verify reduced DMBT1 protein in tumor islands and adjacent histologically normal epithelium, immunohistochemistry was performed (Fig. 6, C and D). Expression in salivary glands is similar in both groups (Fig. 6 D). In normal epithelium, DMBT1 expression is high in the cytoplasm of cells in the basal third of the epithelium (see also Fig. S5, A and B). These data suggest that cancer cells suppress DMBT1 expression in adjacent histologically normal epithelium.

HNSCC reduces DMBT1 expression in adjacent histologically normal epithelium to enhance invasion of HNSCC. (A) LCM to collect normal epithelium (NE; n = 3) and salivary gland (NSG; n = 3) from tissue without cancer. Tumor islands (TI; n = 10), adjacent epithelium (AE; n = 9), and adjacent salivary gland (i.e., no tumor in salivary gland; ASG; n = 5) were isolated from HNSCC tissue sections. Arrows represent tumor islands. Arrowheads show NE, AE, and SGs in corresponding panels. Scale bar = 100 µm. (B) cDNAs were synthesized from RNA collected from NE, NSG, TI, AE, and ASG. Q-RT-PCR was performed (bar graph represents mean ± SD; *, P < 0.001; one-way ANOVA). (C) Immunohistochemistry with anti-DMBT1 on the same HNSCC sections shown in A (n = 6–9/group). (D) DMBT1 immunostaining is scored based on agreement of two independent analyses. (E) HOK16B were treated with complete, blank, or CM from UM-SCC-29 (n = 2). Lysates were collected after 24, 48, and 72 h, and immunoblotted with DMBT1 or actin antibodies. Signal intensity is quantified by densitometric analysis with normalization to actin and then expressed as percent of corresponding control at the same time point (DU). (F) HOK16B were treated with blank or CM from UM-SCC-29, and lysates were collected after 48 h and immunoblotted with DMBT1, E-cadherin, MMP9, and ZEB1 (n = 2). (G) Schematic of the experimental design for H and I. HOK16B were incubated for 96 h with blank medium or CM from UM-SCC-1 or UM-SCC-29. These primed keratinocytes were incubated with fresh blank medium to generate CM. This CM from primed keratinocytes was used for invasion of UM-SCC-1 (H) or UM-SCC-29 on growth factor reduced (GFR) matrigel (I). Scatter plots show the percentage of invasion of HNSCC cells. Each color represents an independent experiment with three replicates in each (n = 3). Data represent mean ± SD (**, P < 0.01; ***, P < 0.001; t test).
HNSCC reduces DMBT1 expression in adjacent histologically normal epithelium to enhance invasion of HNSCC. (A) LCM to collect normal epithelium (NE; n = 3) and salivary gland (NSG; n = 3) from tissue without cancer. Tumor islands (TI; n = 10), adjacent epithelium (AE; n = 9), and adjacent salivary gland (i.e., no tumor in salivary gland; ASG; n = 5) were isolated from HNSCC tissue sections. Arrows represent tumor islands. Arrowheads show NE, AE, and SGs in corresponding panels. Scale bar = 100 µm. (B) cDNAs were synthesized from RNA collected from NE, NSG, TI, AE, and ASG. Q-RT-PCR was performed (bar graph represents mean ± SD; *, P < 0.001; one-way ANOVA). (C) Immunohistochemistry with anti-DMBT1 on the same HNSCC sections shown in A (n = 6–9/group). (D) DMBT1 immunostaining is scored based on agreement of two independent analyses. (E) HOK16B were treated with complete, blank, or CM from UM-SCC-29 (n = 2). Lysates were collected after 24, 48, and 72 h, and immunoblotted with DMBT1 or actin antibodies. Signal intensity is quantified by densitometric analysis with normalization to actin and then expressed as percent of corresponding control at the same time point (DU). (F) HOK16B were treated with blank or CM from UM-SCC-29, and lysates were collected after 48 h and immunoblotted with DMBT1, E-cadherin, MMP9, and ZEB1 (n = 2). (G) Schematic of the experimental design for H and I. HOK16B were incubated for 96 h with blank medium or CM from UM-SCC-1 or UM-SCC-29. These primed keratinocytes were incubated with fresh blank medium to generate CM. This CM from primed keratinocytes was used for invasion of UM-SCC-1 (H) or UM-SCC-29 on growth factor reduced (GFR) matrigel (I). Scatter plots show the percentage of invasion of HNSCC cells. Each color represents an independent experiment with three replicates in each (n = 3). Data represent mean ± SD (**, P < 0.01; ***, P < 0.001; t test).

DMBT1 expression in nonmalignant epithelium. (A and B) Representative images to show immunohistochemical detection of DMBT1 in epithelium of normal tissue. (A) IgG is used as a negative control. (B) Anti-DMBT1 antibody. (C) HOK16B were treated with complete medium, blank medium, or CM from UM-SCC-1. Cell lysates were collected after 24, 48, and 72 h, and immunoblotted with anti-DMBT1 or anti-actin antibodies. Signal intensity was quantified by densitometric analysis with normalization to actin and then expressed as percent of the corresponding control at the same time point (DU). (D) Immunoblot for HOK16B transfected with siNT and siDMBT1 to validate the DMBT1 knockdown. (E) Scatter plots show percent invasion of UM-SCC-29 (bottom) with CM from keratinocytes transfected with siNT or siDMBT1-2, as illustrated in Fig. 7 A. Each color denotes an independent experiment with three replicates in each experiment (n = 3). Data represent mean ± SD (*, P < 0.05; t test; error bars represent SD). (F) Immunoblot for DMBT1 expression in keratinocytes (HOK5973) treated with TNFα, TGFβ1, or IL-10. Cells were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or IL-10 (10 ng/ml). Cell lysates were harvested after 48 h and immunoblotted with anti-DMBT1 and anti-actin antibodies. (G) CM was collected from UM-SCC-1 (n = 2). CM with or without depletion, or with depletion and add-back of TNFα or TGFβ1, was incubated with HOK16B for 72 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. DUs were calculated by normalizing to actin and then to corresponding controls.
