


Genome editing is a powerful technique for delineating complex signaling circuitry and enhancing the functionality of immune cells for immunotherapy. Natural killer (NK) cells are potent immune effectors against cell malignancy, but they are challenging to modify genetically by conventional methods due to the toxicity of DNA when introduced into cells coupled with limited transfection and transduction efficiency. Here, we describe an integrated platform that streamlines feeder-free ex vivo expansion of cryopreserved primary human NK cells and nonviral genome editing by the nucleofection of CRISPR-Cas9 ribonucleoproteins (Cas9 RNPs). The optimized Cas9 nucleofection protocol allows efficient and multiplex gene knockout in NK cells while preserving high cell viability and negligible off-target effects. Cointroduction of a DNA template also enables in-frame gene knock-in of an HA affinity tag and a gfp reporter across multiple loci. This work demonstrates the advantages and flexibility of working with cryopreserved NK cells as potential off-the-shelf engineered therapeutic agents.

Introduction
Adoptive immunotherapy is a promising approach to treat cancers. T and natural killer (NK) cells are popular cell types used in clinical trials owing to their ability to recognize and destroy malignant cells. NK cells are unique from T cells because they do not rely on a matching human leukocyte antigen to function, making allogeneic transfer safe from graft-versus-host diseases (Morvan and Lanier, 2016). Instead, NK cells use an array of activating and inhibitory receptors that form immune synapses with cognate ligands on target cells and modulate the cytotoxicity of NK cells (Morvan and Lanier, 2016). Target lysis can be initiated by endogenous activating receptors or in synergy with an engineered chimeric antigen receptor to increase killing activity and reduce tumor evasion (Guillerey et al., 2016). These unique attributes make NK cell immunotherapy attractive to and more compatible with a broad group of patients (Souza-Fonseca-Guimaraes et al., 2019; Liu et al., 2020).
Genetic engineering is a major driving force in advancing immunotherapy. T cell therapy is a successful example owing to robust viral transduction and CRISPR genome editing (Bailey and Maus, 2019; Roth et al., 2018). In contrast, genetic engineering of NK cells is challenging using conventional methods. NK cells are highly sensitive and resistant to exogenous DNA that encodes the DNA modifications of interest. Retroviral transduction requires high viral titer and poses concerns of insertional mutagenesis and oncogenesis (Imai et al., 2005; Hacein-Bey-Abina et al., 2008). Lentiviral transduction is inconsistent for NK cells even at high multiplicity of infection (Sutlu et al., 2012; Boissel et al., 2012). Plasmid transfection has limited efficiency to express transgenes (Ingegnere et al., 2019; Rautela et al., 2018 Preprint). A more robust and precise genetic toolkit for NK cells is urgently needed.
Recent advances in CRISPR technologies have reinvigorated interest in genome editing of human NK cells (Rautela et al., 2018,Preprint; Lambert et al., 2020; Kararoudi et al., 2019, Preprint; Pomeroy et al., 2020; Nguyen et al., 2020). Cas9 ribonucleoprotein (RNP) is already a popular format for various cell types owing to its transient activity, high efficiency, and multiplex capability (Farboud et al., 2018). Gene KO is mediated by Cas9 DNA cleavage and insertion-deletion mutagenesis (indel) introduced by the nonhomologous end joining (NHEJ) repair pathway. Gene knock-in (KI) is achieved via cointroduction of a DNA repair template for homology-directed repair (HDR) at the Cas9 target site, but the KI efficiency is compounded by high DNA toxicity and poor cell survival (Kararoudi et al., 2019 Preprint). Optimizing the delivery of Cas9 RNP and the DNA template is crucial for efficient genome editing and cell recovery.
Here, we describe a robust and nonviral CRISPR genome editing platform for primary human NK cells that combines the advantages of feeder-free ex vivo expansion of cryopreserved cells and genome editing by Cas9 RNP nucleofection. Cryopreservation simplifies cell storage and transport and makes NK cells more accessible for research applications and the development of off-the-shelf therapeutics. Our protocol generates 2,000-fold expansion on average and completely bypasses the need for co-culture with peripheral blood mononuclear cells or cancer cell lines to avoid mixed cell population and editing results. The expanded NK cells are of high purity, cytotoxicity, and viability for genome editing and functional analyses. We systematically optimized the conditions for Cas9 RNP and DNA nucleofection to achieve robust single and triple KO at >90% and 75%, respectively. Through high-throughput sequencing, we show that single KO is highly precise, but mutagenic chromosomal translocations are evident in multiplex editing. While DNA toxicity remains an obstacle, nonviral gene KI of a human influenza hemagglutinin (HA) affinity tag and a gfp reporter is feasible across multiple genomic loci. Our platform overcomes several technical hurdles in genetic modifications of human NK cells and is readily adoptable for NK cell research and the development of NK cell immunotherapy.
Results
Feeder-free expansion of cryopreserved NK cells is highly robust
Many protocols exist for ex vivo expansion of primary NK cells, but we wanted a more defined condition to investigate the effects of culture medium, cytokine supplementation, and chemical treatment on CRISPR genome editing of NK cells. Although feeder-dependent protocols give higher cell yields, co-culture with peripheral blood mononuclear cells or irradiated cancer cell lines imposes complexity in cell-population and gene-editing analyses. The purification of NK cells before the gene-editing procedure also induces cellular stresses that may influence the editing outcome. To avoid potential pitfalls, we established a feeder-free protocol that allows robust expansion of cryopreserved primary NK cells (Fig. 1 A). To ensure consistency, we independently expanded NK cells from five donors (Table S1) and monitored the expansion rate, surface markers, cell morphology, and in vitro cytotoxicity.

Optimized feeder-free ex vivo expansion enabled robust cell production from cryopreserved primary human NK cells. (A) The expansion protocol consists of two phases, starting with the thawing of cryopreserved NK cells. Expansion rates and NK cell purity in different culture media and IL-2 dosages were analyzed. (B) Expansion rates in Phase I in four different media plus 1,000 U/ml IL-2. (C) Mean percentages and representative flow cytometry plots of CD56+ CD3− cells on day 14. (D) NK cells were nucleofected with CD96-targeting Cas9 RNP using the reference condition (Rautela et al., 2018Preprint) on day 14 to determine cell viability and CD96− cells for quality controls. (E) Expansion rates in Phase II in NK MACS and EL837 media supplemented with two dosages of IL-2. (F) Mean percentages and representative flow cytometry plots of CD56+ CD3− cells on day 28. (G) Cell viability and CD96− cells on day 28. Expansion data are shown as mean ± SD of five donors (n = 5). CD96 KO data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch's unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01. ns, not significant; std, standard for comparison.
Optimized feeder-free ex vivo expansion enabled robust cell production from cryopreserved primary human NK cells. (A) The expansion protocol consists of two phases, starting with the thawing of cryopreserved NK cells. Expansion rates and NK cell purity in different culture media and IL-2 dosages were analyzed. (B) Expansion rates in Phase I in four different media plus 1,000 U/ml IL-2. (C) Mean percentages and representative flow cytometry plots of CD56+ CD3− cells on day 14. (D) NK cells were nucleofected with CD96-targeting Cas9 RNP using the reference condition (Rautela et al., 2018Preprint) on day 14 to determine cell viability and CD96− cells for quality controls. (E) Expansion rates in Phase II in NK MACS and EL837 media supplemented with two dosages of IL-2. (F) Mean percentages and representative flow cytometry plots of CD56+ CD3− cells on day 28. (G) Cell viability and CD96− cells on day 28. Expansion data are shown as mean ± SD of five donors (n = 5). CD96 KO data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch's unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01. ns, not significant; std, standard for comparison.
We divided the expansion procedure into two phases for systematic optimization. Phase I (days 0–14) aimed to improve cell recovery after thawing. NK cells were carefully thawed in four different media (X-VIVO 15, LymphoONE, NK MACS, and EL837; Fig. S1 A). All media were supplemented with 1,000 U/ml IL-2 and antibody-conjugated magnetic beads (anti-NKp46 and anti-CD2) to stimulate growth. We used human platelet lysate (HPL) instead of FBS to keep a xeno-free and clinically compatible procedure. DNase I treatment was needed to prevent cell aggregation and death after thawing (Fig. S1 B). As growth resumed, NK cells formed clusters and transformed from rounded and suspended to irregularly shaped and semiadherent (Fig. S1 C).

The thawing procedure is critical for NK cell viability and expansion. (A) Workflow of the thawing of frozen NK cells as described in Materials and methods. (B) NK cells treated with or without DNase I at 48 h after thawing. (C) Microscopic images of NK cells show the change in cell morphology and formation of large clusters as the expansion progressed. Scale bars indicate 200 µm. Scale bars in the zoomed panels indicate 20 µm.
The thawing procedure is critical for NK cell viability and expansion. (A) Workflow of the thawing of frozen NK cells as described in Materials and methods. (B) NK cells treated with or without DNase I at 48 h after thawing. (C) Microscopic images of NK cells show the change in cell morphology and formation of large clusters as the expansion progressed. Scale bars indicate 200 µm. Scale bars in the zoomed panels indicate 20 µm.
Regarding cell proliferation, NK MACS and EL837 outperformed X-VIVO 15 and LymphoONE, yielding ∼30-fold expansion on day 14 (Fig. 1 B). Donor variation was observed. For instance, in NK MACS, the expansion rates varied from 25- to 45-fold. Regardless, all media maintained high levels of CD56+ CD3− cells at >95% (Fig. 1 C). We selected NK MACS and EL837 for further analyses because of their higher proliferative potential. The NK cells from both media expressed similar profiles of surface receptors, except CD16 (Fig. 2, A and B). The cells were also equally cytotoxic against K562 cells (Fig. 2 C).

