


Cells utilize multiple mechanisms to maintain mitochondrial homeostasis. We recently characterized a pathway that remodels mitochondria in response to metabolic alterations and protein overload stress. This remodeling occurs via the formation of large membranous structures from the mitochondrial outer membrane called mitochondrial-derived compartments (MDCs), which are eventually released from mitochondria and degraded. Here, we conducted a microscopy-based screen in budding yeast to identify factors that regulate MDC formation. We found that two phospholipids, cardiolipin (CL) and phosphatidylethanolamine (PE), differentially regulate MDC biogenesis. CL depletion impairs MDC biogenesis, whereas blocking mitochondrial PE production leads to constitutive MDC formation. Additionally, in response to metabolic MDC activators, cellular and mitochondrial PE declines, and overexpressing mitochondrial PE synthesis enzymes suppress MDC biogenesis. Altogether, our data indicate a requirement for CL in MDC biogenesis and suggest that PE depletion may stimulate MDC formation downstream of MDC-inducing metabolic stress.
Introduction
Mitochondria are hubs for cellular metabolism and signaling (Friedman and Nunnari, 2014; Spinelli and Haigis, 2018). Disrupted mitochondrial homeostasis contributes to aging and numerous metabolic diseases (Wallace, 2005; Kudryavtseva et al., 2016). To maintain mitochondrial function and homeostasis, cells utilize various quality control and signaling pathways, including mitochondrial proteases (Quirós et al., 2015), the ubiquitin-proteasome system (Bragoszewski et al., 2017; Ravanelli et al., 2020), mitophagy (Palikaras et al., 2018; Pickles et al., 2018), mitochondrial-derived vesicles (Sugiura et al., 2014; Picca et al., 2020; König et al., 2021), mitochondrial fission and fusion (Giacomello et al., 2020), the mitochondrial unfolded protein response (Melber and Haynes, 2018), and multiple mitoprotein-induced stress responses (Song et al., 2021; Boos et al., 2020). In recent studies, our group identified a new mechanism by which cells regulate mitochondrial homeostasis—through the formation of mitochondrial-derived compartments (MDCs) (Hughes et al., 2016; English et al., 2020; Schuler et al., 2021). In Saccharomyces cerevisiae, MDCs are dynamic, multilamellar structures generated from the outer mitochondrial membrane (OMM) (Wilson et al., 2023b, Preprint). MDCs can be stimulated by protein overabundance stress in the OMM (Wilson et al., 2023a, Preprint) or metabolic alterations, including elevated intracellular amino acid abundance (Schuler et al., 2021), the latter of which occurs during the progression of aging, metabolic disorders, and neurodegenerative diseases (Wellen and Thompson, 2010; Hughes and Gottschling, 2012; Aliu et al., 2018; Hughes et al., 2020; Ruiz et al., 2020). In previous studies, we identified several perturbations that trigger MDC formation by increasing intracellular amino acids, including impairing vacuolar amino acid storage using the vacuolar H+-ATPase inhibitor concanamycin A (ConcA) and preventing amino acid incorporation into proteins using rapamycin (Rap) or cycloheximide (CHX), which inhibit translation (Schuler et al., 2021; English et al., 2020). The induction of MDCs by ConcA, Rap, and CHX is suppressed by depleting amino acids from the media (Schuler et al., 2021).
Upon formation, MDCs selectively sequester Tom70, a receptor of the mitochondrial outer membrane translocase (TOM) complex (Söllner et al., 1990; Steger et al., 1990), as well as other OMM proteins, diminishing the levels of these proteins in mitochondria (Hughes et al., 2016; Schuler et al., 2021). In contrast, mitochondrial intermembrane space (IMS) proteins, matrix proteins, and mitochondrial inner membrane (IMM) proteins are excluded from MDCs (Hughes et al., 2016; Wilson et al., 2023a, Preprint). After formation, MDCs are separated from the mitochondrial tubule by fission and delivered to the vacuole for degradation by autophagy (Hughes et al., 2016). Thus, MDCs are cargo-selective structures that remodel mitochondria in response to protein-based and metabolic stressors. Failure to form MDCs alters amino acid catabolism and sensitizes cells to amino acid stress (Schuler et al., 2021).
Using super-resolution microscopy, we previously showed that MDCs are closely associated with both mitochondria and the endoplasmic reticulum (ER) (English et al., 2020). MDC generation occurs at the contact sites between the ER and mitochondria, and this process requires proteins that localize to these sites, including the ER–mitochondria encounter structure (ERMES) complex and the ERMES-associated GTPase Gem1 (Kornmann et al., 2009, 2011; English et al., 2020). Various genetic suppressors that rescue defects in ERMES mutants differ in their ability to restore MDC formation (Tan et al., 2013; Lang et al., 2015; John Peter et al., 2017; English et al., 2020). As a result, it remains unclear how ER–mitochondria contacts regulate MDC biogenesis. Additionally, other factors involved in relaying signals and facilitating MDC formation remain unknown.
Here, we present results from a microscopy-based screen of the yeast non-essential deletion collection (Giaever et al., 2002) aimed at uncovering genes involved in MDC formation under metabolic stress. As described below, we identified a number of gene deletions that enhance or suppress MDC formation, which led to discovery of differential roles for the non-bilayer forming phospholipids (PLs) cardiolipin (CL) and phosphatidylethanolamine (PE) in MDC biogenesis.
Results
An imaging-based screen identifies genetic regulators of MDC biogenesis
To identify genetic regulators of MDC biogenesis, we conducted a microscopy-based screen using the yeast non-essential deletion collection (Giaever et al., 2002). We previously showed that MDCs are enriched with OMM proteins, including Tom70, but exclude IMS, IMM, and matrix proteins, such as Tim50 (Yamamoto et al., 2002; Hughes et al., 2016) (Fig. 1 A). Thus, we used the Synthetic Genetic Array (SGA) technique (Tong et al., 2001) to construct a yeast non-essential deletion library in which individual mutants contain endogenously tagged Tom70-GFP and Tim50-mCherry (mCh). Using high-throughput imaging, we examined the formation of Tom70-GFP-positive Tim50-mCh-negative foci (MDCs) in ∼5,000 deletion strains after treatment with the MDC inducer Rap (Schuler et al., 2021) (Fig. 1 B). The percentage of cells with MDCs in each mutant strain was quantified (Table S1). We found that the majority of deletion strains (3,744 out of 4,757 ORFs that were successfully screened) robustly formed MDCs in 20–60% of cells (Fig. 1 C) when treated with Rap in 96-well plates, which is generally lower than what we previously observed when cells were cultured in tubes or flasks (Schuler et al., 2021; English et al., 2020). Gene deletions that led to MDC formation in <20% or >60% of cells were categorized as having potentially “low” and “high” MDC formation rates, respectively. These genes were subjected to gene ontology analysis with additional manual curation and are listed in Fig. 1 D.
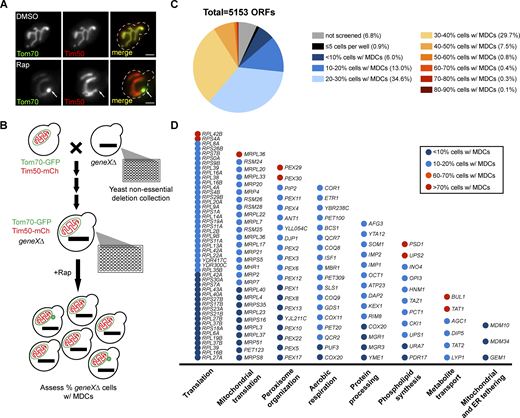
An imaging-based screen identifies genetic regulators of MDC biogenesis. (A) Widefield images of wild-type yeast cells endogenously expressing Tom70-yeGFP (Tom70-GFP) and Tim50-mCherry (Tim50-mCh) treated with DMSO or rapamycin (Rap) for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (B) Schematic of the genome-wide screen to identify genetic regulators of Rap-induced MDC biogenesis. A query strain containing endogenously expressing Tom70-GFP and Tim50-mCh was mated to the yeast non-essential deletion collection to obtain a new collection in which all mutants are labeled with Tom70-GFP and Tim50-mCh after several steps of selection. The collection was treated with Rap in 96-well plates for 2 h and imaged by automated microscopy. MDCs were identified as Tom70-positive, Tim50-negative mitochondria-associated structures. Images were manually assessed and quantified to determine the percentages of cells with MDCs in each well. (C) Fan plot of the MDC screen results showing the ratio of mutants that could not screened (no growth or low image quality), grew poorly (≤5 cells per well), or formed MDCs in the indicated percentages of cells, to the total number of open reading frames (ORFs) contained in the yeast deletion collection. (D) Categories of gene deletions that led to decreased MDC biogenesis (≤20% cells form MDCs) or enhanced MDC biogenesis (≥60% cells form MDCs). For a complete list of all genes, their descriptions, and gene ontology analysis, see Table S1.
An imaging-based screen identifies genetic regulators of MDC biogenesis. (A) Widefield images of wild-type yeast cells endogenously expressing Tom70-yeGFP (Tom70-GFP) and Tim50-mCherry (Tim50-mCh) treated with DMSO or rapamycin (Rap) for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (B) Schematic of the genome-wide screen to identify genetic regulators of Rap-induced MDC biogenesis. A query strain containing endogenously expressing Tom70-GFP and Tim50-mCh was mated to the yeast non-essential deletion collection to obtain a new collection in which all mutants are labeled with Tom70-GFP and Tim50-mCh after several steps of selection. The collection was treated with Rap in 96-well plates for 2 h and imaged by automated microscopy. MDCs were identified as Tom70-positive, Tim50-negative mitochondria-associated structures. Images were manually assessed and quantified to determine the percentages of cells with MDCs in each well. (C) Fan plot of the MDC screen results showing the ratio of mutants that could not screened (no growth or low image quality), grew poorly (≤5 cells per well), or formed MDCs in the indicated percentages of cells, to the total number of open reading frames (ORFs) contained in the yeast deletion collection. (D) Categories of gene deletions that led to decreased MDC biogenesis (≤20% cells form MDCs) or enhanced MDC biogenesis (≥60% cells form MDCs). For a complete list of all genes, their descriptions, and gene ontology analysis, see Table S1.