DMBT1 expression in nonmalignant epithelium. (A and B) Representative images to show immunohistochemical detection of DMBT1 in epithelium of normal tissue. (A) IgG is used as a negative control. (B) Anti-DMBT1 antibody. (C) HOK16B were treated with complete medium, blank medium, or CM from UM-SCC-1. Cell lysates were collected after 24, 48, and 72 h, and immunoblotted with anti-DMBT1 or anti-actin antibodies. Signal intensity was quantified by densitometric analysis with normalization to actin and then expressed as percent of the corresponding control at the same time point (DU). (D) Immunoblot for HOK16B transfected with siNT and siDMBT1 to validate the DMBT1 knockdown. (E) Scatter plots show percent invasion of UM-SCC-29 (bottom) with CM from keratinocytes transfected with siNT or siDMBT1-2, as illustrated in Fig. 7 A. Each color denotes an independent experiment with three replicates in each experiment (n = 3). Data represent mean ± SD (*, P < 0.05; t test; error bars represent SD). (F) Immunoblot for DMBT1 expression in keratinocytes (HOK5973) treated with TNFα, TGFβ1, or IL-10. Cells were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or IL-10 (10 ng/ml). Cell lysates were harvested after 48 h and immunoblotted with anti-DMBT1 and anti-actin antibodies. (G) CM was collected from UM-SCC-1 (n = 2). CM with or without depletion, or with depletion and add-back of TNFα or TGFβ1, was incubated with HOK16B for 72 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. DUs were calculated by normalizing to actin and then to corresponding controls.
To directly investigate whether HNSCC reduces DMBT1 expression, the normal keratinocyte cell line HOK16B was incubated with CM from two unrelated HNSCC cell lines for 24, 48, and 72 h (Fig. 6 E and Fig. S5 C). DMBT1 in keratinocytes was significantly suppressed after exposure to CM from HNSCC. In contrast, keratinocytes incubated with blank medium exhibited high DMBT1. Further investigation showed that the CM-mediated decrease in DMBT1 expression in keratinocytes was accompanied by a decrease in E-cadherin and an increase in MMP9 and ZEB1 (Fig. 6 F).
Since loss of expression of DMBT1 in the tumor microenvironment is associated with microinvasion in mice, we investigated the impact of HNSCC-mediated suppression in adjacent histologically normal epithelium on invasion of HNSCC. Keratinocytes were primed with CM from HNSCC cells, and then fresh medium was added to primed keratinocytes to generate “second-generation” CM (Fig. 6 G). CM from primed keratinocytes enhanced invasion of two HNSCC cell lines, compared with control cells incubated with CM from nonprimed keratinocytes (Fig. 6, H and I).
To directly investigate the impact of suppression of DMBT1 in adjacent histologically normal epithelium on tumor cell invasion, keratinocytes were transfected with siDMBT1, and CM from these cells was used for invasion experiments (Fig. 7 A). Suppression was verified by immunoblot analysis (Fig. 7 B and Fig. S5 D); moreover, E-cadherin was down-regulated and MMP9 and ZEB1 were up-regulated compared with keratinocytes transfected with nontarget siRNA (Fig. 7 B). CM from keratinocytes with suppressed DMBT1 enhanced invasion of UM-SCC-1 compared with UM-SCC-1 incubated with CM from control keratinocytes (Fig. 7 C, top). Similar results were obtained with UM-SCC-29 (Fig. 7 C, bottom; Fig. S5 E). Of note, the keratinocytes and HNSCC cell lines were derived from unrelated patients and do not have the same mutational burden. Taken together, these data support that HNSCC coopts adjacent histologically normal epithelium to promote invasion of HNSCC cells.

TNFα and TGFβ1 down-regulate DMBT1 in adjacent histologically normal epithelium. (A) Schematic for experimental design for B and C. CM from siNT or siDMBT1 HOK16B were used for the invasion assay. (B) siRNA-mediated down-regulation of DMBT1 in HOK16B. Cell lysates were immunoblotted with antibodies against ZEB1, E-cadherin (E-cad), MMP-9, and actin (n = 2). Signal intensity was quantified by densitometric analysis with normalization to actin and then expressed as percent of corresponding control (DU). (C) Scatter plots show percent invasion of UM-SCC-1 (top) and UM-SCC-29 (bottom) with CM from keratinocytes transfected with siNT or siDMBT1. Each color denotes an independent experiment with three replicates in each experiment (n = 2 for UM-SCC-1; n = 3 for UM-SCC-29). Data represent mean ± SD (*, P < 0.05; **, P < 0.01; t test). (D) Heat-map for the expression of IL10, GMCSF, TNFα, and TGFβ1 in normal (N) and cancer (T) tissues from 22 patients from GEO accession no. GSE6631. (E) Dose response of impact of recombinant TNFα and TGFβ1 on DMBT1 expression in HOK16B (n = 3). Cells were incubated with increasing concentrations of TNFα (lanes 2–5, lane 1 as vehicle control) and TGFβ1 (lanes 7–10, lane 6 as vehicle control) for 48 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. (F) Time course of recombinant TNFα and TGFβ1 treatment in HOK16B (n = 3). Cells were incubated with TNFα (10 ng/ml) or TGFβ1 (1 ng/ml). Lysates were harvested at the indicated time and immunoblotted with DMBT1 and actin antibodies. (G) HOK16B were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or vehicle. Lysates were immunoblotted with DMBT1, E-cad, MMP9, ZEB1, and actin antibodies (n = 2). (H) HOK16B transfected with siRNA (NT) or siDMBT1 were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or vehicle. Lysates were immunoblotted with DMBT1, E-cad, MMP9, ZEB1, and actin antibodies (n = 2). GFR, growth factor reduced Matrigel.