The NK cells expanded in NK MACS and EL837 media showed comparable profiles of surface markers and cytotoxicity. (A) Profiles of surface markers from the indicated expansion days and conditions. Data are shown as mean ± SD of five donors (n = 5). (B) Representative flow cytometry plots of surface marker staining, using the cells from donor 3 on day 28 from NK MACS medium plus 1,000 U/ml IL-2. Antibody isotype staining is shown as gray peaks. (C) In vitro cytotoxicity against K562 cells from the indicated expansion days and conditions. (D) Surface markers of the CD96 KO cells that were isolated by FACS and reexpanded for 14 d. The unedited parental cells were processed by FACS and reexpanded for comparison. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. ns, not significant; **, P ≤ 0.01; ***, P ≤ 0.001.
The NK cells expanded in NK MACS and EL837 media showed comparable profiles of surface markers and cytotoxicity. (A) Profiles of surface markers from the indicated expansion days and conditions. Data are shown as mean ± SD of five donors (n = 5). (B) Representative flow cytometry plots of surface marker staining, using the cells from donor 3 on day 28 from NK MACS medium plus 1,000 U/ml IL-2. Antibody isotype staining is shown as gray peaks. (C) In vitro cytotoxicity against K562 cells from the indicated expansion days and conditions. (D) Surface markers of the CD96 KO cells that were isolated by FACS and reexpanded for 14 d. The unedited parental cells were processed by FACS and reexpanded for comparison. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. ns, not significant; **, P ≤ 0.01; ***, P ≤ 0.001.
Phase II (days 15–28) aimed to generate high levels of NK cells. We continued to monitor cell expansion in NK MACS and EL837 (Fig. 1 E). Cell growth in NK MACS plus 1,000 U/ml IL-2 accelerated from day 17 onward to ∼2,000-fold by day 28, but growth reached a stationary phase in the other conditions. Donor variation in the expansion rate became more apparent (1,000- to 3,000-fold). Regardless of medium formulation, all conditions maintained high levels of CD56+ CD3− cells, except that donor 5 in EL837 became mostly CD3+ (Fig. 1 F). Overall, the cell morphology and surface makers in phase II resembled those in phase I (Fig. S1 C and Fig. 2 A). The mean cytotoxicity on day 28 was also similar to that on day 14 (Fig. 2 C). Collectively, our results indicate that NK MACS medium plus 1,000 U/ml IL-2, antibody-conjugated beads, and HPL can robustly expand primary NK cells from cryopreserved stocks.
Our expansion protocol is theoretically capable of generating clinically relevant numbers of primary NK cells (106 to 108 cells per kilogram of patient in one infusion; Liu et al., 2020). A single cryovial contains 5 × 106 to 1 × 107 negatively enriched NK cells. Our mean expansion rates are 500-fold and 2,000-fold on days 21 and 28, respectively. The theoretical yields are 2.5 × 109 to 5 × 109 cells on day 21 and 1 × 1010 to 2 × 1010 cells on day 28, both of which are sufficient for one infusion in an adult patient.
The expanded NK cells could also be refrozen and rethawed and still preserved ∼80% viability after thawing (Fig. S2 A). We harvested cells on day 14 for freezing. We compared CryoStor medium (approved by the US Food and Drug Administration; serum free) and FBS + 10% DMSO (common formulation for laboratory research) and observed no significant difference in cell viability after 3 mo in liquid nitrogen (Fig. S2 A). However, DNase I treatment and 1,000 U/ml IL-2 supplement remained essential after rethawing of NK cells (Fig. S2 B). On day 2, NK cells already formed large clusters, especially in the presence of antibody-conjugated beads (Fig. S2 C). The cells were also capable of reexpansion and exhibited cytotoxicity against K562 similar to that of unfrozen cells from day 14 (Fig. S2, D and E). Although long-term freezing (>3 mo) has not been tested, the ability to refreeze the primary NK cells is advantageous for the storage and application of expanded and gene-edited cells.

The expanded NK cells can be refrozen and thawed with minimal loss in viability and cytotoxicity. (A) Viability of NK cells that were frozen in Cryostor CS10 versus FBS plus 10% DMSO. Cells were analyzed immediately after thawing (day 0) or after 2 d of incubation in NK MACS with 1000 U/ml IL-2. Blue, red, and black indicate three different donors (n = 3). (B) Effect of IL-2 supplement and antibody-conjugated beads on cell viability. (C) Cell morphology in NK MACS with different supplementations. Scale bars indicate 200 µm. (D) Reexpansion rate after thawing. (E) In vitro cytotoxicity against K562 cells on day 2 after rethawing. (F) Workflow of NK cell recovery before gene KO. (G) Comparison of cell viability and CD96− cells between the two recovery periods. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch's unequal variances t test was used to test for statistical significance. ns, not significant; *, P ≤ 0.05.
The expanded NK cells can be refrozen and thawed with minimal loss in viability and cytotoxicity. (A) Viability of NK cells that were frozen in Cryostor CS10 versus FBS plus 10% DMSO. Cells were analyzed immediately after thawing (day 0) or after 2 d of incubation in NK MACS with 1000 U/ml IL-2. Blue, red, and black indicate three different donors (n = 3). (B) Effect of IL-2 supplement and antibody-conjugated beads on cell viability. (C) Cell morphology in NK MACS with different supplementations. Scale bars indicate 200 µm. (D) Reexpansion rate after thawing. (E) In vitro cytotoxicity against K562 cells on day 2 after rethawing. (F) Workflow of NK cell recovery before gene KO. (G) Comparison of cell viability and CD96− cells between the two recovery periods. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch's unequal variances t test was used to test for statistical significance. ns, not significant; *, P ≤ 0.05.
Optimized nucleofection enables efficient delivery of Cas9 RNP and DNA
Nucleofection is a robust method to shuttle Cas9 RNP and DNA into cells, but the combination of nucleofection buffer and pulse setting must be determined experimentally for different cell types. We first examined the NK cells from different expansion conditions to ensure uniform quality for genome editing. On days 14 and 28, we performed Cas9 RNP nucleofection to knock out the CD96 gene and measured CD96− cells by flow cytometry as KO efficiency. For this purpose, we used Solution 2 + mannitol buffer (Sol2) and pulse code CM137 in a Lonza 4D nucleofector (Rautela et al., 2018 Preprint). CD96− cells were comparable at ∼70% in all expansion conditions (Fig. 1, D and G). Our results suggest that the culture medium, cell proliferation rate, IL-2 dosage, and expansion duration (14 d versus 28 d) have no detectable influence on Cas9 gene editing in NK cells. Knowing that the NK cells from days 14 and 28 have comparable editing efficiencies is useful, as this information allows some flexibility in performing KO and KI optimization and analyses.
The nucleofection condition reported by Rautela et al. seemed suboptimal (Rautela et al., 2018 Preprint). We set out to optimize the combination of nucleofection buffer, pulse code, and payload dosages for efficient delivery of Cas9 RNP and DNA (Fig. 3 A). We screened 31 combinations of nucleofection buffer and pulse code, including three reference conditions for parallel comparison (Fig. 3 B and Table S2). To plot the correlation, we independently determined the percentages of CD96− and GFP+ cells (using pmaxGFP reporter plasmid) as the indicators of nucleofection efficiencies for Cas9 RNP and DNA, respectively. We also reduced Cas9 RNP to 40 pmol, which was approximately half of the reported dosages, to sensitize Cas9 editing. Overall, P3 buffer performed better than Sol2 (Fig. 3 C). Conditions 3, 4, and 5 gave more CD96− cells, whereas conditions 2, 3, and 5 gave more GFP+ cells.
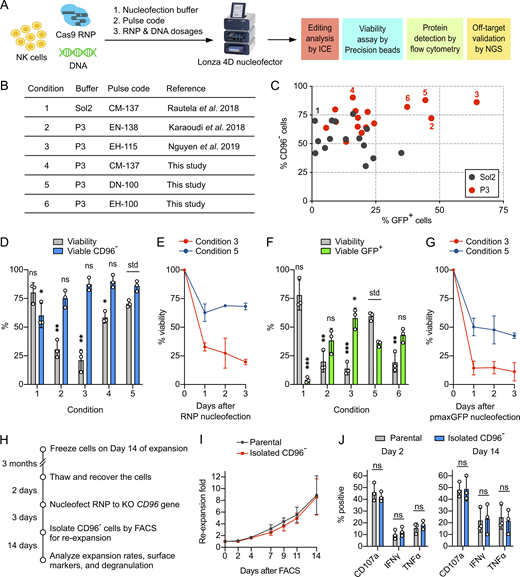
Nucleofection optimization identified conditions for efficient and viable delivery of Cas9 RNP and DNA.(A) Workflow of the nucleofection screening and editing analyses to identify the optimal combination of nucleofection buffer, pulse code, and payload dosages. The screening was performed on NK cells from day 14 of the expansion. (B) Summary of the reference and newly identified conditions. (C) Screening for Cas9 RNP and DNA (using pmaxGFP reporter plasmid) nucleofection conditions in Sol2 and Lonza P3 buffer. CD96− and GFP+ cells were determined independently. The pulse codes, buffers, and raw data are listed in Table S1. Numerically labeled conditions are listed in the summary table. (D) Selected conditions were repeated for Cas9 RNP nucleofection to determine cell viability and viable CD96− cells by Precision beads assay. (E) Cell viability was monitored for 3 d after Cas9 RNP nucleofection. (F) Selected conditions were repeated for pmaxGFP nucleofection. (G) Cell viability after pmaxGFP nucleofection. (H) Workflow of the CD96 KO, FACS, and reexpansion. (I) Reexpansion rates of the FACS-isolated CD96− and parental cells. (J) Analysis of degranulation markers after chemical stimulation. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. ns, not significant; std, standard for comparison.
Nucleofection optimization identified conditions for efficient and viable delivery of Cas9 RNP and DNA.(A) Workflow of the nucleofection screening and editing analyses to identify the optimal combination of nucleofection buffer, pulse code, and payload dosages. The screening was performed on NK cells from day 14 of the expansion. (B) Summary of the reference and newly identified conditions. (C) Screening for Cas9 RNP and DNA (using pmaxGFP reporter plasmid) nucleofection conditions in Sol2 and Lonza P3 buffer. CD96− and GFP+ cells were determined independently. The pulse codes, buffers, and raw data are listed in Table S1. Numerically labeled conditions are listed in the summary table. (D) Selected conditions were repeated for Cas9 RNP nucleofection to determine cell viability and viable CD96− cells by Precision beads assay. (E) Cell viability was monitored for 3 d after Cas9 RNP nucleofection. (F) Selected conditions were repeated for pmaxGFP nucleofection. (G) Cell viability after pmaxGFP nucleofection. (H) Workflow of the CD96 KO, FACS, and reexpansion. (I) Reexpansion rates of the FACS-isolated CD96− and parental cells. (J) Analysis of degranulation markers after chemical stimulation. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. ns, not significant; std, standard for comparison.
Cell survival after nucleofection is an important consideration. We noticed substantial cell debris in some conditions that resulted in poor cell recovery by centrifugation and led to overestimation of viability. We adopted a flow cytometry method using Precision cell count beads to normalize cell density and improve the accuracy of the viability assay (Fig. S3 A). Centrifugation and cell washing steps were eliminated to retain both live cells and dead debris. Using the Precision beads assay, we reexamined Cas9 RNP and DNA nucleofection in the selected conditions and observed a wide range of viability (Fig. S3 B). For RNP nucleofection, conditions 3, 4, and 5 gave more CD96− cells (all >85%) than conditions 1 and 2 (both <75%; Fig. 3 D). However, conditions 3 and 4 resulted in low cell viability at 21 ± 9% and 58 ± 5%, respectively. Only condition 5 maintained high KO efficiency and viability at 86 ± 4% and 70 ± 3%, respectively.