We successfully identified gene deletions that were previously shown to block MDC biogenesis, including ER-mitochondria tethering genes MDM10, MDM34, and GEM1 (Kornmann et al., 2009, 2011; English et al., 2020) (Fig. 1 D). This indicated that the screening approach was capable of identifying genetic regulators of MDC formation. Through ontology analysis, we identified several gene categories that either positively or negatively regulate MDC biogenesis. These include a number of genes that are involved in cytoplasmic and mitochondrial translation, peroxisome organization, aerobic respiration, protein processing, PL biosynthesis, and amino acid transport (Table S1 and Fig. 1 D). We have not yet validated all of the hits from the screen. However, the large amount of gene deletions in each of the categories listed in Fig. 1 D suggests these biological processes likely impact MDC formation. Consistent with previous observations that Rap triggers MDC induction by driving amino acid overabundance in cells (Schuler et al., 2021), we uncovered numerous genes involved in amino acid transport and regulation, as well as amino acid-consuming protein translation, providing additional evidence of connections between MDCs and amino acid homeostasis.
UPS1 and UPS2 regulate MDC biogenesis in an inverse manner
Among genes identified from the screen, we found that UPS1 and UPS2, which encode for IMS-localized intramitochondrial lipid transfer proteins of the PRELI family involved in CL and PE biosynthesis (Dee and Moffat, 2005; Sesaki et al., 2006; Tamura et al., 2009, 2012; Osman et al., 2009; Potting et al., 2010; Connerth et al., 2012; Miyata et al., 2016), regulate MDC biogenesis in opposing directions (Fig. 1 D). CL and PE are the major non-bilayer forming PLs in cells (Osman et al., 2011). Both CL and PE play important roles in mitochondrial structure and function (Acoba et al., 2020; Basu Ball et al., 2018), and genetic depletion of both PLs in mitochondria causes synthetic lethality (Gohil et al., 2005). CL and PE are synthesized in the IMM using substrates obtained from the OMM. The movement of lipid precursors from the OMM to the IMM is mediated by Ups1 and Ups2 (Sesaki et al., 2006; Tamura et al., 2009, 2012; Osman et al., 2009; Potting et al., 2010; Connerth et al., 2012; Miyata et al., 2016; Acoba et al., 2020). Specifically, Ups1 transports phosphatidic acid (PA) from the OMM to IMM for CL synthesis (Connerth et al., 2012), and Ups2 transports phosphatidylserine (PS) from the OMM to IMM for PE synthesis (Miyata et al., 2016; Aaltonen et al., 2016) (Fig. 2 A). Both Ups1 and Ups2 bind to Mdm35, which stabilizes them against proteolytic degradation (Miyata et al., 2016; Connerth et al., 2012; Potting et al., 2010). Deletion of UPS1 blocks mitochondrial CL production (Tamura et al., 2009; Connerth et al., 2012). Similarly, loss of UPS2 prevents PE synthesis in mitochondria (Miyata et al., 2016).

UPS1 and UPS2 regulate MDC biogenesis in opposing directions. (A) Model of intramitochondrial transport of substrates for CL and PE synthesis on the IMM mediated by IMS proteins Ups1 and Ups2. Ups1 transports PA for CL synthesis and Ups2 transports PS for PE synthesis. Ups1 and Ups2 are stabilized against degradation by binding to Mdm35. (B) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates.
UPS1 and UPS2 regulate MDC biogenesis in opposing directions. (A) Model of intramitochondrial transport of substrates for CL and PE synthesis on the IMM mediated by IMS proteins Ups1 and Ups2. Ups1 transports PA for CL synthesis and Ups2 transports PS for PE synthesis. Ups1 and Ups2 are stabilized against degradation by binding to Mdm35. (B) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates.
In the screen, we found that loss of UPS1 inhibited MDC formation, whereas deletion of UPS2 enhanced MDC biogenesis (Fig. 1 D). To verify these preliminary results, we deleted UPS1 and UPS2 and analyzed the lipid profiles and MDC biogenesis rates in these strains. Consistent with previous studies (Miyata et al., 2016; Connerth et al., 2012), deleting UPS1 depleted cellular CL, while loss of UPS2 resulted in reduced cellular PE abundance (Fig. S1 A and Table S2). In alignment with the screen results, MDC biogenesis was impaired in ups1∆ cells in response to MDC inducers Rap, ConcA, and CHX (Fig. 2, B and C; and Fig. S1 B). In contrast, even without the addition of MDC-inducing agents, 40% of ups2∆ cells constitutively formed MDCs, and this percentage further increased when treated with MDC inducers (Fig. 2, B and C; and Fig. S1 B).
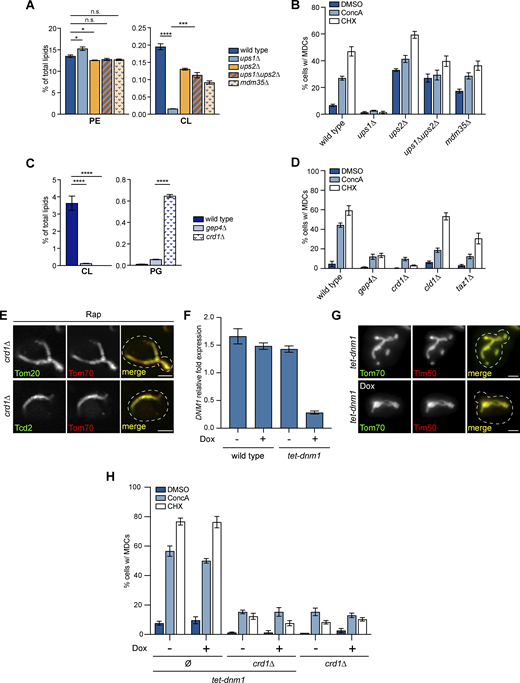
Suppressing CL production inhibits MDC formation (related toFigs. 2 and 3 ). (A) The relative amounts of the indicated phospholipids in whole-cell lysates of wild-type or the indicated mutant cells were determined by mass spectrometry-based lipidomic analysis. Amount of each lipid relative to total lipids was determined. Error bars = SEM of three replicates. Statistical comparison shows the difference to the corresponding wild-type control. n.s., not significant, *P < 0.0332, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (B) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (C) The relative amounts of the indicated phospholipids in whole-cell lysates of wild-type or the indicated mutant cells determined by mass spectrometry-based lipidomic analysis. The amount of each lipid relative to total lipids was determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding wild-type control. ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (D) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tom70-mCh treated with Rap for 2 h. Images show a single focal plane. Scale bar = 2 µm. (F) Quantification of DNM1 mRNA relative abundance by RT-qPCR in wild-type cells or tet-dnm1 mutant cells in the absence or presence of Dox. Error bars = SEM of three replicates. (G) Widefield images of tet-dnm1 mutants endogenously expressing Tom70-GFP and Tim50-mCh in the absence or presence of Dox. Scale bar = 2 µm. (H) Quantification of MDC formation in the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h in the absence or presence of Dox. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
Suppressing CL production inhibits MDC formation (related toFigs. 2 and 3 ). (A) The relative amounts of the indicated phospholipids in whole-cell lysates of wild-type or the indicated mutant cells were determined by mass spectrometry-based lipidomic analysis. Amount of each lipid relative to total lipids was determined. Error bars = SEM of three replicates. Statistical comparison shows the difference to the corresponding wild-type control. n.s., not significant, *P < 0.0332, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (B) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (C) The relative amounts of the indicated phospholipids in whole-cell lysates of wild-type or the indicated mutant cells determined by mass spectrometry-based lipidomic analysis. The amount of each lipid relative to total lipids was determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding wild-type control. ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (D) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tom70-mCh treated with Rap for 2 h. Images show a single focal plane. Scale bar = 2 µm. (F) Quantification of DNM1 mRNA relative abundance by RT-qPCR in wild-type cells or tet-dnm1 mutant cells in the absence or presence of Dox. Error bars = SEM of three replicates. (G) Widefield images of tet-dnm1 mutants endogenously expressing Tom70-GFP and Tim50-mCh in the absence or presence of Dox. Scale bar = 2 µm. (H) Quantification of MDC formation in the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h in the absence or presence of Dox. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
Previous studies showed that UPS2 deletion rescues CL deficiency and mitochondrial import defects in ups1∆ cells (Tamura et al., 2009), possibly through a Ups1-independent CL synthesis pathway activated by the ablation of mitochondrial PE production (Miyata et al., 2017). We constructed ups1∆ups2∆ double mutant yeast and verified that CL abundance was partially replenished in these cells compared with ups1∆ (Fig. S1 A and Table S2). Next, we performed MDC assays on ups1∆ups2∆ yeast. We found that these cells exhibited intermediate MDC formation compared with wild-type, ups1∆, and ups2∆ single mutant strains. Similar to ups2∆, 30% of untreated ups1∆ups2∆ cells formed MDCs (Fig. 2, B and C; and Fig. S1 B). However, upon the addition of MDC inducers, this percentage was not significantly increased and was lower than wild-type and ups2∆ cells—a phenotype that is similar to ups1∆ mutants (Fig. 2, B and C; and Fig. S1 B). In addition, the deletion of MDM35, a gene encoding a protein that binds to and stabilizes both Ups1 or Ups2 (Miyata et al., 2016; Potting et al., 2010), led to similar PL composition (Fig. S1 A and Table S2) and MDC biogenesis phenotypes (Fig. 2, B and C; and Fig. S1 B) as observed in ups1∆ups2∆ cells. These data confirm the results from the screen that UPS1 and UPS2 inversely regulate MDC biogenesis.