TNFα and TGFβ1 down-regulate DMBT1 in adjacent histologically normal epithelium. (A) Schematic for experimental design for B and C. CM from siNT or siDMBT1 HOK16B were used for the invasion assay. (B) siRNA-mediated down-regulation of DMBT1 in HOK16B. Cell lysates were immunoblotted with antibodies against ZEB1, E-cadherin (E-cad), MMP-9, and actin (n = 2). Signal intensity was quantified by densitometric analysis with normalization to actin and then expressed as percent of corresponding control (DU). (C) Scatter plots show percent invasion of UM-SCC-1 (top) and UM-SCC-29 (bottom) with CM from keratinocytes transfected with siNT or siDMBT1. Each color denotes an independent experiment with three replicates in each experiment (n = 2 for UM-SCC-1; n = 3 for UM-SCC-29). Data represent mean ± SD (*, P < 0.05; **, P < 0.01; t test). (D) Heat-map for the expression of IL10, GMCSF, TNFα, and TGFβ1 in normal (N) and cancer (T) tissues from 22 patients from GEO accession no. GSE6631. (E) Dose response of impact of recombinant TNFα and TGFβ1 on DMBT1 expression in HOK16B (n = 3). Cells were incubated with increasing concentrations of TNFα (lanes 2–5, lane 1 as vehicle control) and TGFβ1 (lanes 7–10, lane 6 as vehicle control) for 48 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. (F) Time course of recombinant TNFα and TGFβ1 treatment in HOK16B (n = 3). Cells were incubated with TNFα (10 ng/ml) or TGFβ1 (1 ng/ml). Lysates were harvested at the indicated time and immunoblotted with DMBT1 and actin antibodies. (G) HOK16B were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or vehicle. Lysates were immunoblotted with DMBT1, E-cad, MMP9, ZEB1, and actin antibodies (n = 2). (H) HOK16B transfected with siRNA (NT) or siDMBT1 were incubated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or vehicle. Lysates were immunoblotted with DMBT1, E-cad, MMP9, ZEB1, and actin antibodies (n = 2). GFR, growth factor reduced Matrigel.
HNSCC-secreted TNFα and TGFβ1 suppress DMBT1 in adjacent histologically normal epithelium
HNSCC secretes more TNFα and TGFβ1 than normal oral keratinocytes, and these cytokines decrease upon initiation of therapy (Yamamoto et al., 2003). To investigate the mechanism by which HNSCC suppresses DMBT1 in adjacent histologically normal epithelium, we explored the impact of cytokines that are increased in HNSCC relative to normal epithelium using a published dataset (GEO accession no. GSE6631; Kuriakose et al., 2004), which was selected because of the large collection of paired samples of tumor tissue and clinically uninvolved mucosa from the same patient. Normal and HNSCC clinical samples (22 each), analyzed using the platform Affymetrix Human Genome U95 Version 2 Array and available through GEO (accession no. GSE6631; Kuriakose et al., 2004), were interrogated for changes in expression of IL-10, GM-CSF, TNFα, and TGFβ1. This analysis showed that TNFα and TGFβ1 expressions change between normal and tumor samples, while IL-10 and GM-CSF were unchanged (Fig. 7 D). Therefore, we investigated the impact of TNFα and TGFβ1 on DMBT1 in keratinocytes.
Suppression of DMBT1 in keratinocytes by recombinant TNFα and TGFβ1 was observed at 10 ng/ml and 1 ng/ml, respectively (Fig. 7 E), within 24 h (Fig. 7 F). This suppression was not observed with IL-10 (Fig. S5 F). Moreover, TGFβ1- and TNFα-mediated suppression of DMBT1 in keratinocytes is associated with suppression of E-cadherin and increased expression of MMP9 and ZEB1 (Fig. 7 G). Supporting the suppressive role of these two cytokines, TNFα and TGFβ1 further decreased DMBT1 expression in keratinocytes transfected with siDMBT1 (Fig. 7 H). As expected, this was accompanied by a decrease in E-cadherin and an increase in MMP9; this response was more prominent with TGFβ1 than TNFα (Fig. 7 H). ZEB1 showed no further increase.
To directly investigate whether TNFα and TGFβ1 secreted by HNSCC suppress DMBT1 in keratinocytes, we performed neutralization experiments. CM from UM-SCC-1 was preincubated with anti-TNFα, anti-TGFβ1, or both antibodies, to remove these factors (Fig. 8 A). An equivalent concentration of IgG was added in controls. After removal of antigen–antibody complexes, depletion of cytokines was verified by immunoblot analysis; in CM incubated with IgG, both TNFα and TGFβ1 were detected (Fig. 8 B). However, if either TNFα or TGFβ1 was depleted, the other cytokine was detected; and if both cytokines were depleted, neither was detected (Fig. 8 B). Keratinocytes were incubated for 72 h with TNFα- and/or TGFβ1-depleted CM from cancer cells. Depletion of either cytokine in CM led to an approximately sixfold increase in DMBT1 in keratinocytes (Fig. 8 C). Re-introduction of recombinant TNFα and TGFβ1 to cytokine-depleted CM reversed this effect, i.e., DMBT1 was suppressed. Similar results were observed with CM from UM-SCC-1 (Fig. S5 G). With CM from UM-SCC-29, when both cytokines were depleted (Fig. 8 C, lanes 7 and 8), suppression of DMBT1 was less compared with depletion of each cytokine alone. However, in UM-SCC-1, the effect was greater with the combination than depletion of either cytokine alone. Thus, in UM-SCC-29, some additional factors may be modulating the response. Together, these data suggest that tumor-derived TNFα and TGFβ1 modulate DMBT1 in keratinocytes.

TNFα and/or TGFβ1 from HNSCC suppress DMBT1 in adjacent histologically normal epithelium via E-cadherin (E-cad), MMP9, and ZEB1 modulation. (A) Schematic of procedure for depletion of TNFα and TGFβ1 in CM from HNSCC cells. (B) Immunoblot verification of antibody (ab) depletion of TNFα and TGFβ1 in CM from HNSCC cells (n = 2). (C) CM was collected from UM-SCC-29 (n = 2). CM with or without depletion, or with depletion and add-back of TNFα or TGFβ1, was incubated with HOK16B for 72 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. DUs were calculated by normalizing to corresponding controls. (D) Schematic of siRNA-mediated depletion of TNFα or TGFβ1 in CM from HNSCC cells. (E–H) Validation of down-regulation of TNFα (E) and TGFβ1 (G) in UM-SCC-29 lysate (top) and CM (bottom), respectively. HOK16B were incubated with TNFα-depleted (F) and TGFβ1-depleted (H) CM, and lysates were immunoblotted for DMBT1, E-cad, MMP9, ZEB1, and actin. DUs were calculated by normalizing to corresponding controls (n = 2).