Cell viability assay by Precision cell count beads. (A) Workflow of Precision beads assay. (B) Representative flow cytometry plots of the untreated NK cells and the cells nucleofected with Cas9 RNP in the selected conditions. Viable cells were determined by light scattering as gated in the black circles. FSC, forward scatter; SSC, side scatter.
Cell viability assay by Precision cell count beads. (A) Workflow of Precision beads assay. (B) Representative flow cytometry plots of the untreated NK cells and the cells nucleofected with Cas9 RNP in the selected conditions. Viable cells were determined by light scattering as gated in the black circles. FSC, forward scatter; SSC, side scatter.
For DNA nucleofection, condition 1 was ineffective, giving 4 ± 3% of GFP+ cells (Fig. 3 F). Condition 3 gave the highest number of GFP+ cells (58 ± 9%) but the lowest viability (14 ± 5%; Fig. 3 F). Conditions 2, 5, and 6 showed similar GFP expression (∼40%), but only condition 5 maintained a high viability (60 ± 4%). We did not test condition 4 due to its low GFP expression in the initial screening. We also monitored the viability for 3 d after Cas9 RNP and pmaxGFP nucleofection (Fig. 3, E and G). The majority of NK cells died on day 1, but viability stabilized on days 2 and 3. Overall, condition 5 provided the best balance of nucleofection efficiencies and cell viability.
Gene KO by Cas9 RNP is also effective using the Neon Transfection system
We wanted to see whether Cas9 RNP editing was compatible with other electroporation systems. We screened 41 conditions in Thermo Fisher Scientific’s Neon Transfection system to determine CD96 KO efficiency and cell viability using the same analytic methods (Fig. S4 A and Table S2). The Lonza and Neon systems have distinct reaction setups and optimization schemes, making direct comparison less straightforward. To fit into the 10-µl electroporation tip of the Neon system, we scaled down the cell number and reaction volume by half while keeping the same Cas9 RNP/cell ratio. We selected conditions 7, 8, and 9 from the initial screening for triplicate analysis (Fig. S4 B). Condition 7 was reported for cas9 mRNA electroporation (Pomeroy et al., 2020). It also worked well for Cas9 RNP electroporation, yielding 72 ± 4% of CD96− cells, but at low viability (45 ± 8%). Condition 8 gave more balanced KO efficiency and viability at 51 ± 10% and 64 ± 3%, respectively. Condition 9 was inefficient for KO (28 ± 10%) but preserved high viability (71 ± 2%). While our screening results are promising, further optimization of the pulse setting and buffer composition is needed to improve the editing efficiency. For all subsequent experiments, we used Lonza 4D Nucleofector and condition 5.

Electroporation of Cas9 RNP by Neon Transfection system. (A) We screened 41 pulse settings to electroporate CD96-targeting Cas9 RNP. Condition 7 is a reference condition reported by Pomeroy et al. (2020) for cas9 mRNA. (B) Conditions 7, 8, and 9 were examined in triplicate. Cell viability and viable CD96− cells were determined as described for the Lonza system. The data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01. Std, standard for comparison.
Electroporation of Cas9 RNP by Neon Transfection system. (A) We screened 41 pulse settings to electroporate CD96-targeting Cas9 RNP. Condition 7 is a reference condition reported by Pomeroy et al. (2020) for cas9 mRNA. (B) Conditions 7, 8, and 9 were examined in triplicate. Cell viability and viable CD96− cells were determined as described for the Lonza system. The data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01. Std, standard for comparison.
Gene-KO NK cells can be reexpanded
We envisioned a streamlined protocol in which primary NK cells could be ex vivo expanded, cryopreserved for storage, thawed for gene editing on demand, and reexpanded to increase cell numbers for research and potentially therapeutic applications. Therefore, we assessed several parameters such as recovery time after thawing, reexpansion after gene KO, integrity of surface markers, and degranulation capacity. We froze NK cells on day 14 of the expansion in liquid nitrogen for 3 mo. After thawing, we recovered the cells for 1 or 2 d before Cas9 RNP nucleofection (Fig. S2 F). We did not detect any difference in cell viability and CD96 KO efficiency between the two recovery periods (Fig. S2 G). We chose 2-d recovery to ensure the best stability of NK cells.
Next we isolated the CD96− cells by FACS for reexpansion (Fig. 3 H). To control for the FACS-induced stress, we subjected the unedited parental cells to antibody staining and FACS and collected single cells of the normal dimensions regardless of CD96 expression. Both CD96− and parental cells showed continuous and comparable reexpansion rates to approximately sevenfold in 14 d (Fig. 3 I). However, the reexpansion rates were lower than that of the completely untreated cells, which was ∼17-fold 14 d after thawing (Fig. 3 I versus Fig. S2 D). The results suggest that the isolated NK cells need time to recover from the physical stress induced by FACS.
CD96− and unedited cells displayed similar profiles of surface markers on day 14 (Fig. 2 D). Most of the markers remained highly expressed, except CD16, CD158, PD-1, and the disrupted CD96. The profile of CD96− cells also resembled that of cells from the uninterrupted expansion (Fig. 2 D vs. Fig. 2 A). Finally, CD96− and unedited cells showed similar degranulation activity upon stimulation by phorbol 12-myristate-13-acetate and ionomycin (Fig. 3 J). Collectively, our assessments show that the gene-KO NK cells can be reexpanded and retain normal levels of surface markers and degranulation capacity.
High dosages of Cas9 RNP maintain editing precision but are stressful to cells
We nucleofected increasing dosages of Cas9 RNP to evaluate the effect on cell viability, KO efficiency, and off-target editing. We tested three RNP concentrations, ranging from 20 to 120 pmol, to disrupt the CD96 and SIGLEC-7 genes (Fig. 4, A and B). At both loci, high RNP dosages did not improve KO efficiency but drastically reduced viability. We also performed amplicon-based next-generation sequencing (NGS) to determine the off-target effect at 20, 40, and 120 pmol Cas9 RNP (Fig. 4 C). We analyzed the sequence integrity at the on-target site and top two predicted off-target sites from each of the CD96 and SIGLEC-7 single guide RNAs (sgRNAs). We did not detect any off-target indel at frequencies higher than the untreated cells in all tested conditions (Fig. 4 C). Together, the dosage experiment suggests that 20–40 pmol Cas9 RNP is sufficient for single KO. Furthermore, the off-target analysis suggests that with careful in silico design of sgRNA, gene editing by RNP nucleofection remains precise at up to 120 pmol Cas9.

High dosages of Cas9 RNP reduced cell viability but had a negligible effect on off-target editing. (A) Cell viability and viable CD96− cells at increasing dosages of Cas9 RNP were analyzed by Precision beads assay. Representative flow cytometry histograms were from 40 pmol Cas9 RNP. (B) The results of SIGLEC7 KO. (C) Analysis of CD96 and SIGLEC7 on- and off-target editing by amplicon-based NGS. Mismatches between the on- and off-target sequences are labeled in red. Sequence variations were determined by CRISPResso2 and are presented as indel percentage. The parental cells contained a single-nucleotide polymorphism in the SIGLEC7 on-target site that gave rise to 5% of background indel. Chr, chromosome; PAM, protospacer adjacent motif. (D and E) Influence of different sgRNA preparations on cell viability in the KO of CD96 (D) and SIGLEC7 (E). Data are shown as mean ± SD of three donors (n = 3). The off-target analysis was performed in one donor (n = 1). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. IVT, in vitro transcription; ns, not significant; std, standard for comparison.
High dosages of Cas9 RNP reduced cell viability but had a negligible effect on off-target editing. (A) Cell viability and viable CD96− cells at increasing dosages of Cas9 RNP were analyzed by Precision beads assay. Representative flow cytometry histograms were from 40 pmol Cas9 RNP. (B) The results of SIGLEC7 KO. (C) Analysis of CD96 and SIGLEC7 on- and off-target editing by amplicon-based NGS. Mismatches between the on- and off-target sequences are labeled in red. Sequence variations were determined by CRISPResso2 and are presented as indel percentage. The parental cells contained a single-nucleotide polymorphism in the SIGLEC7 on-target site that gave rise to 5% of background indel. Chr, chromosome; PAM, protospacer adjacent motif. (D and E) Influence of different sgRNA preparations on cell viability in the KO of CD96 (D) and SIGLEC7 (E). Data are shown as mean ± SD of three donors (n = 3). The off-target analysis was performed in one donor (n = 1). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. IVT, in vitro transcription; ns, not significant; std, standard for comparison.
5′ phosphate moiety of the in vitro–transcribed sgRNA is highly toxic
The preparation of sgRNA affects NK cell viability. We observed severe toxicity using the in vitro–transcribed sgRNA, but not the chemically synthesized sgRNA (Fig. 4, D and E). The 5′ triphosphate moiety of the in vitro–transcribed sgRNA is an immunogenic epitope that activates RIG-1 intracellular RNA sensor and triggers inflammatory responses in various cell types (Kim et al., 2018; Wienert et al., 2018). Our results confirm that primary NK cells are also highly sensitive to the 5′ triphosphate group of sgRNA. Removal of the triphosphate group by the treatment of calf intestine phosphatase (CIP) effectively alleviated RNA toxicity and restored cell viability to a level comparable to chemically synthesized sgRNA.
Cas9 RNP nucleofection allows rapid testing of sgRNA in cells
Cas9 RNP nucleofection is a rapid approach to assess sgRNA performance and identify accessible genomic sites in cells. We robustly disrupted TIGIT and CD226 genes, which led to >90% reduction in protein expression of the TIGIT inhibitory and DNAM-1 activating receptors, respectively (Fig. 5). The TIGIT- and CD226-targeting sgRNAs were previously validated in the NK-92 cell line (Huang et al., 2020) and here showed similar efficiencies in primary NK cells. We designed two sgRNAs to target the KLRC1 gene (encoding the NKG2A receptor). The KO efficiency was modest when each sgRNA was applied singly or in combination for double cutting (Fig. 5). We used a similar strategy to target the PDCD1 locus and achieved 98% of PD-1–negative cells, although the parental NK cells were also already ∼75% PD-1 negative (Fig. 5).