CL is required for MDC biogenesis
Reduced MDC formation in ups1∆ mutants suggests that CL may be required for MDC biogenesis. To test this, we analyzed MDC formation in additional mutants that perturb CL biosynthesis. In mitochondria, after PA is translocated from the OMM to the IMM by Ups1, it is converted to CL through several reactions catalyzed by enzymes localized in the IMM (Acoba et al., 2020; Tatsuta et al., 2014) (Fig. 3 A). We deleted non-essential genes encoding mitochondrial CL synthesis pathway components, including GEP4, which catalyzes the formation of phosphatidylglycerol (PG), an intermediate product of CL synthesis (Osman et al., 2010), as well as CRD1, the CL synthase that catalyzes CL formation from PG (Chang et al., 1998). Similar to ups1∆ cells, gep4∆ and crd1∆ strains were depleted of CL (Fig. S1 C and Table S2) and showed impaired MDC biogenesis in response to Rap, ConcA, or CHX (Fig. 3, B and C; and Fig. S1 D).

CL is required for MDC biogenesis. (A) Model of the mitochondrial CL synthesis pathway. After the translocation of PA from the OMM to IMM by the Ups1-Mdm35 complex, CL is produced via several reaction steps. Gep4 catalyzes the synthesis of PG, an intermediate substrate for CL synthesis, and the CL synthase Crd1 converts PG to premature CL (pCL). Cld1 removes one acyl-chain from pCL to generate monolyso-CL (MLCL), followed by the readdition of an acyl-chain to form mature CL (mCL) by Taz1. (B) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tim50-mCh treated with Rap for 2 h. Images show a single focal plane. Scale bar = 2 µm. (E) Quantification of wild-type cells or crd1∆ mutants showing the percentage of cells with larger MDCs, smaller vesicle-like structures that resemble MDCs, or no MDCs by super-resolution microscopy. Microscopy images on the left show examples of these three types. Scale bar = 2 µm. N = 50 cells per replicate, error bars = SEM of three replicates. (F) Quantification of the percentage of cells with MDCs in the indicated mutant yeast treated with DMSO or Rap for 2 h in the absence or presence of Doxycycline (Dox). Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
CL is required for MDC biogenesis. (A) Model of the mitochondrial CL synthesis pathway. After the translocation of PA from the OMM to IMM by the Ups1-Mdm35 complex, CL is produced via several reaction steps. Gep4 catalyzes the synthesis of PG, an intermediate substrate for CL synthesis, and the CL synthase Crd1 converts PG to premature CL (pCL). Cld1 removes one acyl-chain from pCL to generate monolyso-CL (MLCL), followed by the readdition of an acyl-chain to form mature CL (mCL) by Taz1. (B) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tim50-mCh treated with Rap for 2 h. Images show a single focal plane. Scale bar = 2 µm. (E) Quantification of wild-type cells or crd1∆ mutants showing the percentage of cells with larger MDCs, smaller vesicle-like structures that resemble MDCs, or no MDCs by super-resolution microscopy. Microscopy images on the left show examples of these three types. Scale bar = 2 µm. N = 50 cells per replicate, error bars = SEM of three replicates. (F) Quantification of the percentage of cells with MDCs in the indicated mutant yeast treated with DMSO or Rap for 2 h in the absence or presence of Doxycycline (Dox). Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
After synthesis, CLs are further remodeled to generate mature CLs (Acoba et al., 2020). The maturation of CLs requires Cld1, which removes an acyl chain from premature CLs to form monolyso-CLs (Ye et al., 2014), and Taz1, which re-acylates monolyso-CLs to form mature CLs (Gu et al., 2004) (Fig. 3 A). To test whether CL remodeling affects MDC biogenesis, we deleted CLD1 or TAZ1. We found CLD1 deletion inhibited MDC formation induced by ConcA and slightly impaired MDC biogenesis induced by Rap or CHX treatment (Fig. 3, B and C; and Fig. S1 D). In comparison, the deletion of TAZ1 led to reduced MDC biogenesis in response to all MDC inducers (Fig. 3, B and C; and Fig. S1 D). However, the inhibitory effect of TAZ1 deletion was not as strong as the deletion of GEP4 or CRD1, suggesting that CL remodeling has a mild impact on MDC biogenesis but is not as critical as the production of CL from PA.
To test whether CL depletion blocks MDC formation and does not just alter the cargo selectivity of MDCs, we examined the localization and behavior of other known MDC cargoes in crd1∆ mutants. Indeed, when CRD1 was deleted, we did not observe MDCs when visualizing mistargeted carrier proteins or other outer membrane proteins (Fig. 3 D; and Fig. S1 E) that we previously showed localized to MDCs (Hughes et al., 2016; Schuler et al., 2021; Wilson et al., 2023a, Preprint). In addition, we recently showed that we can observe both small and larger-sized MDCs in cells and that altering the fusion/fission equilibrium of mitochondria regulates MDC size (Wilson et al., 2023b, Preprint). We considered the possibility that CL loss only prevents the formation of large MDCs. To test this, we employed super-resolution microscopy to quantify the numbers of both small and large MDCs in cells. We found that both wild-type and crd1Δ cells contained small and large MDCs. Importantly, ∼70% of crd1∆ cells form neither of these structures (Fig. 3 E). This suggests that CL is necessary for normal levels of MDC formation and does not just affect MDC size. Consistent with this observation, we used the tetracycline-regulated promoter system to effectively deplete DNM1, an OM fission GTPase required for mitochondrial fission and MDC separation from mitochondrial tubules (Bleazard et al., 1999; Hughes et al., 2016), in crd1∆ cells (Fig. S1, F and G). We still did not observe MDCs at normal levels in these strains (Fig. 3 F and Fig. S1 H). Altogether, these results indicate that depleting CL inhibits MDC formation.
PE depletion constitutively activates MDC biogenesis
In addition to the CL synthesis pathway, we also investigated how deficient mitochondrial PE production affects MDC biogenesis. Similar to UPS2, we found that PSD1, which encodes for the predominantly IMM and partially ER-localized decarboxylase that catalyzes PE synthesis from PS (Tatsuta et al., 2014; Friedman et al., 2018) (Fig. 4 A), was also identified as a potential negative regulator of MDC formation in the screen (Fig. 1 D). Deletion of PSD1 reduced PE abundance in whole-cell lysates and mitochondria-enriched membrane fractions (Fig. S2, A and B; and Table S2). Like ups2∆ strains, 35% of untreated psd1∆ cells constitutively formed MDCs (Fig. 4, B and C), indicating that the MDC pathway is activated in the absence of mitochondrial PE synthesis. MDCs that constitutively formed in psd1∆ mutants also contained other previously reported MDC cargoes (Hughes et al., 2016; Schuler et al., 2021) (Fig. 4 D and Fig. S2 C). In psd1∆ cells, MDC formation is further increased with the addition of MDC inducers (Fig. S2 D).

PE depletion constitutively activates MDC biogenesis. (A) Model of PE synthesis pathways. PS, the substrate for PE synthesis, is synthesized from PA on the ER membrane. The last step of PS synthesis is catalyzed by ER-localized Cho1. After transfer from the OMM to the IMM by the Ups2-Mdm35 complex, PS is converted to PE by an IMM-localized decarboxylase Psd1. PE produced in mitochondria is further transferred to the ER for PC synthesis, which is mediated by Cho2 and Opi3 in the ER. Psd1 can also dually localize to the ER membrane. Two alternative PE synthesis pathways include (1) PE production mediated by Psd2 which is localized on the endosomal membrane and (2) PE generation from exogenous ethanolamine (Etn) via the Kennedy pathway. (B) Widefield images of wild-type cells or psd1∆ mutants endogenously expressing Tom70-GFP and Tim50-mCh. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Super-resolution images of psd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tim50-mCh. White arrows mark the positions of MDCs. Images show single focal plane. Scale bar = 2 µm. (E) Widefield images of wild-type cells or tet-cho1 mutants endogenously expressing Tom70-GFP and Tim50-mCh in the absence or presence of Dox. White arrows mark the positions of MDCs. Scale bar = 2 µm. (F) Quantification of D showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (G) Quantification of MDC formation in wild-type cells and the indicated mutant yeast treated with DMSO or Rap for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (H) Quantification of MDC formation in wild-type cells and the indicated mutant yeast. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or psd1∆ mutants in the presence or absence of 5 mM Etn. N > 100 cells per replicate, error bars = SEM of three replicates. (J) Widefield images of psd1∆ mutants containing an empty vector (EV) or the indicated Psd1-FLAG constructs endogenously expressing Tom70-GFP and Tim50-mCh. White arrows mark the positions of MDCs. Scale bar = 2 µm. (K) Quantification of (J) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates.