TNFα and/or TGFβ1 from HNSCC suppress DMBT1 in adjacent histologically normal epithelium via E-cadherin (E-cad), MMP9, and ZEB1 modulation. (A) Schematic of procedure for depletion of TNFα and TGFβ1 in CM from HNSCC cells. (B) Immunoblot verification of antibody (ab) depletion of TNFα and TGFβ1 in CM from HNSCC cells (n = 2). (C) CM was collected from UM-SCC-29 (n = 2). CM with or without depletion, or with depletion and add-back of TNFα or TGFβ1, was incubated with HOK16B for 72 h. Cell lysates were immunoblotted with DMBT1 and actin antibodies. DUs were calculated by normalizing to corresponding controls. (D) Schematic of siRNA-mediated depletion of TNFα or TGFβ1 in CM from HNSCC cells. (E–H) Validation of down-regulation of TNFα (E) and TGFβ1 (G) in UM-SCC-29 lysate (top) and CM (bottom), respectively. HOK16B were incubated with TNFα-depleted (F) and TGFβ1-depleted (H) CM, and lysates were immunoblotted for DMBT1, E-cad, MMP9, ZEB1, and actin. DUs were calculated by normalizing to corresponding controls (n = 2).
To investigate the mechanism of TNFα- and TGFβ1-mediated suppression of DMBT1 in nonmalignant keratinocytes, transcript expression at multiple time points was investigated. TNFα (Fig. 9 A) and TGFβ1 (Fig. 9 B) suppressed DMBT1 transcript expression in keratinocytes at multiple time points and was significantly different from control cells at 24, 48, and 72 h after initiation of treatment. To investigate the mechanism of decreased transcript expression, we focused on transcript stability and transcription. An investigation of transcript stability in the presence of actinomycin D showed that TNFα (Fig. 9 C) did not modify transcript stability, whereas TGFβ1 showed a transient increase (Fig. 9 D). To assess transcriptional activity, assays were performed in keratinocytes transfected with a DMBT1 promoter luciferase reporter construct. Both TNFα (Fig. 9 E) and TGFβ1 (Fig. 9 F) suppressed transcription of DMBT1 within 8 h after initiation of treatment and continued to decrease until 72 h. Together, these data show that TNFα and TGFβ1 suppress DMBT1 transcription in keratinocytes.

TNFα and TGFβ1 suppress transcription of DMBT1 in adjacent histologically normal epithelium. (A and B) HOK16B were incubated with 10 ng/ml TNFα (A), 1 ng/ml TGFβ1 (B), or vehicle control for the indicated duration. Q-RT-PCR was performed with primer pairs specific to DMBT1 (DMBT1 GE-TRV2-7) and actin, and quantified using the relative quantification method. Data are from two independent experiment with three replicates in each experiment. Data represent mean ± SD (*, P < 0.05; ****, P < 0.0001; one-way ANOVA). (C and D) HOK16B were incubated with TNFα or vehicle for 16 h (C) or TGFβ1 or vehicle for 8 h (D) and then treated with actinomycin D for the indicated time. Q-RT-PCR was performed with primer pairs specific to DMBT1 (DMBT1 GE-TRV2-7) and actin. Data are representative of two independent experiments with three replicates in each experiment. (E and F) HOK16 B were transfected with a DMBT1 promoter reporter construct and then incubated with 10 ng/ml TNFα (E) or 1 ng/ml TGFβ1 (F). Luciferase activity was quantified at the indicated times. Data are representative of two independent experiments with three replicates in each experiment.
TNFα and TGFβ1 suppress transcription of DMBT1 in adjacent histologically normal epithelium. (A and B) HOK16B were incubated with 10 ng/ml TNFα (A), 1 ng/ml TGFβ1 (B), or vehicle control for the indicated duration. Q-RT-PCR was performed with primer pairs specific to DMBT1 (DMBT1 GE-TRV2-7) and actin, and quantified using the relative quantification method. Data are from two independent experiment with three replicates in each experiment. Data represent mean ± SD (*, P < 0.05; ****, P < 0.0001; one-way ANOVA). (C and D) HOK16B were incubated with TNFα or vehicle for 16 h (C) or TGFβ1 or vehicle for 8 h (D) and then treated with actinomycin D for the indicated time. Q-RT-PCR was performed with primer pairs specific to DMBT1 (DMBT1 GE-TRV2-7) and actin. Data are representative of two independent experiments with three replicates in each experiment. (E and F) HOK16 B were transfected with a DMBT1 promoter reporter construct and then incubated with 10 ng/ml TNFα (E) or 1 ng/ml TGFβ1 (F). Luciferase activity was quantified at the indicated times. Data are representative of two independent experiments with three replicates in each experiment.
Discussion
Patients with HNSCC are very susceptible to recurrent or new tumors, which are the main cause of death (Plavc et al., 2020). Tumor recurrence was attributed to lateral invasion of small tumor nests beneath the surface epithelium, but the mechanism of lateral invasion is unclear. We identified a mechanism by which cancer cells subvert adjacent histologically normal epithelium to promote lateral invasion. Suppression of DMBT1 within cancer cells leads to aggressive invasion via ZEB1-mediated repression of E-cadherin and up-regulation of MMP9. Importantly, cancer cells also release TGFβ and TNFα, which in turn suppress DMBT1 in adjacent histologically normal epithelium, coopting it to release factors that encourage small clusters of cancer cells to invade laterally. This lateral invasion is associated with a high recurrence rate and poor survival. Due to its pivotal role in cancer progression, we suggest that the adjacent histologically normal epithelium is an active component of the tumor microenvironment. Novel therapeutic strategies that ablate invasion could reduce recurrence of oral cancer; if the adjacent histologically normal epithelium can be primed to promote tumor progression, then it could be targeted to reduce tumor progression.
Lateral invasion is the spread of small foci of cancer cells in the submucosa under grossly normal surface epithelium. Tumor islands under surface epithelium led to the concept of field cancerization to explain recurrent and new tumors in patients already treated for HNSCC (Curtius et al., 2018; Slaughter et al., 1953). According to this concept, each discrete tumor island beyond the bulk of the tumor could give rise to cancer, which eventually coalesces (recurrence) or remains separate (second primary tumor). More recent studies showed that these tumor islands are genetically related to the original tumor, i.e., rogue tumor cells that have “broken away” from the tumor to invade laterally (Bedi et al., 1996; Califano et al., 1996). This expansion of a mutant clone to sparsely populate an entire field is the current concept of field cancerization in HNSCC (Curtius et al., 2018). Field cancerization occurs in multiple cancers at different sites (Curtius et al., 2018).