Cas9 RNP nucleofection allowed rapid testing of sgRNA in cells. Six sgRNAs were designed to target TIGIT, CD226, KLRC1, and PDCD1 at the marked exons. Representative flow cytometry plots show the mean percentages of negative cells. In KLRC1 and PDCD1 KO, the distances between the two sgRNAs are labeled. ICE analysis was performed in KLRC1 and PDCD1 KO to reveal the discrepancy between DNA sequencing and flow cytometry readouts. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; ***, P ≤ 0.001. FSC, forward scatter; ns, not significant.
Cas9 RNP nucleofection allowed rapid testing of sgRNA in cells. Six sgRNAs were designed to target TIGIT, CD226, KLRC1, and PDCD1 at the marked exons. Representative flow cytometry plots show the mean percentages of negative cells. In KLRC1 and PDCD1 KO, the distances between the two sgRNAs are labeled. ICE analysis was performed in KLRC1 and PDCD1 KO to reveal the discrepancy between DNA sequencing and flow cytometry readouts. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05; ***, P ≤ 0.001. FSC, forward scatter; ns, not significant.
Strikingly, we observed a discrepancy between the protein and DNA analyses by flow cytometry and inference of CRISPR edits (ICE), respectively. In the KLRC1 KO, the indel percentages of sgRNA 5 and sgRNA 5+6 by ICE analysis were both >80%, in sharp contrast to the <25% of NKG2A− cells detected by flow cytometry (Fig. 5). The indel percentage induced by sgRNA 7+8 in the PDCD1 KO was also surprisingly higher than the individual sgRNA alone. Although Cas9 RNP nucleofection allows rapid assessment of sgRNA performance, our findings highlight the need for complementary analyses at both the protein and DNA levels to confirm the editing efficiency.
Multiplex KO by Cas9 RNP nucleofection is highly efficient
KO of multiple genes is needed to dissect a complex biological process or study a combinatory effect. However, disrupting multiple genes one at a time is less feasible for primary cells when the ex vivo life span is limited. The Cas9 RNP approach allows the pooling of multiple Cas9 complexes, each with unique target specificity at a precise molar ratio, to simultaneously target multiple loci in a single nucleofection reaction. This multiplexing approach is more challenging in DNA- or mRNA-based CRISPR editing due to the difficulty of cointroducing multiple sgRNAs. To demonstrate this in primary NK cells, we first performed double KO of surface receptors by combining two Cas9 RNP complexes to a total of 80 pmol. Double KO of TIGIT and CD96 was highly robust, converting 89% of double-positive parental cells to 86% double negative (Fig. 6 A). Viability was maintained at ∼60%, as seen in the RNP dosage experiment. Similar KO efficiency and viability were obtained in the double KO of TIGIT and CD226 (Fig. 6 B). We then combined the TIGIT-, CD96-, and CD226-targeting RNP complexes for triple KO. Surprisingly, triple KO was just as efficient as double KO, converting 73% of triple-positive parental cells to 76% triple negative (Fig. 6 C). For simplicity, we did not quantitate the single- and double-positive populations. Cell viability dropped to ∼35% due to high combined RNP dosage (40 pmol × 3). Reducing the RNP dosage rescued the viability without changing the triple-KO efficiency, but large donor variation renders the improvement insignificant (Fig. 6 C). Our results demonstrate that multiplex KO by Cas9 RNP nucleofection is an efficient approach in primary NK cells. Fine-tuning of the RNP ratio could improve KO efficiency and cell viability.

Multiplex KO was highly efficient by Cas9 RNP nucleofection. (A) Double KO of TIGIT and CD96 by combining two doses of 40 pmol Cas9 RNP complexes in one nucleofection reaction. Representative flow cytometry plots show the mean percentages of positive and negative cells. (B) Double KO of TIGIT and CD226 (encoding DNAM-1). (C) Triple KO of TIGIT, CD96, and CD226 by combining three doses of 40 pmol Cas9 RNP complexes in one nucleofection reaction. Q2 and Q4 of the representative flow cytometry plots were gated to determine triple-positive and triple-negative cells, respectively. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. FSC, forward scatter; ns, not significant.
Multiplex KO was highly efficient by Cas9 RNP nucleofection. (A) Double KO of TIGIT and CD96 by combining two doses of 40 pmol Cas9 RNP complexes in one nucleofection reaction. Representative flow cytometry plots show the mean percentages of positive and negative cells. (B) Double KO of TIGIT and CD226 (encoding DNAM-1). (C) Triple KO of TIGIT, CD96, and CD226 by combining three doses of 40 pmol Cas9 RNP complexes in one nucleofection reaction. Q2 and Q4 of the representative flow cytometry plots were gated to determine triple-positive and triple-negative cells, respectively. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. FSC, forward scatter; ns, not significant.
Double KO suggests that CD96 is not crucial for NK cell activation
The role of CD96 in human NK cell activation is unclear. Studies using blocking antibodies suggest that CD96 has inhibitory or no function (Sun et al., 2019; El-Sherbiny et al., 2007; Carlsten et al., 2007). However, blocking antibodies of different clones are known to give conflicting results in murine NK cells (Roman Aguilera et al., 2018). We sought to gain more insight using the genetic approach. Since CD96 and DNAM-1 recognize the same ligand CD155, we asked how single and double KO affected NK cell activity against CD155+ human cancer cell lines (breast cancer MDA-MB-231 and malignant glioblastoma U-251 MG; Fig. 7 A). We isolated the single- and double-KO cells and performed cytotoxicity and degranulation assays (Fig. 7, B and C). The CD96-KO cells showed slight decrease in cytotoxicity against MDA-MB-231, but not U-251 MG (Fig. 7 D). In comparison, the CD226-KO cells lost nearly half of the cytotoxicity against both cancer cell lines, confirming its importance in NK cell activation. The double KO did not show further reduction in cytotoxicity compared with the single CD226 KO (Fig. 7 D). Large donor variation in the degranulation assay rendered most of the differences insignificant, except that CD226 KO showed a reduced CD107a level (Fig. 7 E). Our results suggest that CD96 is not crucial for NK cell activation and agree with the findings by El-Sherbiny et al. (2007) and Carlsten et al. (2007).

Double KO revealed that CD96 was not crucial for NK cell activation. (A) Cytotoxicity assay was performed using MDA-MB-231 and U-251 MG cancer cell lines. Both cells expressed high levels of CD155, which is a ligand for CD96 and DNAM-1 (encoded by the CD226 gene). (B) Representative flow cytometry plots show the distributions of single- and double-KO cells. (C) Workflow of the double-KO nucleofection, FACS, and functional assays. (D) Cytotoxicity of the KO cells against the cancer cells at the indicated ratios. Data are shown as mean ± SD of three donors and each in triplicate (n = 9). (E) Degranulation markers of the KO cells after incubating with the cancer cells. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. ns, not significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Double KO revealed that CD96 was not crucial for NK cell activation. (A) Cytotoxicity assay was performed using MDA-MB-231 and U-251 MG cancer cell lines. Both cells expressed high levels of CD155, which is a ligand for CD96 and DNAM-1 (encoded by the CD226 gene). (B) Representative flow cytometry plots show the distributions of single- and double-KO cells. (C) Workflow of the double-KO nucleofection, FACS, and functional assays. (D) Cytotoxicity of the KO cells against the cancer cells at the indicated ratios. Data are shown as mean ± SD of three donors and each in triplicate (n = 9). (E) Degranulation markers of the KO cells after incubating with the cancer cells. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. ns, not significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Multiplex KO poses a risk of chromosomal translocation
We previously reported in the NK-92 human NK cell line that multiplex KO led to chromosomal translocation (Huang et al., 2020), likely due to misligation by NHEJ between multiple Cas9-induced double-strand breaks. Chromosomal translocation is a highly deleterious mutation, but it is often overlooked by standard gene-editing analyses. We used the same PCR assay to detect chromosomal translocation in triple-KO primary NK cells. CD96 and TIGIT are both on chromosome 3, and CD226 is on chromosome 18. We used 15 combinations of PCR primers to probe various inter- and intrachromosomal translocation patterns across the target sites (Fig. 8 A). The parental cells were negative in all 15 patterns, as shown by DNA gel electrophoresis and highly sensitive SYRB Gold staining (Fig. 8 B). Even after 35 cycles of over-amplification, we observed only minor nonspecific PCR products. In contrast, chromosomal translocation was evident in the triple-KO cells. We detected several patterns of chromosomal arrangement that matched the expected fragment sizes at various band intensities (Fig. 8 B). Our results show that chromosomal translocation is a risk of multiplex gene editing but can be detected by a simple PCR assay.

Chromosomal translocation was detected in the TIGIT-CD96-CD226 triple-KO cells. (A) Schematics of the TIGIT, CD96, and CD226 loci show the PCR primers, sgRNA target positions, and the expected sizes of PCR fragments. (B) We used a PCR-based assay and 15 primer sets to probe various patterns of chromosomal translocation. PCR was performed for 28 and also 35 cycles to increase the detection sensitivity. All DNA gels were loaded with the same amount of DNA ladder (marked as M) for parallel comparison and visualized by SYBR Gold staining.
Chromosomal translocation was detected in the TIGIT-CD96-CD226 triple-KO cells. (A) Schematics of the TIGIT, CD96, and CD226 loci show the PCR primers, sgRNA target positions, and the expected sizes of PCR fragments. (B) We used a PCR-based assay and 15 primer sets to probe various patterns of chromosomal translocation. PCR was performed for 28 and also 35 cycles to increase the detection sensitivity. All DNA gels were loaded with the same amount of DNA ladder (marked as M) for parallel comparison and visualized by SYBR Gold staining.
Plasmid-based gene editing is completely ineffective
Plasmid-based CRISPR gene editing is a standard approach for many cell types, but it failed to work in NK-92 cells (Huang et al., 2020). Here, we discovered that plasmid-based editing was also ineffective in primary NK cells. We suspected that DNA toxicity was the main issue. Using pmaxGFP as a reporter, we saw a rapid decline in cell viability at higher DNA concentrations, but donor variation remained large (Fig. 9 A). Then we constructed three CRISPR plasmids, each encoding the same CD96, TIGT or KLRC1-target sgRNA sequence validated by the RNP approach. These plasmids produced high % indels across the three loci in HEK293T cells, but gave no detectable indel in NK cells (Fig. 9 B). Similar to NK-92 cells, Cas9 expression could not be detected by Western blotting against the FLAG fusion tag (Fig. 9 C). The expression of sgRNA in primary NK cells was also lower than in HEK293T as detected by RT-PCR (Fig. 9 C). The results suggest that the lack of Cas9 expression is likely the reason that plasmid-based editing failed in primary NK cells.