PE depletion constitutively activates MDC biogenesis. (A) Model of PE synthesis pathways. PS, the substrate for PE synthesis, is synthesized from PA on the ER membrane. The last step of PS synthesis is catalyzed by ER-localized Cho1. After transfer from the OMM to the IMM by the Ups2-Mdm35 complex, PS is converted to PE by an IMM-localized decarboxylase Psd1. PE produced in mitochondria is further transferred to the ER for PC synthesis, which is mediated by Cho2 and Opi3 in the ER. Psd1 can also dually localize to the ER membrane. Two alternative PE synthesis pathways include (1) PE production mediated by Psd2 which is localized on the endosomal membrane and (2) PE generation from exogenous ethanolamine (Etn) via the Kennedy pathway. (B) Widefield images of wild-type cells or psd1∆ mutants endogenously expressing Tom70-GFP and Tim50-mCh. White arrows mark the positions of MDCs. Scale bar = 2 µm. (C) Quantification of (B) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Super-resolution images of psd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tim50-mCh. White arrows mark the positions of MDCs. Images show single focal plane. Scale bar = 2 µm. (E) Widefield images of wild-type cells or tet-cho1 mutants endogenously expressing Tom70-GFP and Tim50-mCh in the absence or presence of Dox. White arrows mark the positions of MDCs. Scale bar = 2 µm. (F) Quantification of D showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (G) Quantification of MDC formation in wild-type cells and the indicated mutant yeast treated with DMSO or Rap for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (H) Quantification of MDC formation in wild-type cells and the indicated mutant yeast. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or psd1∆ mutants in the presence or absence of 5 mM Etn. N > 100 cells per replicate, error bars = SEM of three replicates. (J) Widefield images of psd1∆ mutants containing an empty vector (EV) or the indicated Psd1-FLAG constructs endogenously expressing Tom70-GFP and Tim50-mCh. White arrows mark the positions of MDCs. Scale bar = 2 µm. (K) Quantification of (J) showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates.
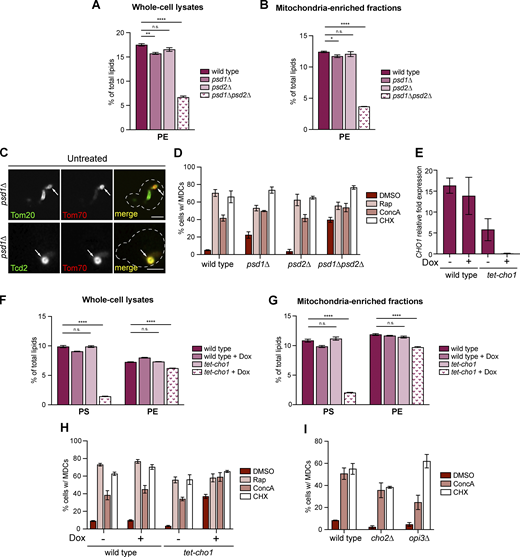
Defective mitochondrial PE synthesis activates MDC biogenesis (related toFig. 4 ). (A and B) The relative amounts of PE in whole-cell lysates (A) or mitochondria-enriched membrane fractions (B) isolated from wild-type or the indicated mutant cells were determined by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding wild-type control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (C) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tom70-mCh treated with Rap for 2 h. White arrows mark positions of MDCs. Images show single focal plane. Scale bar = 2 µm. (D) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Quantification of CHO1 mRNA abundance by RT-qPCR in wild-type cells or tet-cho1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates. (F and G) The relative amounts of the indicated phospholipids in whole-cell lysates (F) or mitochondria-enriched membrane fractions (G) isolated from wild-type or tet-cho1 mutants in the absence or presence of Dox determined by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of four replicates. Statistical comparison shows difference to the corresponding wild-type control. n.s., not significant, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (H) Quantification of MDC formation in wild-type cells or tet-cho1 cells treated with DMSO, Rap, ConcA, or CHX for 2 h in the absence or presence of Dox. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates.
Defective mitochondrial PE synthesis activates MDC biogenesis (related toFig. 4 ). (A and B) The relative amounts of PE in whole-cell lysates (A) or mitochondria-enriched membrane fractions (B) isolated from wild-type or the indicated mutant cells were determined by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding wild-type control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (C) Super-resolution images of crd1∆ mutants endogenously expressing MDC cargos tagged with GFP and Tom70-mCh treated with Rap for 2 h. White arrows mark positions of MDCs. Images show single focal plane. Scale bar = 2 µm. (D) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Quantification of CHO1 mRNA abundance by RT-qPCR in wild-type cells or tet-cho1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates. (F and G) The relative amounts of the indicated phospholipids in whole-cell lysates (F) or mitochondria-enriched membrane fractions (G) isolated from wild-type or tet-cho1 mutants in the absence or presence of Dox determined by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of four replicates. Statistical comparison shows difference to the corresponding wild-type control. n.s., not significant, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (H) Quantification of MDC formation in wild-type cells or tet-cho1 cells treated with DMSO, Rap, ConcA, or CHX for 2 h in the absence or presence of Dox. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates.
To test whether constitutive MDC biogenesis in psd1∆ cells could be a result of the accumulation of the Psd1 substrate PS on the OMM (Miyata et al., 2017) (Fig. 4 A), we reduced the levels of CHO1, which encodes the enzyme that catalyzes the last step of PS synthesis on the ER membrane (Miyata et al., 2017). In response to doxycycline (Dox)-dependent inhibition, tet-CHO1 mutants showed depleted CHO1 mRNA abundance (Fig. S2 E) and reduced cellular and mitochondrial PS abundance (Fig. S2, F and G; and Table S2). In tet-CHO1 mutants, cellular and mitochondrial PE levels were also decreased (Fig. S2, F and G; and Table S2) due to the lack of the PE synthesis substrate PS. Similar to deleting UPS2 or PSD1, inhibiting CHO1 expression also caused constitutive MDC formation (Fig. 4, E and F; and Fig. S2 H), indicating that MDC biogenesis is not a consequence of PS accumulation on the OMM. We also tested whether inhibiting mitochondrial PE production induces MDC formation by limiting substrates for PC synthesis on the ER membrane (Tatsuta et al., 2014). We deleted CHO2 and OPI3, two enzymes that catalyze PC generation from PE (Kodaki and Yamashita, 1987, 1989). Unlike ups2∆ and psd1∆ mutants, untreated cho2∆ and opi3∆ mutants did not form MDCs constitutively (Fig. 4 G), indicating that MDC biogenesis triggered by deletion of UPS2 and PSD1 is not caused by impaired PC production. On the contrary, deletions of CHO2 and OPI3 slightly reduced MDC levels in response to Rap and ConcA compared with wild-type cells (Fig. 4 G and Fig. S2 I). Interestingly, in cho2∆ and opi3∆ mutants, mitochondrial PE is elevated (Schuler et al., 2016), which may explain the modest reduction in MDC formation in these strains. Taken together, our results suggest that lack of mitochondrial PE, but not alterations in the abundance of PS or PC, stimulates MDC biogenesis.
In yeast, PE synthesis is not merely dependent on Psd1. In psd1∆ cells, PE produced by other cellular pathways partially supplies mitochondrial PE (Trotter and Voelker, 1995; Birner et al., 2001; Bürgermeister et al., 2004; Gulshan et al., 2010; Gibellini and Smith, 2010; Acoba et al., 2020). From previous studies (Schuler et al., 2016) and as verified here, the PE level in psd1∆ mutants is only slightly reduced compared with wild-type cells (Fig. S2 A and B; and Table S2). To test whether MDC biogenesis is exacerbated by the inhibition of redundant cellular PE production pathways, we deleted PSD2, which catalyzes PE synthesis on endosomal membranes (Tatsuta et al., 2014) (Fig. 4 A). The deletion of PSD2 itself did not affect total cellular PE levels (Fig. S2, A and B; and Table S2) or MDC biogenesis (Fig. 4 H and Fig. S2 D). However, in whole-cell lysate and mitochondria-enriched membrane fractions, PE abundance was severely reduced in psd1∆psd2∆ double mutants (Fig. S2, A and B; and Table S2). We found that 45% of psd1∆psd2∆ cells constitutively formed MDCs, which was higher than the rate in psd1∆ single mutants (Fig. 4 H). In the presence of MDC inducers, psd1∆psd2∆ cells showed similar MDC levels compared with psd1∆ single mutants (Fig. S2 D). In addition to Psd2, cells also generate PE from exogenous Ethanolamine (Etn) via the Kennedy Pathway (Gibellini and Smith, 2010) (Fig. 4 A). Etn can rescue growth defects of psd1∆psd2∆ cells on synthetic media (Fig. S3 A) (Friedman et al., 2018; Calzada et al., 2019). We found that Etn addition slightly reduces MDC formation but is incapable of blocking constitutive MDC biogenesis in psd1∆ mutants (Fig. 4 I). These results indicate that MDC formation is activated by lowering PE produced in mitochondria, but not other cellular compartments.

PE synthesized in the IMM is required for MDC biogenesis (related to Figs. 4 and 5,). (A) Fivefold serial dilutions of wild-type cells or the indicated mutant cells on YPAD, SD, or SD+ 5 mM Etn agar plates. (B) Schematic of the indicated Psd1-FLAG constructs containing TMD of Psd1 (orange), Sec66 (light blue), Tom70 (turquoise), or Mic60 (purple) used in Fig. 4, J and K. (C) Widefield images of indirect immunofluorescence staining against the FLAG epitope and endogenous Tom70 in psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs. Nucleus is stained with NucBlue. White arrow marks perinuclear ER. Images show a single focal plane. Scale bar = 2 μm. (D) Fivefold serial dilutions of psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs on SD agar plates. (E) Quantification showing the percentage of cells with tubular, aggregated, or fragmented mitochondrial morphology in psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs. Microscopy images on the left show examples of these three mitochondrial morphologies. Scale bar = 2 μm. N > 100 cells per replicate, error bars = SEM of three replicates. (F) Quantification of MDC formation in psd1∆ mutants containing an empty vector (EV) or the indicated Psd1-FLAG constructs treated with Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (G) Quantification of MDC formation in mmm1∆ VPS13(D716H) cells or mmm1∆ VPS13(D716H) cells with the indicated gene deletions treated with DMSO, ConcA, or CHX for 2 h. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates. (H) Quantification of MDC formation in gem1∆ mutant cells or gem1∆ cells with the indicated gene deletions treated with DMSO, ConcA, or CHX for 2 h. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Tetrads of psd1∆/+ crd1∆/+ diploid cells dissected on YPAD or YPAD + 5 mM Etn. (J) Quantification of PSD1 mRNA abundance by RT-qPCR in wild-type cells or tet-psd1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates.