The mechanism of lateral spread of tumor islands and recurrence remains largely uncharacterized, although suggestions include shedding of cancer cells into saliva and reimplantation at a site of erosion in adjacent histologically normal epithelium, or transport within the surface epithelium (Bedi et al., 1996). Lateral invasion of small tumor islands is grossly undetectable. Of particular concern is that small clusters of tumor cells can be microscopically undetectable on routine pathology stains (Fig. 5 F, top) used to evaluate surgical margins. Essentially, encouraged by subverted adjacent histologically normal epithelium, these tumor islands burrow undetected because they are small enough to blend with surrounding tissues. Consequently, a surgical margin could inadvertently be evaluated as “free” of tumor. This spread beyond the field of treatment could explain the high rate of recurrent or new tumors observed. In our study, cytokeratin immunohistochemistry was used to detect laterally invasive tumor islands (Fig. 5 F, bottom), which were associated with a significantly higher rate of recurrence.
We found that DMBT1 was selectively down-regulated in adjacent histologically normal epithelium, whereas it was unchanged in salivary glands regardless of the presence of cancer. We identified that TGFβ1 and TNFα from HNSCC reduce DMBT1 expression in adjacent histologically normal epithelium. It is possible that this reduction is due to an overlap in molecular changes. However, we showed that suppression of DMBT1 was observed in adjacent histologically normal epithelium cells that were treated with CM from two unrelated HNSCC cell lines, i.e., genetically unrelated cells. This reduction was reversed when TGFβ1 and TNFα were depleted in the CM, and was rescued when recombinant TGFβ1 and TNFα were reintroduced in the depleted media. TGFβ1-mediated regulation of homeostasis in differentiated cells is cell type–specific; in keratinocytes, it regulates AP1, FOXO1, E2F4, and ATF3 (David and Massagué, 2018; Massagué, 2012). Similar to our findings showing that TGFβ1 inhibits transcription of DMBT1, TGFβ1 acts as a transcriptional repressor (of ID1) in keratinocytes (David and Massagué, 2018; Massagué, 2012).
Intriguingly, low doses of TNFα induced DMBT1 expression, but high doses suppressed expression. It is possible that TNFα has a biphasic effect on DMBT1; a similar effect has been reported in cardiomyocytes and synovial cells. In cardiomyocytes, low TNFα enhances nuclear translocation of Nrf2 and subsequent expression of its targets (Shanmugam et al., 2016). Similarly, in synovial cells, low TNFα increased metalloproteinase expression, and high TNFα suppressed metalloproteinases (Ahmadzadeh et al., 1990).
Modulation of DMBT1 in normal tissue has been reported previously, but the significance was not investigated. For example, consistent with our findings, DMBT1 was lower in normal tissue from cancer patients than in normal tissue from cancer-free patients (Blackburn et al., 2007). In contrast, an earlier study reported induction of DMBT1 in tissue adjacent to breast cancer (Mollenhauer et al., 2004). Similarly, in lung cancer, DMBT1 is up-regulated in tumor-adjacent epithelium (Mollenhauer et al., 2002). The presence of hyperplastic epithelium adjacent to tumor may have contributed to up-regulation of DMBT1 adjacent to tumor (Mollenhauer et al., 2004). Other possibilities may account for this ostensible contradiction; for example, DMBT1 may be decreased due to protein redistribution. Consistent with this possibility, in lung adenocarcinomas, DMBT1 redistributes from luminal localization to the extracellular matrix (Mollenhauer et al., 2002). Similarly, in kidney epithelial cells that function in trans-epithelial transport, DMBT1 moves from within the cell to the extracellular matrix (Vijayakumar et al., 1999). These findings suggest that regulation of DMBT1 in surrounding tissues is an important part of the pathology, underscoring the inclusion of the adjacent histologically normal epithelium in the tumor microenvironment.
In summary, down-regulation of DMBT1 in HNSCC promotes invasion and metastasis. Moreover, TGFβ1 and TNFα secreted by HNSCC suppress DMBT1 in adjacent histologically normal epithelium, to coopt it to promote lateral microinvasion. Taken together, this study shows that the definition of the HNSCC tumor microenvironment should be expanded to include adjacent histologically normal epithelium. This could have important implications for tumor progression and recurrence in patients with HNSCC. This work opens new possibilities for therapeutic strategies that reduce tumor recurrence by modulation of the adjacent histologically normal epithelium in the tumor microenvironment.
Materials and methods
Human tissue samples
De-identified tissue sections of human HNSCC were obtained from the University of Michigan Head and Neck Cancer Specialized Program of Research Excellence (HNSPORE)/Head and Neck (HN) Oncology Program. University of Michigan Institutional Review Board (IRB) approval and patient consent were obtained by the University of Michigan HNSPORE/HN Oncology Program before specimen collection (Schmitd et al., 2018a). Of these 71 patients, using slides previously stained with H&E and cytokeratin (Schmitd et al., 2018a), only 58 had adjacent surface epithelium and were used in this study to assess lateral invasion (Table S5). Among the 58 patients, 57 were surgically treated, and 1 was excluded because treatment was palliative and never definitive. 31 of the 57 had lateral invasion. There were 12 recurrences and 16 deaths within 5 yr of diagnosis (8 from disease, 3 cause unknown, and 5 other causes).
In vivo mouse xenograft model
All animal experiments were conducted in compliance with the National Institutes of Health guidelines for animal research and approved by the Institutional Animal Care and Use Committee of the University of Michigan. Stable UM-SCC-29-pCMV6 and UM-SCC-29-DMBT1 cells were injected subcutaneously in athymic nude mice (Crl:NU(NCr)-Foxn1nu, age 5–6 wk, male, n = 10/group) at 1 × 106 cells in each mouse. Tumors were monitored and measured at least three times a week. A linear mixed effects statistical model was used to analyze the data, and a P value <0.05 was considered to be significant. The mice were euthanized at predetermined end points, and tumors were harvested. Tumor tissue was processed and sectioned. Tissue sections were stained with H&E, and histopathologic analysis was performed. A modified histological malignancy grading system established for human tumors (Bryne et al., 1992) was used to score nuclear pleomorphism, mitoses, pattern of invasion, and degree of differentiation in the xenografts. The score for each feature was used to develop a total malignancy score (Bryne et al., 1992). The mitotic count and field of view were standardized as recommended (Meuten et al., 2016). Attempts to induce UM-SCC-29 xenografts in female athymic nude mice (Crl:NU(NCr)-Foxn1nu, age 5–6 wk) were unsuccessful, possibly because this cell line was developed from a male patient (Brenner et al., 2010).