Plasmid-based CRISPR gene editing was completely ineffective in primary NK cells. (A) Effect of pmaxGFP dosage on cell viability and GFP expression. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05. ns, not significant; NT, untreated cells; std, standard for comparison. (B) Plasmid-based gene editing was performed to target CD96, TIGIT, and KLRC1 genes in NK cells and the HEK293T cell line as a positive control. No indel was detected by ICE analysis in NK cells from three donors (n = 3). HEK293T results were from three independent experiments (n = 3). (C) Western blotting and RT-PCR were performed to detect the expression of Cas9-FLAG and sgRNA, respectively. Cell lysates were sampled immediately (time = 0) and 24 h after nucleofection. GAPDH protein and β-tubulin cDNA served as internal controls. The results were from one donor (n = 1).
Plasmid-based CRISPR gene editing was completely ineffective in primary NK cells. (A) Effect of pmaxGFP dosage on cell viability and GFP expression. Data are shown as mean ± SD of three donors (n = 3). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05. ns, not significant; NT, untreated cells; std, standard for comparison. (B) Plasmid-based gene editing was performed to target CD96, TIGIT, and KLRC1 genes in NK cells and the HEK293T cell line as a positive control. No indel was detected by ICE analysis in NK cells from three donors (n = 3). HEK293T results were from three independent experiments (n = 3). (C) Western blotting and RT-PCR were performed to detect the expression of Cas9-FLAG and sgRNA, respectively. Cell lysates were sampled immediately (time = 0) and 24 h after nucleofection. GAPDH protein and β-tubulin cDNA served as internal controls. The results were from one donor (n = 1).
Conucleofection of a DNA template enabled site-specific gene KI
Gene KI by Cas9-mediated HDR is a useful strategy to site-specifically insert DNA sequences of interest into the genome to provide novel gene functions. However, the endogenous activity of HDR is tightly regulated in cells, and the frequency of HDR is low. KI efficiency is further confined by toxicity associated with the introduction of synthetic DNA into cells. This is a known problem in human primary NK cells and cell lines (Kararoudi et al., 2019, Preprint; Huang et al., 2020). To determine the KI efficiency in our setup, we synthesized sgRNAs and DNA templates for in-frame insertion of the coding sequences of HA affinity tag and GFP protein into the CD96, ACTB, and RAB11A loci (Fig. S5 A).
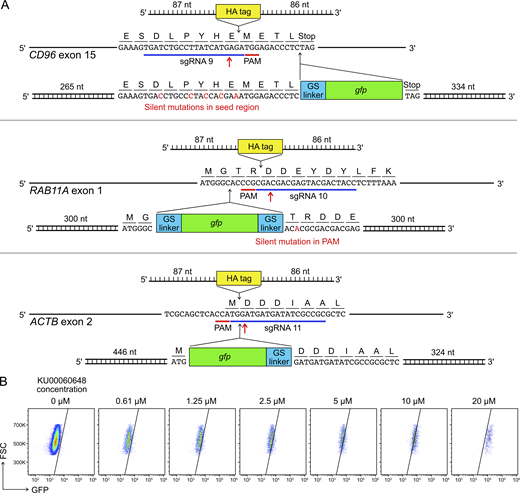
HDR template design and KU00060648 treatment.(A) Single-stranded DNA ultramers were used for the KI of HA tag; whereas PCR-generated, double-stranded DNA templates were used for the KI of gfp gene. Red arrows mark Cas9 cleavage sites. GS indicates a glycine-serine linker. The length of homology arms is indicated next to the insert. In the CD96 HDR template, silent mutations were introduced in the sgRNA seed region to avoid Cas9 targeting. In the RAB11A HDR template, a single-base silent mutation was created to disrupt the PAM sequence. The HDR strategies for RAB11A and ACTB were adopted from Roberts et al. (2017) and Roth et al. (2018). The full DNA sequences are listed in Table S5. (B) Treatment of KU00060648 alone, which is a DNA-dependent protein kinase inhibitor and also marketed as a HDR enhancer, produced false GFP signal in a dosage-dependent manner. FSC, forward scatter; PAM, protospacer adjacent motif.
HDR template design and KU00060648 treatment.(A) Single-stranded DNA ultramers were used for the KI of HA tag; whereas PCR-generated, double-stranded DNA templates were used for the KI of gfp gene. Red arrows mark Cas9 cleavage sites. GS indicates a glycine-serine linker. The length of homology arms is indicated next to the insert. In the CD96 HDR template, silent mutations were introduced in the sgRNA seed region to avoid Cas9 targeting. In the RAB11A HDR template, a single-base silent mutation was created to disrupt the PAM sequence. The HDR strategies for RAB11A and ACTB were adopted from Roberts et al. (2017) and Roth et al. (2018). The full DNA sequences are listed in Table S5. (B) Treatment of KU00060648 alone, which is a DNA-dependent protein kinase inhibitor and also marketed as a HDR enhancer, produced false GFP signal in a dosage-dependent manner. FSC, forward scatter; PAM, protospacer adjacent motif.
Our expanded NK cells were capable of HDR, but the KI efficiency varied significantly between different target sites and donor cells. The HA KI was most efficient at the CD96 locus at 16.5 ± 8.1% frequency (Fig. 10, A and B). The detection of the HA tag by ICE and flow cytometry analyses gave comparable KI efficiencies. However, the same analyses at the ACTB and RAB11A loci yielded very different results, as ICE failed to detect the presence of the HA sequence (Fig. 10 A). This finding again stresses the need for complementary editing analyses. The gfp KI efficiencies were similar across all three sites, but the viability was the lowest at RAB11A (Fig. 10, C and D). As seen in the HA and gfp KI experiments, both single- and double-stranded DNA templates were toxic to primary NK cells.