PE synthesized in the IMM is required for MDC biogenesis (related to Figs. 4 and 5,). (A) Fivefold serial dilutions of wild-type cells or the indicated mutant cells on YPAD, SD, or SD+ 5 mM Etn agar plates. (B) Schematic of the indicated Psd1-FLAG constructs containing TMD of Psd1 (orange), Sec66 (light blue), Tom70 (turquoise), or Mic60 (purple) used in Fig. 4, J and K. (C) Widefield images of indirect immunofluorescence staining against the FLAG epitope and endogenous Tom70 in psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs. Nucleus is stained with NucBlue. White arrow marks perinuclear ER. Images show a single focal plane. Scale bar = 2 μm. (D) Fivefold serial dilutions of psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs on SD agar plates. (E) Quantification showing the percentage of cells with tubular, aggregated, or fragmented mitochondrial morphology in psd1∆ mutants expressing an empty vector (EV) or the indicated Psd1-FLAG constructs. Microscopy images on the left show examples of these three mitochondrial morphologies. Scale bar = 2 μm. N > 100 cells per replicate, error bars = SEM of three replicates. (F) Quantification of MDC formation in psd1∆ mutants containing an empty vector (EV) or the indicated Psd1-FLAG constructs treated with Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (G) Quantification of MDC formation in mmm1∆ VPS13(D716H) cells or mmm1∆ VPS13(D716H) cells with the indicated gene deletions treated with DMSO, ConcA, or CHX for 2 h. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates. (H) Quantification of MDC formation in gem1∆ mutant cells or gem1∆ cells with the indicated gene deletions treated with DMSO, ConcA, or CHX for 2 h. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Tetrads of psd1∆/+ crd1∆/+ diploid cells dissected on YPAD or YPAD + 5 mM Etn. (J) Quantification of PSD1 mRNA abundance by RT-qPCR in wild-type cells or tet-psd1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates.
To further test this, we constructed chimeric Psd1-FLAG plasmids with that express Psd1 with the transmembrane domains (TMDs) of ER-localized Sec66 or IMM-localized Mic60 as previously described (Friedman et al., 2018) (Fig. S3 B). We also constructed plasmids expressing Psd1 with the TMD of OMM-localized Tom70 (Millar and Shore, 1994) to test whether PE produced in the OMM (Calzada et al., 2019) impacts MDC biogenesis (Fig. S3 B). We verified the localization of different Psd1-FLAG constructs by immunofluorescence staining (Fig. S3 C). We found that the expression of Psd1-FLAG and Psd1Mic60(1-57)-FLAG in psd1∆ mutants fully rescued growth and mitochondrial morphology (Fig. S3, D and E) and blocked MDC formation (Fig. 4, J and K). Interestingly, Psd1Sec66(1-60)-FLAG partially rescued growth and mitochondrial morphology (Fig. S3, D and E) of psd1∆ mutants, and MDC biogenesis was unaffected in psd1∆ yeast (Fig. 4, J and K), indicating that PE production on the ER likely does not affect MDC formation. This is consistent with previous findings that the PE exchange between mitochondria and the outside is limited (Gohil et al., 2005). When treated with MDC inducers, psd1∆ mutants expressing Psd1-FLAG, Psd1Mic60(1-57)-FLAG, or Psd1Sec66(1-60)-FLAG form MDCs comparable with the empty vector control (Fig. S3 F). Psd1Tom70(1-36)-FLAG, however, severely affected cellular growth and mitochondrial morphology, and blocked MDC formation even in the presence of MDC inducers (Fig. 4, J and K; and Fig S3, D–F). These results suggest that MDCs are most responsive to PE produced locally in the IMM.
PSD1 deletion partially restores MDC formation in ERMES/Gem1 mutants
We previously showed that proteins at ER–mitochondria contacts are required for MDC biogenesis (English et al., 2020). Deletion of individual ERMES components or its regulatory factor GEM1 prevents MDC formation (English et al., 2020). However, it was unclear whether the role of the ERMES complex in MDC biogenesis was linked to its function in PL homeostasis (Kornmann et al., 2009; Tatsuta et al., 2014; AhYoung et al., 2015; Kojima et al., 2016; Jeong et al., 2017). Given that defective mitochondrial PE synthesis constitutively activates MDC biogenesis, we tested whether PE depletion could bypass the requirement for ERMES/Gem1 in MDC formation. To do this, we deleted UPS2 or PSD1 in ERMES mutants expressing VPS13(D716H), which is a mutant version of VPS13 that has been shown to rescue mitochondrial morphology, but not ERMES complex assembly (Lang et al., 2015) or MDC biogenesis (English et al., 2020) in strains lacking ERMES component Mmm1. In the absence of MDC-inducing agents, PSD1 deletion led to constitutive MDC formation in 25% of mmm1∆ VPS13(D716H) cells. This percentage increased to 60% with the addition of Rap or CHX but not ConcA (Fig. 5, A and B; and Fig. S3 G). Deleting UPS2 in mmm1∆ VPS13(D716H) cells only moderately restored MDC biogenesis when cells were treated with Rap or CHX (Fig. 5, A and B; and Fig. S3 G). We also tested whether deletion of UPS2 or PSD1 restored MDC formation in mutants lacking GEM1, the GTPase that regulates the ERMES complex (Kornmann et al., 2011). Deletion of GEM1 is one of the most robust MDC inhibitors identified so far as all common suppressors of ERMES mutants fail to rescue MDC formation in gem1∆ mutants (English et al., 2020). We found that with the addition of Rap, ConcA, or CHX, PSD1 deletion rescued MDC formation in <20% of gem1∆ mutants (Fig. 5, C and D; and Fig. S3 H), and UPS2 deletion failed to rescue MDC formation in gem1∆ mutants (Fig. 5, C and D; and Fig. S3 H). These results suggest that genetically inhibiting mitochondrial PE synthesis can bypass the requirement for ERMES in the MDC pathway, indicating that the role of the ERMES complex in MDC formation is likely linked to its function in PL homeostasis. Because restoration of MDC formation in gem1∆ mutants was not as robust, it is possible that Gem1 plays additional roles in MDC production.

PSD1 depletion partially restores MDC biogenesis in ERMES/Gem1 mutants but is hypostatic of CRD1 deletion. (A) Widefield images of mmm1∆ VPS13(D716H) cells or mmm1∆ VPS13(D716H) cells with the indicated gene deletions endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (B) Quantification of A showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (C) Widefield images of gem1∆ cells or gem1∆ cells with the indicated gene deletions endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (D) Quantification of C showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Widefield images of tet-psd1 mutants with or without CRD1 deletion endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h in the absence or presence of Dox. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (F) Quantification of (E) showing the percentage of cells with MDCs. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
PSD1 depletion partially restores MDC biogenesis in ERMES/Gem1 mutants but is hypostatic of CRD1 deletion. (A) Widefield images of mmm1∆ VPS13(D716H) cells or mmm1∆ VPS13(D716H) cells with the indicated gene deletions endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (B) Quantification of A showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (C) Widefield images of gem1∆ cells or gem1∆ cells with the indicated gene deletions endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (D) Quantification of C showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Widefield images of tet-psd1 mutants with or without CRD1 deletion endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h in the absence or presence of Dox. Ø, no gene is deleted. White arrows mark the positions of MDCs. Scale bar = 2 µm. (F) Quantification of (E) showing the percentage of cells with MDCs. Ø, no gene is deleted. N > 100 cells per replicate, error bars = SEM of three replicates.
CRD1 deletion prevents MDC formation caused by PSD1 depletion
Genetically inhibiting mitochondrial CL and PE production appears to have opposite effects on MDC biogenesis. To test the genetic epistasis between CL and PE in MDC formation, we attempted to create a mutant strain lacking both CRD1 and PSD1. As previously reported (Gohil et al., 2005), deleting both CRD1 and PSD1 leads to synthetic lethality, which cannot be rescued by adding exogenous Etn (Fig. S3 I). Therefore, we used the Tet-regulated promoter inhibition system to conditionally suppress PSD1 expression. Upon the addition of Dox, PSD1 mRNA levels were efficiently depleted (Fig. S3 J). Consistent with our observations in psd1∆ yeast, tet-PSD1 yeast constitutively formed MDCs in the presence of Dox (Fig. 5, E and F). Deletion of CRD1 blocked MDC biogenesis in Dox-treated tet-PSD1 mutants (Fig. 5, E and F). Thus, CRD1 deletion is epistatic to PSD1 depletion, suggesting that CL is indispensable for MDC formation.
Cellular and mitochondrial PE decline in response to MDC-inducing agents
In addition to investigating how genetically altered mitochondrial PL synthesis affects MDC biogenesis, we also considered the possibility that lipid changes may play a role in activating MDCs by known MDC-inducing agents, including ConcA, Rap, and CHX. Through mass spectrometry-based lipidomic analysis on whole-cell lipid extracts, we found that all three MDC inducers triggered a decrease in whole-cell PS and PE abundance (Fig. 6 A and Table S2). Cellular PC levels were unaffected by ConcA and slightly reduced by Rap and CHX treatment (Fig. 6 A and Table S2). ConcA, but not the other two inducers, increased cellular PG abundance (Fig. 6 A and Table S2). Whole-cell CL levels, however, were not significantly affected in response to all three inducers (Fig. 6 A and Table S2).