Dmbt1−/− knockout mouse model
Dmbt1−/− mice were generated from cryopreserved mouse sperm (B6.129X1-Dmbt1tm1Kumc/Mmmh; 030289-MU-SPERM, MMRRC; De Lisle et al., 2008) that was used for in vitro fertilization in C57BL/6 mice (University of Michigan Transgenic Animal Model Core). Genotype was verified according to the supplier’s protocol (MMRRC 30289). Dmb1+/+ and Dmb1−/− mice were used for experiments.
Chorioallantoic membrane assay
For loss-of-function studies, UM-SCC-29-shEZH2 cells transfected with siDMBT1 or siNT were seeded on the CAM of chick embryos (day 10) as described (Liu et al., 2013) for the DMBT1 loss-of-function study (n = 5). For invasion assays, the CAM was harvested on day 14, sectioned, and stained with H&E or immunostained for collagen IV (antibody obtained from Dr. Weiss, University of Michigan, Ann Arbor, MI, and Dr. Linsenmeyer, Tufts University, Medford, MA) and DAPI (Invitrogen) to highlight the basement membrane and nuclei, respectively (Liu et al., 2013). Images for GFP, collagen IV, and DAPI were merged in NIS-Elements (Nikon). Invasive islands were quantified in five randomly selected fields from control and siDMBT1 CAMs (n = 5); the average was graphed. For metastasis assays, the lower CAM and liver were harvested on day 17, and Alu-PCR was performed as described (Liu et al., 2013). For pCMV6 and DMBT1 studies, the number of invasive islands was quantified from the entire section of CAMs (n > 8). For metastasis assays, the lower CAM and liver were harvested on day 17, and Alu-PCR was performed as described (Liu et al., 2013). The number of metastatic cells per microgram of DNA was quantified; one metastatic cell/µg of DNA was considered as the cutoff for quantifying the absolute number shown in Fig. 1 F and Fig. 2 H.
Cell culture
HNSCC cells were cultured as described (Banerjee et al., 2014). UM-SCC cell lines (T. Carey, University of Michigan, Ann Arbor, MI) were genotyped (sequencing core, University of Michigan) before and after completion of studies. Genotype was verified against published sequences for UM-SCC cell lines (Brenner et al., 2010). Primary oral keratinocytes (HOK5973) were cultured according to the supplier’s instructions (ScienCell Research Laboratories). MOC1 cells were obtained from Dr. R. Uppaluri (Dana Farber Cancer Institute, Boston, MA). Immortalized oral keratinocytes (HOK16B; N.H. Park, University of California, Los Angeles, Los Angeles, CA) were cultured as described (Van Tubergen et al., 2011).
Cell transfection and transduction
UM-SCC-1 and UM-SCC-29 cells were transfected with empty vector (pCMV6) or DMBT1 plasmids (#RC215454; OriGene). Four siRNAs targeting E-cadherin (Dharmacon) were tested, and two were used for further experiments (#J-003877-08-0020 and #J-003877-10-0020).
MMP9 or ZEB1 (OHS6085-213579104, OHS6269-213586263; Dharmacon) was transiently up-regulated in UM-SCC-29 (pCMV6 and DMBT1) cell lines by transduction with lentiviral particles (Dharmacon) with appropriate empty vector control (pLX304, 25890; Addgene) for 48 h.
For stable down-regulation of EZH2, HNSCC cells were transduced with lentiviral particles of control shRNA (shVSVG; #RHS4346) or shEZH2 (clone ID: V2LHS_17507; Dharmacon) with GFP reporters and selected with puromycin (UM-SCC-29, 30 µg/ml; UM-SCC-47, 25 µg/ml). After selection, cells were maintained with 20 µg/ml puromycin. For doxycycline-inducible EZH2 down-regulation (UM-SCC-1), Expression Arrest TRIPZ Lentiviral shRNAmir GFP constructs were from Open Biosystems (#RHS4696-200; clone ID: V2THS_17507). An oligonucleotide construct in pTRIPZ vector targeting EZH2 and control scrambled shRNA was used to make lentiviral particles (University of Michigan Vector Core). After standardization, lentiviral particles were used to make stable UM-SCC-1 cells with inducible down-regulation of EZH2.
UM-SCC-29-shEZH2 and doxycycline-inducible UM-SCC-1-shEZH2 cells pretreated with doxycycline for 24 h were used for experiments with down-regulation of endogenous DMBT1. Four siRNAs targeting DMBT1 (Dharmacon) were tested, and two (siDMBT1-1 #J-007883-07-0020 and siDMBT1-2 #J-007883-08-0020) were selected for subsequent experiments. The doxycycline-inducible UM-SCC-1-shEZH2 cells were maintained in doxycycline during the experiment.
Histology and immunohistochemistry
Mouse tumors were removed with 1 cm of adjacent tissue, fixed overnight in 4% paraformaldehyde, sectioned as shown (Fig. 5 A, right), and embedded in paraffin. 4-µm tissue sections were stained with H&E for histological analysis. For quantification of satellite lesions, tumor islands that were separate from the main tumor were identified on H&E-stained slides of representative sections of the entire tumor. Brightfield images were acquired using an Eclipse TU2000-U microscope connected to a cooled color CCD camera (RTKE Diagnostic Instruments) using NSI-Elements (Nikon).
After approval of exempt status by the University of Michigan IRB, preexisting, de-identified, paraffin-embedded human tissue sections were deparaffinized in xylene. Endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide in methanol. Antigen retrieval was performed by boiling slides in citrate buffer (10 mM, pH 6.0, 15 min). Slides were rinsed in Tris-buffered saline–Tween 0.05% and blocked for 30 min with 1% fatty acid–free BSA. Primary antibodies and biotinylated secondary antibodies were diluted in 1% BSA. Slides were incubated overnight at 4°C with primary antibodies and for 25 min at room temperature with secondary antibodies. Subsequently, slides were incubated with peroxidase-conjugated avidin for 20 min and 3,3′-diaminobenzidine (Dako) as a chromogen for 5 min and counterstained with hematoxylin. Staining intensity was compared with the control (IgG). If >50% of epithelial cells stained positively for DMBT1, then the sample was considered positive, and 0–50% was considered negative. A complete list of antibodies is provided in Table S6.