Nonviral gene KI is feasible in primary NK cells. (A) HA tag KI efficiencies at the CD96, ACTB, and RAB11A loci were determined by ICE and flow cytometry. The percent viability was analyzed by Precision beads assay. (B) Representative flow cytometry plots with the mean KI rates ± SD. The positions of HA tag KI are indicated. (C) The gfp KI efficiencies as determined by flow cytometry. (D) Representative flow cytometry plots with the mean KI rates ± SD. The positions of gfp KI are indicated. (E) Reexpansion rate of the FACS-isolated GFP+ cells that carried the gfp KI at the CD96 locus. (F) Representative flow cytometry histogram shows the expression of GFP after 14 d of reexpansion. (G) Screening of HDR enhancers to increase the gfp KI efficiency at the CD96 locus. (H) Flow cytometry plots show the percentage of GFP+ cells from different enhancer treatments. Cell density was normalized by Precision beads count to reveal reduction in the cell number in some treatments. Data are shown as mean ± SD of three donors (n = 3), except that HDR enhancers were tested in one donor (n = 1). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05. FSC, forward scatter; ns, not significant.
Nonviral gene KI is feasible in primary NK cells. (A) HA tag KI efficiencies at the CD96, ACTB, and RAB11A loci were determined by ICE and flow cytometry. The percent viability was analyzed by Precision beads assay. (B) Representative flow cytometry plots with the mean KI rates ± SD. The positions of HA tag KI are indicated. (C) The gfp KI efficiencies as determined by flow cytometry. (D) Representative flow cytometry plots with the mean KI rates ± SD. The positions of gfp KI are indicated. (E) Reexpansion rate of the FACS-isolated GFP+ cells that carried the gfp KI at the CD96 locus. (F) Representative flow cytometry histogram shows the expression of GFP after 14 d of reexpansion. (G) Screening of HDR enhancers to increase the gfp KI efficiency at the CD96 locus. (H) Flow cytometry plots show the percentage of GFP+ cells from different enhancer treatments. Cell density was normalized by Precision beads count to reveal reduction in the cell number in some treatments. Data are shown as mean ± SD of three donors (n = 3), except that HDR enhancers were tested in one donor (n = 1). Two-tailed Welch’s unequal variances t test was used to test for statistical significance. *, P ≤ 0.05. FSC, forward scatter; ns, not significant.
We also isolated the GFP+ cells by FACS and monitored the reexpansion rate (Fig. 10 E). The gfp KI cells showed minimal proliferation (twofold), unlike the gene-KO cells (sevenfold). Despite not proliferating, the isolated cells maintained robust GFP signal after 14 d of reexpansion (Fig. 10 F). The results show that Cas9-mediated gene KI downstream of a steady endogenous promoter (of the CD96 gene in this case) is a useful strategy to stably express foreign genes in NK cells. However, the FACS process appeared stressful and reduced the growth of the sorted parental cells in both KO and KI experiments.
To improve the KI efficiency, we treated cells with a panel of HDR enhancers and repeated the gfp KI at the CD96 locus (Fig. 10 F). None of the enhancers showed clear improvement on the KI efficiency. In fact, several enhancers were toxic to NK cells (Fig. 10 F). The toxicity could be due to cross-reactivity with other cellular pathways, as seen with the DNA-dependent protein kinase inhibitor KU-00060648. This inhibitor also blocks phosphatidylinositol 3-kinases, a family of enzymes essential for IL-2 signaling and NK cell survival (Kawauchi et al., 2005; Munck et al., 2012). Strangely, treatment with KU-00060648 alone also gave a false GFP signal in a dosage-dependent manner (Fig. S5 B). Taken together, our results show that nonviral gene KI, via the conucleofection of Cas9 RNP and synthetic DNA templates, is feasible in primary NK cells. However, chemical treatment is not a direct approach to enhance the KI efficiency and may lead to unexpected effects on NK cells.
Discussion
Recent advances in CRISPR genome editing technology create exciting new possibilities to genetically study NK cell immunology and engineer NK cells to enhance anticancer capabilities. In this work, we demonstrate a robust CRISPR genome editing platform for primary human NK cells that combines the advantages of feeder-free ex vivo expansion and Cas9 RNP nucleofection to replace inefficient plasmid transfection and viral transduction. There are many opportunities and challenges to extend our proof-of-concept demonstrations toward therapeutic applications of NK cell.
We provide the first demonstration (to the best of our knowledge) of robust feeder-free ex vivo expansion of cryopreserved primary human NK cells. Our protocol has many advantages. First, the use of cryopreserved cells streamlines cell storage and experimental logistics and creates more research opportunities for laboratories that do not have routine access to fresh donor blood. Second, the elimination of feeder cells simplifies the manufacturing of cell-based therapeutics and provides a defined growth condition to study NK cell functionalities. Third, our protocol can theoretically produce clinically relevant numbers of highly pure and cytotoxic NK cells for therapeutic applications and research. The expanded cells also retain high viability to sustain the stress induced by the nucleofection procedure. Forth, the expanded cells can be frozen again. Upon rethawing, the cells preserved high viability, cytotoxicity, and ability to continue to expand. Taken together, our expansion protocol provides great flexibility not only for genome editing but also for the development of NK cells as off-the-shelf therapeutics.
The cell expansion rate can be improved. Culture media can be completely replaced in every passage to ensure maximal nutrients instead of being diluted with fresh media to maintain cell density. Gas-permeable culture plates are recommended for high-density cell culture to improve gas exchange. Incorporating an IL-15 regimen into the expansion could promote NK cell growth (Felices et al., 2018; Wagner et al., 2017). The antibody-conjugated beads are critical for NK cell expansion but are added as a one-time treatment at the initial thawing of NK cells, as suggested by the manufacturer. Because the beads are diluted continuously as the cells grow, replenishing them may help maintain the same level of growth stimulation. Although costly, these modifications are straightforward to increase the expansion rate of NK cells and shorten the expansion duration to avoid the decline in NK cell cytotoxicity.
Cas9 RNP nucleofection is an increasingly popular approach for cell types that are difficult to manipulate genetically using conventional plasmid and viral methods. Cas9 protein and sgRNA can be synthesized and validated in advance and stably stored in the freezer for nucleofection on demand. Cas9 RNP is highly robust, capable of multiplexing, free of genetic materials, and quickly degraded in cells to avoid residual editing activity. These attributes are ideal for the manufacture of genome-engineered NK cells that complies with good manufacturing practice standards for clinical use. We tested the Lonza 4D Nucleofector and Thermo Fisher Scientific Neon Transfection systems to show that Cas9 RNP editing is applicable using different electroporation systems. We prefer the Lonza 4D Nucleofector because of its semi-high-throughput capability and nontoxic carbon-based electrodes for better cell viability. Since the composition of Lonza nucleofection buffers and the pulse code setting are propriety, condition screening is necessary to optimize the codelivery of Cas9 RNP and DNA and preserve cell viability. It is possible to explore other formulations of nucleofection buffer, culture media, and additives to improve cell viability during and after nucleofection.
Using the optimized nucleofection condition and properly prepared sgRNA, single and multiplex gene KO is highly effective in primary NK cells. Cas9 RNP nucleofection is a rapid approach to assess target sites and sgRNA performance in cells, where computational prediction may not always be reliable, as seen in NK-92 cells (Huang et al., 2020). While multiplex KO is straightforward to set up, the risk of chromosomal translocation is a concern. NHEJ can mis-ligate Cas9-induced double-strand breaks when multiple breaks are induced simultaneously. This is evident in our multiplex KO experiment, where various patterns of intra- and interchromosomal translocations have been detected by a simple PCR assay. Although occurring at lower frequency than the faithful repair, chromosomal translocations are highly mutagenic and potentially tumorigenic. More thorough DNA sequencing and functional validations are needed to ensure the genome integrity of the edited cells.
Primary NK cells are capable of site-specific gene KI via Cas9-mediated HDR. We demonstrate the feasibility to insert DNA sequences of various lengths to label endogenous proteins of interest with an affinity tag and a fluorescent reporter gene. Although the KI efficiency is suboptimal and cannot be straightforwardly enhanced by chemical treatment, our KI procedure is still able of generate a sufficient number of cells for research purposes. Although optimizing nucleofection conditions may increase DNA delivery and potentially KI efficiency, strategies are needed to overcome DNA toxicity to improve the recovery and reexpansion of viable KI cells.
In summary, we demonstrate that CRISPR genome editing by Cas9 RNP nucleofection works robustly in primary human NK cells. From the standpoint of cell manufacturing and immunotherapy, the efficiency of gene editing should not be based solely on gross KO or KI efficiencies. The problems with chromosomal translocation, DNA toxicity, and donor variation must be overcome to ultimately ensure the purity and safety of gene-edited cell therapeutics.
Materials and methods
Maintenance of cell lines
All cell culture reagents and media were purchased from Gibco (Thermo Fisher Scientific) unless stated otherwise. All cell lines were of human origin, maintained in 37°C incubator with 5% CO2 in the specified media, and routinely tested for mycoplasma contamination using the EZ-PCR detection assay kit (Biological Industries). The chronic myelogenous leukemia cell line K562 (ATCC) was maintained in RPMI-1640 (ATCC modification) supplemented with 12.5% heat-inactivated FBS, 25 mM Hepes, 1× GlutaMAX, and 1× penicillin-streptomycin. K562 cells were maintained at 105 to 106 cells/ml to avoid cell differentiation. The embryonic kidney cell line HEK293T (ATCC), the breast cancer cell line MDA-MB-231 (ATCC), and the glioblastoma cell line U-251 MG (ATCC) were maintained in DMEM with high glucose (HyClone) supplemented with 15% heat-inactivated FBS, 25 mM Hepes, 1× GlutaMAX, and 1× penicillin-Sstreptomycin.
Ex vivo expansion of primary human NK cells
Cryopreserved human peripheral blood NK cells, which were negatively enriched, were purchased from Lonza and Cellero. Donor information is summarized in Table S1. All experiments on human cells were conducted according to the human experimental guidelines approved by the Institutional Review Board on Biomedical Science Research, Academia Sinica. The following thawing procedure is highly critical to NK cell viability and expansion, and an overview is illustrated in Fig. S1 A. First, the vial of frozen NK cells was incubated in a 37°C water bath for 2 min, without mixing, until the ice was partially melted. The vial was placed in the palm of the hand for ∼1 min, without mixing, to completely melt the ice. Immediately after melting, the cell suspension was transferred to a 15-ml tube by gentle pipetting. 9 ml ice-cold RPMI-1640 was slowly added and mixed by swirling into the cell suspension to dilute out DMSO in the cryopreservation reagent. The cells were pelleted by centrifugation at room temperature at 200 g for 10 min, during which time the medium gradually warmed to room temperature. The cell pellet was then resuspended at 106 cells/ml in X-VIVO 15 (Lonza), LymphoONE (Takara Bio), NK MACS (Miltenyi Biotec), or EL837 (EliteCell) medium, which was prewarmed to room temperature. All media were supplemented with 5% EliteGro-Adv HPL (EliteCell), 1,000 U/ml of IL-2 (PeproTech), MACSiBeads conjugated with anti-human NKp46 and anti-CD2 antibodies (Miltenyi Biotec), and 10 U/ml DNase I (Sigma-Aldrich). NK cells were maintained in a 24-well plate at 1 ml per well in a 37°C incubator with 5% CO2 and left as undisturbed as possible for the first 4 d. DNase I is necessary to prevent irreversible cell aggregation and death. IL-2 powder was reconstituted in 100 mM acetic acid (Sigma-Aldrich) to 5 × 106 U/ml, strictly as described by the manufacturer, and diluted to 106 U/ml in Dulbecco's PBS (DPBS) + 0.1% BSA (Fraction V and molecular-biology grade from Gold Biotechnology). It is also important to note that the BSA-containing solutions must be prepared freshly for the preparation of MACSiBeads and dilution of IL-2.