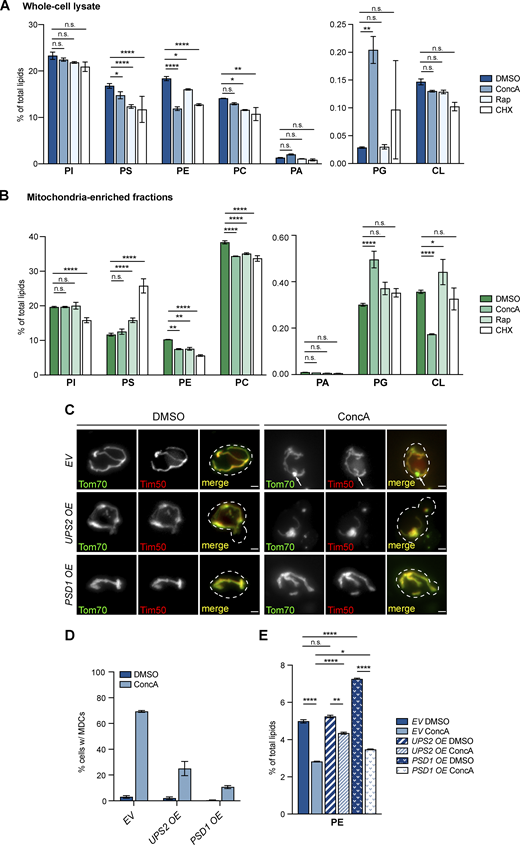
Cellular and mitochondrial PE decline in response to MDC-inducing agents. (A and B) The relative amounts of the indicated phospholipids in whole-cell lysates (A) or mitochondria-enriched membrane fractions (B) isolated from yeast cells treated with DMSO, ConcA, Rap, or CHX for 2 h detected by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For the absolute abundance of each phospholipid species detected by mass spectrometry-based lipidomic analysis, see Table S2. (C) Widefield images of yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or ConcA for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (D) Quantification of C showing the percentage of cells with the indicated integrated vectors with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (E) The relative amounts of PE in whole-cell lysates from yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO or ConcA for 2 h detected by mass spectrometry-based lipidomic analysis. Amounts of PE relative to total lipids were determined. Error bars = SEM of four replicates. The statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2.
Cellular and mitochondrial PE decline in response to MDC-inducing agents. (A and B) The relative amounts of the indicated phospholipids in whole-cell lysates (A) or mitochondria-enriched membrane fractions (B) isolated from yeast cells treated with DMSO, ConcA, Rap, or CHX for 2 h detected by mass spectrometry-based lipidomic analysis. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For the absolute abundance of each phospholipid species detected by mass spectrometry-based lipidomic analysis, see Table S2. (C) Widefield images of yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or ConcA for 2 h. White arrows mark the positions of MDCs. Scale bar = 2 µm. (D) Quantification of C showing the percentage of cells with the indicated integrated vectors with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (E) The relative amounts of PE in whole-cell lysates from yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO or ConcA for 2 h detected by mass spectrometry-based lipidomic analysis. Amounts of PE relative to total lipids were determined. Error bars = SEM of four replicates. The statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2.
Consistent with total cellular PE levels, we also observed lower PE in mitochondria-enriched membrane fractions isolated from cells treated with ConcA, Rap, or CHX (Fig. 6 B and Table S2). Mitochondrial PS levels, in contrast to whole-cell PS, were unaffected or increased (Fig. 6 B and Table S2). With all three inducers, mitochondrial PC abundance decreased Fig. 6 B and Table S2), which could be explained by the lack of PE, the substrate for PC synthesis (Tatsuta et al., 2014). Mitochondrial PG abundance was significantly increased by ConcA, but not Rap or CHX (Fig. 6 B and Table S2). The mitochondrial CL levels exhibit distinct alterations in response to different MDC-inducing agents, which were decreased by ConcA, increased by Rap, and unaffected by CHX (Fig. 6 B).
Our genetic results showed that severely depleting CL inhibits MDC biogenesis (Fig. 3). However, we did not observe any consistent changes in CL abundance across different MDC inducers. This suggests that MDC biogenesis requires the presence of CL, but that the changes in CL levels under MDC-inducing conditions are not likely to contribute to the regulation of MDCs. In contrast, all three MDC-inducing agents caused a decline in both cellular and mitochondrial PE abundance, which is consistent with the result that MDC biogenesis is activated by genetically impairing mitochondrial PE synthesis (Fig. 2, B and C; Fig. 4, B and C). These data suggest that reduced mitochondrial PE levels could be involved in triggering MDC formation in response to ConcA, Rap, and CHX.
To further explore this possibility, we next analyzed whether known mutants with elevated or reduced MDC biogenesis exhibit differences in whole-cell PE levels upon Rap treatment. We found that in mutants with deficient CL synthesis, including ups1∆, gep4∆, and crd1∆ yeast, PE depletion by Rap was similar to wild-type cells (Fig. S4, A and B), indicating that the requirement for CL in MDC biogenesis is unlikely linked to changes in PE. This result is also consistent with the epistatic effect of CRD1 deletion on inhibiting MDC biogenesis in strains with repressed PSD1 expression (Fig. 5, E and F). In contrast, yeast with defective mitochondrial PE production, including ups2∆, ups1∆ups2∆, mdm35∆, and psd1∆, which have constitutively elevated MDC formation (Fig. 2 B and C; and Fig. 3, B and C), exhibited an attenuated decrease in cellular PE levels induced by Rap compared with wild-type cells (Fig. S4, A and C). This suggests that genetically inhibiting mitochondrial PE synthesis and Rap treatment share some redundancy in activating MDC formation. Interestingly, psd2∆ mutants exhibited strong PE depletion upon Rap treatment compared with wild-type cells, and this depletion was not further exacerbated in psd1∆psd2∆ mutants (Fig. S4 C), indicating that a decline of PE synthesis by Psd1, but not Psd2, is triggered by MDC-inducing stress. In addition to mitochondrial PL synthesis pathway mutants, we found that PE decline was blunted in gem1∆ cells (Fig. S4 D), which coincides with the result that deletion of GEM1 prevents MDC biogenesis.

MDC-inducing conditions trigger PE depletion (related to Figs. 6, 7, and 8 ). (A–D) The relative amounts of PE in whole-cell lysate of wild-type cells or the indicated mutant yeast cells treated with DMSO or Rap for 2 h. The average amounts of PE in DMSO-treated cells were set to 100%. Error bars = SEM of three replicates. Statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. (E) Quantification of MDC formation in cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (F) Quantification of MDC formation in cells with genomic integration of an empty vector (EV) or CRD1 overexpressing vector (CRD1 OE) treated with DMSO, Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (G) The relative amounts of MMPE and DMPE in whole-cell lysate of yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO or ConcA for 2 h. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. (H) Quantification of MDC formation in yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, or minimal media that excludes amino acids (indicated as High, Low, and −, respectively) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or the indicated mutant cells grown in synthetic media that contains low levels of amino acid (Low AA) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (J) Quantification of MIM1 mRNA abundance by RT-qPCR in wild-type cells or tet-mim1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates. (K) Quantification of MDC formation in wild-type cells or tet-mim1 mutants treated with DMSO, ConcA, or CHX for 2 h in the absence or presence of Dox. N > 100 cells per replicate, error bars = SEM of three replicates. (L) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV) or MIM1 overexpressing vector (MIM1 OE) treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates.
MDC-inducing conditions trigger PE depletion (related to Figs. 6, 7, and 8 ). (A–D) The relative amounts of PE in whole-cell lysate of wild-type cells or the indicated mutant yeast cells treated with DMSO or Rap for 2 h. The average amounts of PE in DMSO-treated cells were set to 100%. Error bars = SEM of three replicates. Statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. (E) Quantification of MDC formation in cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (F) Quantification of MDC formation in cells with genomic integration of an empty vector (EV) or CRD1 overexpressing vector (CRD1 OE) treated with DMSO, Rap, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (G) The relative amounts of MMPE and DMPE in whole-cell lysate of yeast cells with genomic integration of an empty vector (EV), UPS2 overexpressing vector (UPS2 OE), or PSD1 overexpressing vector (PSD1 OE) treated with DMSO or ConcA for 2 h. Amounts of each lipid relative to total lipids were determined. Error bars = SEM of three replicates. Statistical comparison shows difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. (H) Quantification of MDC formation in yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, or minimal media that excludes amino acids (indicated as High, Low, and −, respectively) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (I) Quantification of MDC formation in wild-type cells or the indicated mutant cells grown in synthetic media that contains low levels of amino acid (Low AA) treated with DMSO, Rap, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (J) Quantification of MIM1 mRNA abundance by RT-qPCR in wild-type cells or tet-mim1 mutants in the absence or presence of Dox. Error bars = SEM of three replicates. (K) Quantification of MDC formation in wild-type cells or tet-mim1 mutants treated with DMSO, ConcA, or CHX for 2 h in the absence or presence of Dox. N > 100 cells per replicate, error bars = SEM of three replicates. (L) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV) or MIM1 overexpressing vector (MIM1 OE) treated with DMSO, ConcA, or CHX for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates.
Overexpressing UPS2 and PSD1 inhibits MDC biogenesis
To further investigate whether PE decline could trigger MDC biogenesis downstream of metabolic MDC activators, we tested how genetically upregulating mitochondrial PE synthesis pathway components impacted MDC formation induced by Rap, CHX, and ConcA. Indeed, overexpressing UPS2 or PSD1 inhibited MDC formation in response to ConcA (Fig. 6, C and D). In cells treated with Rap or CHX, MDC biogenesis was prevented by UPS2 overexpression and was partially impaired by PSD1 overexpression (Fig. S4 E). In comparison, genetic overexpression of CRD1 did not affect MDC biogenesis (Fig. S4 F). Analysis of the lipid composition of cells overexpressing UPS2 revealed that PE decline was blunted in these cells (Fig. 6 E). Cells overexpressing PSD1, on the other hand, exhibited higher baseline PE levels than wild-type cells (Fig. 6 E). Although PSD1 overexpression did not prevent PE decline upon ConcA treatment, the final cellular PE level remained higher than in cells expressing an empty vector (Fig. 6 E). Notably, levels of monomethyl-PE and dimethyl-PE were elevated in both untreated and treated PSD1 overexpressing cells (Fig. S4 G), suggesting these downstream PE products could also play a role in MDC regulation. Altogether, these results indicate that boosting the expression of mitochondrial PE synthesis pathway components prevents MDC formation induced by metabolic alterations.