Laser capture microdissection (LCM)
After University of Michigan IRB approval of exempt status, preexisting, de-identified human tissue specimens were used. Surface epithelium and salivary gland from nontumor tissue, and tumor islands, surface epithelium adjacent to HNSCC, and salivary glands from HNSCC tissue specimens were isolated by LCM using 8-µm sections of formalin-fixed paraffin-embedded tissue on glass PEN membrane slides (11505158; Leica) using the LCM V 4.0.3.7 system (Leica). Total RNA was isolated with the RNeasy FFPE Kit (QIAGEN), and cDNA was synthesized (SuperScript III; Invitrogen). Q-RT-PCR was performed.
Q-RT-PCR
Total RNA was isolated from keratinocytes and UM-SCC grown to ∼60–70% confluence using the miRNAeasy kit (QIAGEN). cDNA was synthesized using SuperScript III (Invitrogen), and Q-RT–PCR was performed with Power SYBR Green Master Mix on a StepOne Plus Real-Time PCR System (Applied Biosystems). Data were analyzed by the relative quantification method with normalization to GAPDH. Screening of transcription factors was performed by Q-RT-PCR. The forward and reverse primers for DMBT1, CDH1, MMP9, ZEB1, NRF2, β-catenin, VDR, YY1, STAT1, STAT3, c-Jun (AP1), c-Fos (AP1), SP1, TP53, EGR1, KLF4, NFATC1 (NFAT), and HNF1 (HNF1A) are listed in Table S3. Each sample was assayed in triplicate, and average fold change was determined.
Immunoblot analysis
Immunoblotting was performed as described (Mitra et al., 2003). Primary antibodies were anti-DMBT1 (1:500; #SAB2700429; Sigma-Aldrich), anti-EZH2 (1:1,000; #5246), anti-MMP2 (1:1,000; #4022), anti-MMP9 (1:1,000; #3852), anti-TNFα (1:1,000; #3707), anti-TGFβ1 (1:1,000; #3907; Cell Signaling Technology), anti–E-cadherin (1:2,000; #610182), anti-actin (1:2,000; #612656; BD Biosciences), anti–ZEB-1 (1:500; #21544-1-AP; Proteintech), and GAPDH (1:5,000; #MAB374; Millipore). Secondary antibodies were HRP-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Jackson ImmunoResearch). Immunoreactive proteins were visualized by SuperSignal West Pico Chemiluminescent Substrate. Signal intensity was quantified using ImageJ and expressed as densitometric units normalized to the control.
The high molecular weight of DMBT1 protein is due to glycosylation at multiple sites (Martínez et al., 2011). Changes in molecular mass of DMBT1 may result from variations in the number of SRCR domains or glycosylation (Reichhardt et al., 2017). Glycosylation may have contributed to multiple bands or “smeared” appearance observed on some immunoblots in the present study. The multiple bands or entire smear was quantified when present.
Depletion of TNFα and TGFβ1
For neutralization studies, UM-SCC-1 and UM-SCC-29 were cultured for 48 h, washed, and incubated with basal medium (EpiLife; #MEPICF500; Invitrogen) for 16 h. Medium was collected and centrifuged, and the supernatant (CM) was incubated with HOK16B. To deplete TNFα and TGFβ1, CM was incubated with anti-TNFα (0.5 µg/ml) or anti-TGFβ1 (1 µg/ml) or both at room temperature (1 h) and centrifuged (#UFC805096; Amicon) to remove antigen–antibody complexes. To verify depletion, medium was concentrated 100× (#UFC800396; Amicon) and immunoblotted with anti-TNFα and anti-TGFβ1.
For siRNA-mediated down-regulation of TNFα (siTNFα-1, SR30002; and siTNFα-2, SR30003; from main kit #SR322065; Origene) and TGFβ1 (siTGFβ1-1, #LQ-012562-07; and siTGFβ1-2, #LQ-012562-09; Dharmacon), UM-SCC-29 was transfected with siRNAs for 48 h. After this, the medium was replaced with blank medium for collection of CM. Depletion was verified by immunoblot analysis. HOK16B were treated with HNSCC-derived CM for 48 h followed by immunoblot analysis of cell lysates.
ChIP
ChIP was performed with the EZ-Magna ChIP A/G system (17–10086; Millipore) according to the manufacturer’s protocol. HNSCC cells (∼60% confluent) were cross-linked with 1% formaldehyde and quenched with 0.125 M glycine. Cells were lysed, and chromatin was fragmented to an average size of 500 bp followed by overnight incubation with antibodies and protein A or G magnetic beads. Cross-links were reversed by incubating chromatin with proteinase K at 62°C for 2 h, and DNA was isolated. ChIP antibodies were anti-ZEB1 (#21544-1-AP; Proteintech), anti–c-JUN (#9165; Cell Signaling Technology), anti-RNA Pol II, and mouse and rabbit IgGs. Purified DNA was analyzed by Q-RT-PCR to determine fold enrichment relative to input DNA. Primer sequences for the promoter regions of CDH1, MMP9, and DMBT1 are provided in Table S4.
Invasion assay
Invasion was quantified 48–72 h after transfecting cells using Transwell inserts (3422; Corning) coated with Matrigel (#354230; Corning, BD Biosciences). Uncoated inserts were used as migration controls. Invasion was normalized to migration according to the manufacturer’s instructions by dividing the absorbance of cells that invaded through Matrigel by the total number of cells that invaded through the control insert as described (Banerjee et al., 2014).
Gelatin zymography
CM was collected as described (Henson et al., 2005) and concentrated in Centrifugal Filter Units (Millipore). Gelatin enzyme activity for MMP9 and MMP2 was evaluated by zymography (Mitra et al., 2008).
Transcript stability
UM-SCC-29-pCMV6, UM-SCC-29-DMBT1, UM-SCC-1-pCMV6, and UM-SCC-1-DMBT1 were treated with actinomycin D (1 µg/ml) and lysed with Qiazol at 0 h (no treatment, control cells), 1 h, 2 h, 4 h, and 6 h. Total RNA was prepared (#217004; QIAGEN) and cDNA synthesized (#11904018). Q-RT-PCR was performed with primers targeting MMP9, CDH1, and GAPDH using SYBR Green PCR Master Mix (#4309155; Applied Biosystems) on a StepOne plus Real-time PCR machine (Applied Biosystems). Data were analyzed by a relative quantification method normalized to GAPDH expression and then to corresponding control at 0 h. Primers are shown in Table S3.