For subsequent cell passage in Phase I, all media were prewarmed to room temperature and freshly supplemented with 1,000 U/ml IL-2, but no MACSiBeads or DNase I was added. On day 5, half of the media was removed from top of the well and replenished with fresh media. On day 7, NK cells were gently resuspended by pipetting for cell counting, and diluted in fresh media to 0.5 × 106 cells/ml. During days 7–10, NK cells started to expand rapidly, although the expansion rates varied among different donors. On day 10 and onward, the cell density was maintained at 0.5 × 106 cells/ml and was diluted in fresh media as frequently as necessary. Starting in Phase II, the NK cells were maintained at 106 cells/ml in NK MACS and EL837 supplemented with 5% EliteGro-Adv and either 100 or 1,000 U/ml IL-2. Cell density and viability were measured by Trypan blue staining in Countess II cell counter (Thermo Fisher Scientific) throughout the expansion. Cell images were taken on an Olympus CKX41 inverted microscope and processed by ImageJ.
Cryopreservation and thawing of expanded NK cells
The expanded NK cells were frozen at 107 cells/ml per cryogenic tube. First, MACSiBeads were removed from the cell suspension by capturing on a DynaMag-15 magnetic stand (Thermo Fisher Scientific). The cells were then pelleted at 200 g for 10 min and resuspended in 1 ml of ice-cold CryoStor CS10 (Sigma-Aldrich) or 90% heat-inactivated FBS plus 10% BloodStor 100 (BioLife Solution), which is essentially cell culture–grade DMSO. The cell suspension was then aliquoted into cryogenic vials and prechilled at 4°C for 10 min. After prechilling, the cryogenic vials were frozen at −80°C in CoolCell (Corning) overnight, and then stored in liquid nitrogen for more than 2 wk before the second thawing experiment. The thawing process was as described above. The cells were resuspended at 106 cells/ml in room-temperature NK MACS medium supplemented with 1,000 U/ml IL-2 and 10 U/ml DNase I. The cells were then maintained using the standard culture method as described above.
Antibodies
All the antibodies were purchased from BioLegend unless specifically indicated. The following antibodies were used for flow cytometry: APC anti-CD3 (OKT3), PE anti-CD56 (5.1H11), APC anti-CD96 (NK92.39), PE anti-NKG2A (REA110; Miltenyi Biotec), PerCp-Cy5.5 anti-TIGIT (A15153G), PE anti–PD-1 (EH12.2H7), PE anti–Siglec-7 (QA-79; Invitrogen), PE anti–DNAM-1 (11A8), APC anti-NKp46 (9E02), APC anti-NKG2D (1D11), PE and PerCP-Cy5.5 anti-CD16 (3G8), APC anti-HA.11 Epitope-Tag (16B12), Alexa Fluor 488 anti–TNF-α (MAb11), Alexa Fluor 647 anti–IFN-γ (H4A3), Pacific Blue anti-CD107a (4SB3), PE anti-CD155 (SKII.4), APC Mouse IgG1κ Isotype (MOPC-21), APC Mouse IgG2b Isotype (MOPC-173), PE Mouse IgG1κ Isotype (MOPC-21), PE Mouse IgG2b Isotype (MPC-11), PerCP-Cy5.5 Mouse IgG1κ Isotype (MOPC-21) and PE REA control (REA293; Miltenyi Biotec). The following antibodies were used for Western blotting: anti-FLAG M2 (Sigma-Aldrich), anti-GAPDH (Proteintech), and anti-mouse IgG, HRP-linked (Cell Signaling).
Flow cytometry analyses and cell sorting
All experiments were performed using CytoFLEX (Beckman Coulter) and FACSAria III (BD Biosciences) in the flow cytometry core facilities at the Institute of Biological Chemistry and the Institute of Biomedical Sciences at Academia Sinica. Ice-cold FACS buffer (DPBS supplemented with 2% FBS, 25 mM Hepes, and 0.5 mM EDTA) was used for cell washing, resuspension, and antibody dilution. To preserve the integrity of cell surface proteins, adherent cell lines were detached and dissociated by PBS-based Cell Dissociation buffer (Thermo Fisher Scientific). For the analysis of surface markers, NK cells were pelleted at 500 g for 5 min, washed once, and stained in the antibody solution (diluted as per the manufacturer’s recommended ratios) in the dark for 20 min on ice. After staining, NK cells were washed once, resuspended in 200 µl FACS buffer, and kept on ice before analysis. For cell sorting, NK cells were pelleted at 200 g for 10 min and stained with antibodies by the same method. The final resuspension was gently filtered through a 35-µm nylon mesh cell strainer (Corning) and kept in 5 ml polypropylene tube on ice until sorting. NK MACS medium supplemented with 1,000 U/ml IL-2 and 10 U/ml DNase I was used as the collection medium. The sorted NK cells were pelleted at 200 g for 10 min, resuspended in NK MACS medium supplemented with 1,000 U/ml IL-2, and kept in a 37°C incubator until analysis. For intracellular staining, NK cells were pelleted at 500 g for 5 min. The cells were then fixed and permeabilized by Cyto-Fast Fix/Perm Buffer Set (BioLegend), stained with antibodies at room temperature for 20 min in the dark, and processed similarly for flow cytometry. The data were analyzed with FlowJo (BD Biosciences) and CytExpert (Beckman Coulter).
Precision beads viability assay
The Precision beads assay was performed as described previously (Huang et al., 2020), with modifications. Briefly, the NK cell suspension was gently filtered through 35-µm nylon mesh cell strainer (Corning). Stock solution of Precision cell count beads (BioLegend) was thoroughly resuspended before use by vortexing at high speed for 40 s. 20 μl Precision beads was added to each filtered cell sample and vortexed at low speed for 5 s. The samples were kept on ice and gently vortexed immediately before analysis by flow cytometry. 1,000 events of Precision beads were counted by APC and PB450 signals as an internal standard to quantitate the cell density (gated as P1 and shown as the red rectangle in Fig. S3 B). Forward and side scattering were used to gate viable cells (gated as P2 and shown as the black circle in Fig. S3 B). KO cells (negative for the target protein) or GFP+ cells were determined within the viable cell population. The following equations were used to calculate percent viability, percentage of viable KO cells, and percentage of GFP+ cells:
Calcein-AM cytotoxic assay
MDA-MB-231 and U-251 MG cells were detached by PBS-based Cell Dissociation buffer, pelleted at 300 g for 5 min, and washed with 10 ml DPBS. K562 cells were pelleted at 300 g for 3 min and washed with 10 ml DPBS. After washing, 106 cells were resuspended in 1 ml DPBS with 10 µM Calcein-AM (BioLegend) and incubated at 37°C for 30 min. The stained cells were washed three times with 37°C RPMI-1640 and resuspended to 105 cells/ml in 37°C RPMI-1640. Primary NK cells were pelleted at 200 g for 10 min and resuspended in RPMI-1640 to 4 × 105 cells/ml. 100 μl primary NK cells per well was added into a U-bottom 96-well plate, and serial dilution in RPMI-1640 was performed to adjust different NK cell/target ratios. 100 μl of the stained target cells was then added into the wells containing NK cells. The plate was centrifuged at 200 g for 3 min to facilitate cell contact and then incubated in 37°C incubator for 4 h. The plate was centrifuged at 200 g for 3 min, and 100 µl of the supernatant was transferred to a 96-well Opti-plate (Basic Life) to determine the experimental release of Calcein-AM. Spontaneous release of Calcein-AM from the target cells was measured in the absence of NK cells. Maximal release was measured by complete lysis of the target cells in RPMI-1640 with 2% Triton-X100. The fluorescence intensity at 488/520 nm was recorded using M1000 pro (Tecan). The cytotoxicity assay against K562 cells was performed on days 14 and 28 of the expansion procedure on NK cells from five donors. For the refreezing experiment, the cytotoxicity assay was performed on three donor cells. Cytotoxicity of the gene-KO cells was performed 48 h after cell sorting. Timing details are indicated in the figure legends. The data point of each donor represents the mean from a triplicate experiment. Cytotoxicity was calculated using the following equation:
Degranulation assay
NK cells were activated by 1000 U/ml of IL-2 for 48 h before assay. The adherent target cells were detached by PBS-based Cell Dissociation buffer to preserve cell surface ligands. The NK cells and target cells were individually pelleted at 200 g for 10 min and resuspended in room-temperature RPMI-1640 to cell density of 106 cells/ml. To start the experiment, 50,000 NK cells (50 µl) were incubated with 50,000 target cells (50 µl) at 1:1 cell ratio in an U-bottom 96-well plate. Anti-CD107a antibody was added into each well at a final concentration of 20 µg/ml. The plate was centrifuged at 200 g for 3 min to facilitate cell contact and then incubated in 37°C incubator for 1 h. Brefeldin A and Monensin (BioLegend) were then added into each well at final concentrations of 1×. The plate was incubated in 37°C incubator for an additional 3 h. After incubation, all cells were fixed and permeabilized by Cyto-Fast Fix/Perm Buffer Set (BioLegend) per the manufacturer’s protocol. The fixed and permeabilized cells were costained by anti–TNF-α and anti–IFN-γ antibodies at room temperature for 20 min. The stained cells were washed once and resuspended in ice-cold FACS buffer for analysis. To chemically stimulate degranulation, NK cells were treated with phorbol 12-myristate-13-acetate and ionomycin using 1× Cell Activation Cocktail (with Brefeldin A) from BioLegend. Briefly, NK cells were pelleted at 200 g for 10 min, resuspended in NK MACS medium supplemented with 1× Cell Activation Cocktail plus 20 µg/ml anti-CD107a antibody, and incubated in 37°C incubator for 4 h. The activated NK cells were processed and analyzed by the same standard methods.
Synthesis of sgRNA
The sgRNA sequences were designed using CRISPR Design Tool on the Benchling website (http://www.benchling.com). On-target efficiencies and off-target scores were predicted in silico by the same tool. The sgRNAs were enzymatically synthesized by in vitro transcription using T7 polymerase as described previously (Lin et al., 2014). The DNA oligonucleotides for sgRNA synthesis are listed in Table S3. The transcribed sgRNAs were resolved by denaturing Urea-PAGE, and the full-length sgRNA was excised and extracted from the gel. The sgRNAs were subsequently treated with CIP (New England Biolabs) to remove the 5′ phosphate group to prevent innate immune responses (Kim et al., 2018). After phenol/chloroform extraction and ethanol precipitation, the final sgRNAs were dissolved in sgRNA buffer (20 mM Hepes at pH 7.5, 150 mM KCl, 10% glycerol, 1 mM β-mercaptoethanol, and 1 mM MgCl2) to 48 µM as determined by the absorbance at OD280nm in NanoDrop Lite (Thermo Fisher Scientific). The chemically synthesized Dharmacon sgRNAs were from Horizon Discovery. The chemically synthesized sgRNAs were also dissolved in sgRNA buffer to 48 µM. All sgRNAs, both enzymatically and chemically synthesized, were refolded in sgRNA buffer by incubating at 60°C for 3 min and gradual cooling to room temperature. The final sgRNA products were stored as 48-µM aliquots at −80°C.
Cas9 RNP preparation