Cellular and mitochondrial PE abundance correlates with amino acid levels
Thus far, one possible hypothesis to explain our data is that PE acts as a downstream signal to stimulate MDCs upon amino acid elevation. A prediction of this hypothesis is that PE loss normally triggered by ConcA, Rap, and CHX would be expected to be blunted in cells grown in low amino acids, a condition that blocks MDC formation (Schuler et al., 2021) (Fig. 7 A and Fig. S4 H). Indeed, we found that when cells are grown in synthetic media containing low levels of amino acids or in minimal media that completely exclude amino acids, the initial cellular and mitochondrial PE abundance was higher than in cells cultured in high amino acid media (Fig. 7, B–E). Although we still observed a decrease in cellular and mitochondrial PE abundance with ConcA treatment in synthetic media or minimal media, the final levels of PE after treatment in low amino acid media are comparable with the initial cellular and mitochondrial PE levels in cells growing in high amino acid media (Fig. 7, B–E). These results suggest that mitochondrial PE levels are sensitive to amino acids’ abundance in the media. However, these data also indicate that the relationship between MDC-stimulating agents and PE is complex and that MDC-inducing compounds may stimulate MDCs through additional mechanisms beyond alterations in PE.
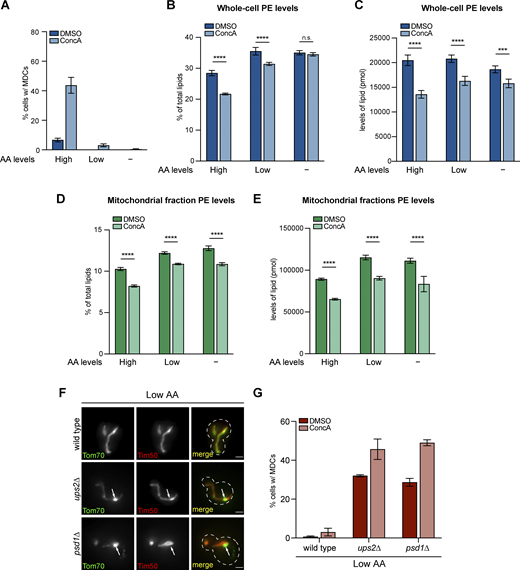
Cellular and mitochondrial PE abundance correlates with amino acid levels. (A) Quantification of MDC formation in yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, and minimal media that exclude amino acids (indicated as High, Low, and −, respectively) treated with DMSO or ConcA for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (B–E) The relative amounts (B and D) or absolute abundance (C and E) of PE in whole-cell lysates (B and C) or mitochondria-enriched membrane fractions (D and E) isolated from yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, and minimal media that exclude amino acids (indicated as High, Low, and −, respectively) treated with DMSO or ConcA for 2 h detected by mass spectrometry-based lipidomic analysis. For relative amounts of PE, PE abundance relative to total lipids were determined. The absolute amounts are normalized to spiked internal standards used by mass spectrometry-based lipidomic analysis, indicating pmol lipid per mg sample for whole-cell lysates, or pmol lipid per mg protein for mitochondria-enriched membrane fractions. Error bars = SEM of four replicates. The statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (F) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh grown in media containing low levels of amino acids. White arrows mark the positions of MDCs. Scale bar = 2 µm. (G) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO or ConcA. N > 100 cells per replicate, error bars = SEM of three replicates.
Cellular and mitochondrial PE abundance correlates with amino acid levels. (A) Quantification of MDC formation in yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, and minimal media that exclude amino acids (indicated as High, Low, and −, respectively) treated with DMSO or ConcA for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (B–E) The relative amounts (B and D) or absolute abundance (C and E) of PE in whole-cell lysates (B and C) or mitochondria-enriched membrane fractions (D and E) isolated from yeast cells grown in amino acid-rich media, synthetic media that contains low levels of amino acid, and minimal media that exclude amino acids (indicated as High, Low, and −, respectively) treated with DMSO or ConcA for 2 h detected by mass spectrometry-based lipidomic analysis. For relative amounts of PE, PE abundance relative to total lipids were determined. The absolute amounts are normalized to spiked internal standards used by mass spectrometry-based lipidomic analysis, indicating pmol lipid per mg sample for whole-cell lysates, or pmol lipid per mg protein for mitochondria-enriched membrane fractions. Error bars = SEM of four replicates. The statistical comparison shows a difference to the corresponding DMSO control. n.s., not significant, *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001, two-way ANOVA with Holm-Šídák test. For a complete profile of all major phospholipid species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis, see Table S2. (F) Widefield images of wild-type cells or the indicated mutant yeast endogenously expressing Tom70-GFP and Tim50-mCh grown in media containing low levels of amino acids. White arrows mark the positions of MDCs. Scale bar = 2 µm. (G) Quantification of MDC formation in wild-type cells or the indicated mutant cells treated with DMSO or ConcA. N > 100 cells per replicate, error bars = SEM of three replicates.
Deletion of UPS2 or PSD1 triggers MDC formation regardless of amino acid levels
Another prediction of a working hypothesis that PE decline promotes MDC biogenesis downstream of elevated amino acids is that genetic impairment of PE production would be expected to stimulate MDCs independently of cellular amino acid levels. To test this, we assessed MDC formation rates in ups2∆ and psd1∆ yeast grown in low amino acid media, where MDCs normally do not form. We found that 30% of ups2∆ and psd1∆ mutants constitutively formed MDCs in synthetic low amino acid media (Fig. 7, F and G). This percentage is comparable with ups2∆ and psd1∆ mutants cultured in amino acid-rich media (Fig. 2 C and Fig. 4 C), suggesting that lowering amino acid abundance is dispensable for MDC biogenesis induced by defective mitochondrial PE production. Interestingly, the percentages of ups2∆ and psd1∆ cells that formed MDCs were further increased by the addition of MDC-inducing agents (Fig. 7 G and Fig. S4 I), again suggesting that MDC-inducing compounds may also stimulate MDCs by means other than altering mitochondrial PE.
CRD1 and PSD1 may impact MDC formation through effects on the OMM
Regardless of whether altered PE serves as a downstream regulatory cue in the MDC pathway or functions as an amino acid-independent mechanism of MDC induction, an important question raised by our results is how PE and CL impact MDC formation. PE and CL are often thought to have redundant and overlapping roles in supporting various aspects of mitochondrial biology (Acoba et al., 2020; Basu Ball et al., 2018). However, they have opposing effects on MDC biogenesis. Thus, in considering possible mechanisms by which these lipids could modulate MDC formation, we turned to the literature to identify mitochondrial processes in which CL and PE have opposing effects. Notably, CL and PE are reported to exhibit distinct capabilities in affecting protein import into the OMM (Becker et al., 2013; Sauerwald et al., 2015). Specifically, CL is required for targeting multispan integral mitochondrial proteins to the OMM (Sauerwald et al., 2015). In comparison, PE impairs import of β-barrel, but not α-helical proteins to the OMM, while loss of PE enhances the import of α-helical proteins into the OMM (Becker et al., 2013).
Interestingly, MDC cargoes identified from previous studies are mostly multispan or single pass α-helical domain-containing proteins, including native OMM proteins as well as mitochondrial carrier proteins mistargeted to the OMM (Hughes et al., 2016; Schuler et al., 2021; Wilson et al., 2023a, Preprint). Furthermore, overexpression of these types of proteins induces MDC biogenesis in the absence of amino acid stress (Schuler et al., 2021; Wilson et al., 2023a, Preprint). Importantly, Tom70, which is required for the import of multispan proteins to the OMM (Wiedemann and Pfanner, 2017), is indispensable for MDC formation (Hughes et al., 2016; Schuler et al., 2021). Based on this data, it is plausible that the impact of CL and PE on MDC biogenesis is tied to their roles in modulating OMM protein load. Consistent with this idea, deleting CRD1 blocked MDC formation triggered by carrier protein overexpression, whereas inhibiting PSD1 further increased the percentage of cells forming MDCs when overexpressing carrier proteins (Fig. 8 A). We also tested whether the import of multispan proteins to the OMM is required for MDC formation by using Tet-dependent downregulation of Mim1, a protein that is required for targeting multispan α-helical proteins to the OMM (Becker et al., 2011). By qPCR, we found that insertion of the Tet-promoter upstream of MIM1 partially suppressed MIM1 expression even without the addition of Dox, and this inhibition was further exacerbated in the presence of Dox (Fig. S4 J). In tet-MIM1 yeast without Dox treatment, although mitochondrial morphology was unaffected, MDC formation was blunted in response to different MDC inducers (Fig. 8, B and C; and Fig. S4 K). In the presence of Dox, tet-MIM1 yeast had aggregated mitochondrial morphology, and Tom70 was mislocalized to unknown cellular structures not connected to the mitochondria (Fig. 8 B). In addition, MDC formation was severally blocked in tet-MIM1 mutants treated with Dox (Fig. 8, B and C), indicating that the import of multispan proteins to the OMM is required for MDC biogenesis.

CRD1 and PSD1 may impact MDC formation through effects on the OMM. (A) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV), AAC1 overexpressing vector (AAC1 OE), or OAC1 overexpressing vector (OAC1 OE). N > 100 cells per replicate, error bars = SEM of three replicates. (B) Widefield images of wild-type cells or tet-mim1 mutants endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h in the absence or presence of Dox. White arrows mark the positions of MDCs. Cyan arrows mark mislocalized Tom70. Scale bar = 2 µm. (C) Quantification of B showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV) or MIM1 overexpressing vector (MIM1 OE) treated with DMSO or Rap for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Model of MDC formation regulated by CL and PE in response to MDC-inducing stress.