For DMBT1 transcript stability assays, HOK16B were treated with TNFα (10 ng/ml, 16 h), TGFβ1 (1 ng/ml, 8 h), or vehicle. After addition of actinomycin D (500 ng/ml), cells were harvested at multiple time points up to 2 h. Cells were washed once with PBS and lysed with Qiazol followed by RNA extraction, cDNA synthesis, and Q-RT-PCR.
Luciferase assay
UM-SCC-29-pCMV6, UM-SCC-29-DMBT1, UM-SCC-1-pCMV6, and UM-SCC-1-DMBT1 cells were cotransfected with E-cadherin (#42081; Addgene) and MMP9 (#53434; Addgene) promoter-firefly luciferase reporter and Renilla luciferase constructs (normalization control) with Lipofectamine 2000. After 48 h, cell extracts were used to assay promoter activity with the Dual-Luciferase Reporter System on LMaxx384 (Molecular Devices). Firefly luciferase activity was expressed as relative luminescence units normalized to Renilla luciferase activity.
DMBT1 promoter (#S718344) was cloned in the LightSwitch Reporter vector (#S718295; Active Motif) and transfected in HOK16B. After 48 h, transfected cells were stimulated with TNFα (10 ng/ml), TGFβ1 (1 ng/ml), or vehicle. Promoter activity was assayed using RenSP luciferase assay system (#32032) according to the manufacturer’s instructions and normalized to corresponding controls.
Meta-analysis of HNSCC datasets
These studies were performed using Oncomine (http://Oncomine.org). Datasets used for this study were Pyeon Floor of the Mouth, TCGA (The Cancer Genome Atlas) Head-Neck, Talbot Lung, Pyeon Oropharyngeal, Estilo Head-Neck, Pyeon Tongue, Kuriakose Head-Neck, Cromer Head-Neck, Toruner Head-Neck, Peng Head-Neck, Ye Head-Neck, Ginos Head-Neck, Pyeon Oral Cavity, Sengupta Head-Neck, and Peng Head-Neck 2 (Table S1). P values were generated using one-sample t tests at the 0.05 and 0.001 levels. The associated t-statistics and degrees of freedom are also shown in that table. Survival functions were estimated by Kaplan–Meier methods using DMBT1 classified as high or low based on mean or median expression values.
Detection of transcription factor binding to the promoter region
The datasets used in the analyses are parts of Transfac Professional and Genome Trax databases available at http://www.biobase-international.com/.
Statistical analysis
Statistical analysis was performed using a Student t test or one-way ANOVA.
Study approval
University of Michigan IRB approval and patient consent were obtained by the University of Michigan HNSPORE/HN Oncology Program before specimen collection.
Online supplemental material
Fig. S1 shows that DMBT1 is down-regulated in HNSCC, and overexpression of DMBT1 suppresses invasion and causes tumor progression in additional cell lines. Fig. S2 shows suppression of DMBT1 down-regulates E-cadherin, up-regulates MMPs, and promotes invasion in different cell lines. Fig. S3 shows that overexpression of DMBT1 regulates mRNA stability and transcription of MMP9 and E-cadherin. Fig. S4 shows lateral (sub-epithelial) invasion in patients with HNSCC. Fig. S5 shows DMBT1 expression in nonmalignant epithelium. Table S1 provides meta-analysis of DMBT1 expression in head and neck cancer versus normal tissue datasets. Table S2 displays the total malignancy score for xenograft tumors with control (empty vector) or DMBT1-overexpressing cells. Table S3 lists sequences for gene expression primers. Table S4 lists sequences for ChIP primers. Table S5 lists demographic and clinical characteristics of patients evaluated. Table S6 provides a detailed list of reagents and resources used for this study.
Acknowledgments
We are grateful to the University of Michigan Vector Core for generating stable and inducible constructs. We thank Mutant Mouse Regional Resource Center U42OD010918 (Columbia, MO) for providing the mouse strain 030289-MU [B6.129X1-Dmbt1tm1Kumc/Mmmh]. We also thank the University of Michigan Transgenic Animal Model Core for in vitro fertilization with the frozen sperm from Mutant Mouse Regional Resource Center. We thank Ms. Theresa Cody for tissue sectioning and Ms. Victoria Zakrzewski for help with the illustration. The authors thank many investigators in the University of Michigan HNSPORE/HN Cancer Program for their contributions to patient recruitment, assistance in data collection, and encouragement, including Carol R. Bradford, MD, Thomas E. Carey, PhD, Douglas B. Chepeha, MD, Sonia Duffy, PhD, Avraham Eisbruch, MD, Joseph Helman, DDS, Kelly M. Malloy, MD, Jonathan McHugh, MD, Scott A. McLean, MD, Tamara H. Miller, RN, Jeff Moyer, MD, Mark E. Prince, MD, Nancy Rogers, RN, Matthew E. Spector, MD, Nancy E. Wallace, RN, Heather Walline, PhD, Brent Ward, DDS, and Francis Worden, MD. We greatly thank patients and their families who tirelessly participated in survey and specimen collections.
This work was supported by grants from the National Institute of Dental and Craniofacial Research (DE022567 and DE027551 to N.J. D’Silva). The HNSPORE grant no. was P50CA097248.
Author contributions: Concept, design and overall supervision were performed by N.J. D’Silva. P. Singh, R. Banerjee, S. Piao, M. Liu, M. Costa de Medeiros, E. Danella, and D. Damodaran P.V. designed and performed experiments and analyzed and interpreted data. N.J. D’Silva, P. Singh, and L.B. Schmitd did tissue analysis. N. Russo did antibody optimization for immunoblots, and R.C. Inglehart did analysis of Oncomine datasets of HNSCC. P. Singh and E. Bellile did statistical analysis. N.J. D’Silva and P. Singh wrote the manuscript. G. Wolf/HNSPORE provided patient samples. D.M. Wellik and K.M. Pineault helped with the genetically modified mouse model. G. Wolf and D.M. Wellik gave helpful suggestions and were involved in methods and data discussion.