Recombinant Cas9 protein was expressed from plasmid pMJ915 (#69090; Addgene) in Escherichia coli BL21 (DE3) and purified as described previously (Lingeman et al., 2017). The Cas9 protein was stored in Cas9 RNP buffer (20 mM Hepes at pH 7.5, 150 mM KCl, 10% glycerol, and 1 mM β-mercaptoethanol) as 40-µM aliquots at −80°C. The protein was thawed immediately before use and never refrozen. Cas9 RNP was assembled within 1 h before nucleofection. Equal volumes of 40-µM Cas9 protein and 48-µM refolded sgRNA were mixed at molar ratio of 1:1.2 by slowly pipetting and swirling Cas9 protein into sgRNA, but not in the opposite order, which is more prone to aggregation. The mixture was then incubated at 37°C for 15 min. The assembled Cas9 RNP was defined as 20 µM and kept at room temperature until nucleofection.
HDR template construction and preparation
The DNA sequences of the HDR templates and the related PCR primers are listed in Table S5. Single-stranded Ultramer Oligos (Integrated DNA Technologies) were dissolved in molecular-grade water to 100 µM and used for KI of HA tag. The HDR templates for the gfp KI experiments were PCR-generated, double-stranded linear DNA consisting of a 5′-homology arm, a 3′-homology arm, and a gfp reporter gene. They were PCR-amplified from Addgene and our plasmid using KAPA HiFi PCR Kit (KAPA Biosystems). The RAB11A-GFP and ACTB-GFP templates were amplified from pTR143 (#112012; Addgene) and AICSDP-15 (#87425; Addgene) plasmids, respectively, as described previously (Roberts et al., 2017). The PCR products of RAB11A-GFP and ACTB-GFP templates were purified by 0.55× of AMPure XP beads (Beckman Coulter) according to the manufacturer’s protocol. For the construction of CD96-GFP plasmid, the homology arms were amplified from NK cell genomic DNA by KAPA HiFi HotStart PCR kit (KAPA Biosystems). The gfp gene was amplified from pTR143 plasmid (#112012; Addgene) using the KAPA HiFi PCR Kit. PCR fragments were purified using the Zymoclean Gel DNA Recovery Kit (Zymo Research) and assembled into the pUC19 vector using NEBuilder HiFi DNA Assembly (New England Biolabs). Subsequently, Round-the-Horn PCR mutagenesis was performed to introduce silent mutations into the sgRNA seed region to prevent targeting of the HDR template by Cas9 RNP. The plasmid sequences were validated by Sanger sequencing. The CD96-GFP template was then amplified from the CD96-GFP plasmid, and purified by QIAquick PCR Purification Kit (Qiagen). All PCR-generated templates were subsequently precipitated by isopropanol, washed with 70% ethanol, and dissolved in molecular-grade water to 2 µg/µl. All templates were stored at −20°C until usage.
Screening of Cas9 RNP and DNA nucleofection conditions
NK cells were activated with fresh IL-2 at 1,000 U/ml 48 h before nucleofection. Fresh NK MACS medium was prepared on the day of nucleofection with 1,000 U/ml IL-2 and prewarmed in a 37°C incubator. MACSiBeads were removed from the cell suspension by capturing on a DynaMag-15 magnetic stand. The cells were then pelleted at 200 g for 10 min. A nucleofection reaction consisted of 5 × 105 of NK cells in 20 µl nucleofection buffer and 2 µl Cas9 RNP or pmaxGFP plasmid (Lonza). The concentrations of Cas9 RNP and pmaxGFP are 40 pmol and 0.4 µg, respectively, unless specified otherwise. The nucleofection buffer was P3 (Lonza) or Sol2, which consisted of 150 mM sodium phosphate buffer (pH 7.2), 5 mM KCl, 15 mM MgCl2, 15 mM Hepes, and 50 mM mannitol (Chicaybam et al., 2013). Sol2 was stored at 4°C, never frozen to avoid precipitation, and replaced every month. The cell mixture was then transferred into 16-well strip-format nucleofection cuvettes. Nucleofection was performed in 4D Nucleofector X-unit (Lonza) using the specified pulse code summarized in Table S2. Immediately after nucleofection, 100 µl prewarmed culture medium was added into each well to allow cell recovery in the 37°C incubators for 15 min. Afterwards, the cells were transferred into culture plates and maintained by the standard culture method. All gene-editing results were analyzed 72 h after nucleofection unless indicated otherwise. The expression of turboGFP was analyzed by flow cytometry 24 h after nucleofection.
Electroporation of Cas9 RNP using Neon Transfection system
NK cells were prepared by the standard method. The cells were pelleted at 200 g for 10 min and washed by DPBS once before electroporation. An electroporation reaction consisted of 2.5 × 105 NK cells in 9 µl Buffer T and 1 µl of Cas9 RNP. The electroporation was conducted in 10-µl Neon electroporation tip in Buffer E according to the manufacturer’s protocol. Buffer E was replaced between experiments of different donors. Immediately after electroporation, NK cells were transferred to 200 µl prewarmed 37°C NK MACS medium with 1,000 U/ml IL-2 in a 96-well culture plate. Cells were kept in 37°C incubator for 72 h before CD96 KO analysis by flow cytometry. Table S2 shows a complete list of pulse settings and KO efficiencies.
Nucleofection of Cas9 RNP and HDR template for KI
NK cells were prepared using the standard method. A nucleofection reaction consisted of 5 × 105 of NK cells in 20 µl P3 buffer, 2 µl Cas9 RNP, and 1 µl of HDR templates (either 2 µg double-stranded PCR template or 100 pmol single-stranded IDT DNA ultramer). Nucleofection was performed using condition 5 with pulse code DN-100. The cells were then recovered and cultured by standard methods. All KI results were analyzed 72 h after nucleofection. KI of HA tag was detected by intracellular immunostaining and flow cytometry as described.
HDR enhancer screening
DiscoveryPak HDR Enhancers Set (BioVision) was used for this screening. 1 h before nucleofection, NK cells were pretreated with 0.2 µM Brefeldin A, 20 µM L-755507, 20 µM KU-0060648, 20 µM NU-7441, 20 µM SCR-7, or 20 µM RS-1. The nucleofection procedure was as described. After nucleofection, NK cells were first recovered in the enhancer-free NK MACS medium with 1,000 U/ml IL-2 for 15 min and then transferred into the prewarmed medium containing the same enhancer at the same dosage. After 6 h of incubation, the medium was replaced to remove the enhancer. HDR efficiencies were analyzed 72 h after nucleofection.
CRISPR plasmid nucleofection
For plasmid-based gene editing, 5 × 105 NK cells were resuspended in 20 µl P3 buffer and then mixed with 2 µg all-in-one CRISPR-Cas9 and sgRNA dual-expressing plasmid. Construction and purification of CRISPR plasmids targeting the CD96, TIGIT, and KLRC1 genes was described previously (Huang et al., 2020). HEK293T cells were used as a positive control. A quarter million HEK293T cells per reaction were pelleted at 90 g for 10 min and resuspended in 20 µl SF nucleofection buffer (Lonza). Pulse codes DN-100 and CM-130 were used for the nucleofection of NK and HEK293T cells, respectively. Immediately after nucleofection, prewarmed culture medium was added into each well, and cells were allowed to recover in a 37°C incubator for 15 min. After recovery, cells were maintained using the standard method. Editing efficiencies were determined at 72 h after nucleofection.
Validation of Cas9 and sgRNA expression from CRISPR plasmids
RT-PCR was performed to detect sgRNA expression. Cells were lysed by TRIzol reagent (Thermo Fisher Scientific). Total RNA was extracted using a Direct-zol RNA Microprep kit (Zymo Research) and eluted in molecular-grade water. The RNA extract was treated with DNase I to deplete most of the CRISPR plasmid. The presence of sgRNA was detected by reverse transcription using SuperScript IV Reverse transcription (Thermo Fisher Scientific) and PCR amplification using the KAPA HiFi HotStart PCR kit with sgRNA scaffold–specific primers (Table S4). cDNA of sgRNA was resolved on 2% agarose gel in Tris-acetate-EDTA buffer and visualized by SYBR Gold staining (Thermo Fisher Scientific). Western blotting was performed to detect Cas9 expression. Primary NK and HEK293T cells were pelleted at 500 g for 5 min, lysed in SDS loading buffer (80 mM Tris-HCl, pH 6.8, 15% glycerol, 4% SDS, and 2% β-mercaptoethanol), and then boiled at 95°C for 5 min. The protein lysate was resolved by gel electrophoresis in 10% SDS-PAGE gel and then transferred to a polyvinylidene difluoride membrane at 250 mA for 100 min for blotting. The membrane was blocked in 5% skim milk for 60 min at room temperature, incubated with the primary antibodies in 5% skim milk overnight at 4°C, and then with the secondary antibody for 60 min at room temperature. The membrane was washed three times in between antibody staining in 1× TBST for 5 min each. Plus-ECL (PerkinElmer) chemiluminescence detection kit was used to visualize target proteins.
Gene-editing analysis by DNA sequencing
The edited cells were collected at 500 g for 5 min and washed once with DPBS. Cell pellets were lysed by QuickExtraction solution (Lucigen) at 65°C for 15 min and 98°C for 5 min to extract genomic DNA. Target regions were amplified by PCR using KAPA HiFi HotStart PCR kit and the primer sets in Table S4. The PCR products were purified by QIAquick PCR Purification Kit (Qiagen) and eluted in molecular-grade water for Sanger Sequencing in DNA Sequencing Core Facility at the Institute of Biomedical Sciences at Academia Sinica. The indel percentage was analyzed online at the Synthego website by an ICE tool (https://www.synthego.com/products/bioinformatics/crispr-analysis).
Deep-sequencing analyses of on-target and off-target efficiency
Chromosome translocation analysis
Chromosomal translocations between the three Cas9 cleavage sites were detected by a PCR assay (Jeong et al., 2019). Genomic DNA of CD96-TIGIT-CD226 triple-negative cells was extracted by QuickExtraction solution. Fifteen combinations of forward and reverse primers were used to probe different translocation patterns. Genomic PCR was performed using KAPA HiFi HotStart PCR kit and the following thermocycle setting: 28 or 35 cycles of 98°C for 20 s, 70°C for 10 s, and 72°C for 15 s. The PCR products were then resolved in 2% agarose gel in Tris-acetate-EDTA buffer and stained by SYBR Gold (Thermo Fisher Scientific) for visualization.
Statistical analyses
The nucleofection screening was performed in the NK cells from a single donor. All other data were collected from multiple donors or multiple independent experiments, as specified in Materials and methods and the figure legends, to determine mean values ± SD. Two-tailed Welch’s unequal variances t test was used to test for statistical significance in all experiments. Statistical analyses were performed using GraphPad Prism 8.
Online supplemental material
Fig. S1 shows the thawing procedure of the cryopreserved NK cell and the morphological change during ex vivo expansion. Fig. S2 shows that the expanded NK cells can be refrozen and thawed with minimal loss in viability and cytotoxicity. Fig. S3 shows the cell viability assay by Precision cell count beads. Fig. S4 shows the screening of Cas9 RNP electroporation conditions in the Neon Transfection system. Fig. S5 shows the design of single-stranded and double-stranded HDR template and the effect of KU00060648 treatment on NK cells. Table S1 provides the donor information of the commercial, cryopreserved primary NK cells. Table S2 lists the conditions tested in the Lonza 4D Nucleofector and Thermo Fisher Neon Transfection systems. Table S3 lists the sgRNA target sequences and DNA oligonucleotides used for the synthesis of sgRNA templates. Table S4 lists the primer sequences for PCR, NGS analysis, and plasmid construction. Table S5 lists the sequences of the HDR templates and the related primers.
Acknowledgments
We thank members of the Lin laboratory for helpful discussions on this manuscript. We thank the High Throughout Genomics Core at Biodiversity Research Center at Academia Sinica for conducting the NGS library preparation and sequencing.
We thank the Program for Translational Innovation of Biopharmaceutical Development - Technology Supporting Platform Axis (https://tspa.sinica.edu.tw; grant AS-KPQ-106-TSPA) and Academia Sinica (https://www.sinica.edu.tw) for funding support. Both funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: R.S. Huang, M.C. Lai, H.A. Shih, and S. Lin conceived and designed this study. R.S. Huang, M.C. Lai, and H.A. Shih performed the experiments. R.S. Huang analyzed the NGS results. R.S. Huang and S. Lin wrote the manuscript.