CRD1 and PSD1 may impact MDC formation through effects on the OMM. (A) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV), AAC1 overexpressing vector (AAC1 OE), or OAC1 overexpressing vector (OAC1 OE). N > 100 cells per replicate, error bars = SEM of three replicates. (B) Widefield images of wild-type cells or tet-mim1 mutants endogenously expressing Tom70-GFP and Tim50-mCh treated with DMSO or Rap for 2 h in the absence or presence of Dox. White arrows mark the positions of MDCs. Cyan arrows mark mislocalized Tom70. Scale bar = 2 µm. (C) Quantification of B showing the percentage of cells with MDCs. N > 100 cells per replicate, error bars = SEM of three replicates. (D) Quantification of MDC formation in wild-type cells or the indicated mutant yeast with genomic integration of an empty vector (EV) or MIM1 overexpressing vector (MIM1 OE) treated with DMSO or Rap for 2 h. N > 100 cells per replicate, error bars = SEM of three replicates. (E) Model of MDC formation regulated by CL and PE in response to MDC-inducing stress.
We further tested the relationship between lipids, MDCs, and OMM protein content by determining whether overexpressing MIM1 impacts MDC biogenesis in crd1∆ or psd1∆ yeast. We found that MIM1 overexpression slightly increased MDC formation in wild-type and psd1∆ cells, and partially restored MDC biogenesis in crd1∆ mutants (Fig. 8 D and Fig. S4 L). Together, these results raise the possibility that the distinct roles of CL and PE in MDC biogenesis may be driven by their regulation of OMM protein content.
Discussion
We previously showed that the MDC pathway is activated by elevated intracellular amino acid levels as well as protein overloading in the OMM (Schuler et al., 2021; Wilson et al., 2023a, Preprint) and that MDCs form at ER–mitochondria contacts in an ERMES/Gem1-dependent manner (English et al., 2020). However, it remained unclear from these studies how amino acid changes or OMM protein load initiate MDC biogenesis, as well as whether PL transport, which is a major function of the ERMES complex (Tatsuta et al., 2014), affects the generation of MDCs. In this study, we used an unbiased genome-wide imaging screen to identify genetic regulators of the MDC pathway. This approach identified numerous gene deletions that impact MDC biogenesis, suggesting that the MDC pathway is influenced by a variety of cellular processes. Amongst gene hits, we focused on further characterizing a set of factors linked to mitochondrial PL homeostasis, given the known role of ERMES in MDC formation (English et al., 2020). Through a combination of genetics and mass-spectrometry-based lipidomic analyses, we uncovered distinct roles for CL and PE in MDC formation (Fig. 8 E). We found that CL is required for MDC biogenesis as the deletion of genes required for CL synthesis, including UPS1, GEP4, and CRD1 prevented MDC formation (Fig. 2, B and C; Fig. 3, B and C). In contrast, we showed that PE levels declined upon treatment of cells with MDC inducers Rap, CHX, and ConcA (Fig. 6, A and B; and Fig. 7, A–E). We also found that genetic impairment of mitochondrial PE synthesis by deleting UPS2 or PSD1 was sufficient to trigger MDC formation regardless of the amino acid abundance in the media (Fig. 2, B and C; Fig. 4, B and C; and Fig. 7, F and G). In addition, we showed that overexpression of PE synthesis components UPS2 and PSD1 blunted MDC formation (Fig. 6, C and D). Based on these results, we propose that PE decline triggers MDC formation and may function as a key intermediate step in MDC formation in response to amino acid overabundance stress (Fig. 8 E).
As outlined in Fig. 8 E, it is important to note that our data suggest that the interplay between PE, CL, amino acids, and protein overabundance in the OMM is complex and that all of these factors can influence the magnitude of MDC formation in cells. Currently, we cannot exclude the possibility that amino acids and MDC activators ConcA, CHX, and Rap can trigger MDCs through a PE-independent mechanism. Additionally, CHX, ConcA, and Rap may also have amino acid–independent effects on PE levels. It also remains unclear whether MDCs triggered by amino acids and protein overabundance stress share any activation or regulation mechanisms. Given the results presented in Fig. 8 and the known opposite effects that CL and PE have on OMM protein import, it is intriguing to speculate that amino acids may ultimately trigger protein overabundance stress in the OMM by causing alterations in PE. This remains an open question for future investigation. Notably, recent work in mammalian cells also showed that OMM protein import stress triggers remodeling of the OMM by the Structure Positive for Outer Mitochondrial Membrane (SPOT) pathway in response to Toxoplasma gondii infection (Li et al., 2022). Although MDCs and SPOTs differ in their dependence on the conserved GTPase Gem1 (yeast) or MIRO (mammals) to form (Li et al., 2022; Schuler and Hughes, 2022; Schuler et al., 2020, Preprint), these studies together suggest a tight connection between OMM import and OMM membrane remodeling.
It is interesting that CL and PE have different effects on the MDC pathway, given their somewhat similar roles in mitochondria, including promoting respiratory chain activity, mitochondrial membrane potential, and mitochondrial import (Gohil and Greenberg, 2009; Osman et al., 2011; Basu Ball et al., 2018; Acoba et al., 2020). Additionally, deleting both CRD1 and PSD1 leads to synthetic lethality in yeast (Gohil et al., 2005). While it is possible that their known opposing effects on OMM protein import are sufficient to explain their contrasting effects on the MDC pathway, other possibilities remain. For example, it is possible CL plays a direct role in MDC membrane formation, which would explain its strict requirement for MDC biogenesis. Likewise, loss of PE may trigger MDCs by altering IMM and OMM connections or through additional unknown mechanisms.
Regardless of the mechanism by which PE and CL regulate MDC formation, our results provide a step forward in understanding how amino acid surplus triggers alterations in mitochondrial structure and composition via the MDC pathway. Upon treatment with three different inhibitors that induce intracellular amino acid accumulation, PE levels decline in the mitochondria. Our data show that the removal of amino acids prevents PE decline (Fig. 7, A–E), suggesting that elevated amino acids, but not vacuole impairment or translation inhibition per se, cause PE reduction. Moreover, genetic depletion of PE is epistatic to amino acids, as deleting UPS2 or PSD1 triggers MDCs regardless of amino acid status in the media (Fig. 7, F and G). Interestingly, the concept of PE functioning as a lipid-based signal downstream of amino acid perturbation to drive MDC biogenesis exhibits similarities to a recently published study in mammals, which showed that alterations in mitochondrial PE levels stimulate mitochondrial remodeling during hypoxia or impairment of the mTOR pathway (MacVicar et al., 2019). In that study, the authors found that mTOR inhibition decreased PE levels by stimulating the activity of LIPIN1, which drives the conversion of PA to diacylglycerol and triacylglycerol, thus lowering PL levels. They also showed that a decline in PE triggered changes in mitochondrial protein abundance by activating the mitochondrial-localized protease, YME1L1 (MacVicar et al., 2019). Of note, our preliminary screen results did identify the YME1L1 homolog Yme1 (Leonhard et al., 1999; Potting et al., 2010; Song et al., 2007; MacVicar et al., 2019) as a protein required for MDC formation (Fig. 1 D). Given the impact of PE levels on Yme1L1 activity in mammals (MacVicar et al., 2019), it will be interesting to determine in future studies whether a PE–protease axis controls MDC biogenesis in yeast. Overall, the results we presented here enhanced our understanding of the MDC pathway and its regulation. These results further support the concept of PE functioning as a key signaling node coupling cellular metabolic status to mitochondrial composition and function.
Materials and methods
Reagents
Plasmids, chemicals, and antibodies used in this study are listed in Table S3.
Yeast strains
All yeast strains are derivatives of S. cerevisiae S288c (BY) (Brachmann et al., 1998) and are listed in Table S4. Strains expressing fluorescently tagged TOM70, TIM50, OAC1, ALO1, TOM20, or TCD2 from their native loci were created by one-step PCR-mediated C-terminal endogenous epitope tagging using standard techniques and oligo pairs listed in Table S5. Plasmid templates for fluorescent epitope tagging were from the pKT series of vectors (Sheff and Thorn, 2004). Correct integrations were confirmed by correctly localized expression of the fluorophore by microscopy. For strains bearing a deletion of CHO2, CLD1, CRD1, GEM1, GEP4, MDM35, OPI3, PSD1, PSD2, TAZ1, UPS1, or UPS2, one copy of the gene was deleted by one-step PCR-mediated gene replacement with the indicated selection cassette using standard techniques and oligo pairs listed in Table S5 to create a heterozygous diploid. Plasmid templates for gene replacement were from the pRS series of vectors (Sikorski and Hieter, 1989; Andersen et al., 2008). Correct insertion of the selection cassette into the target gene was confirmed by colony PCR across the chromosomal insertion site. The heterozygous diploids bearing gene deletions were subsequently sporulated and dissected to obtain haploid mutants. Strains bearing the tetO7-TATACYC1 promoter allele upstream of CHO1 or MIM1 were pulled out from the Yeast Tet-promoters Hughes collection (Mnaimneh et al., 2004). For strains bearing the tetO7-TATACYC1 promoter allele upstream of DNM1 or PSD1, the tetO7-TATACYC1 promoter was integrated as described previously (Mnaimneh et al., 2004). Briefly, the tetO7-TATACYC1 promoter allele was amplified from tet-cho1 mutant genomic DNA by PCR using oligo pairs designed previously (Mnaimneh et al., 2004), which are also listed in Table S5. The tetO7-TATACYC1 promoter allele was then integrated by PCR-mediated gene replacement with the indicated selection cassette using standard techniques. Yeast strains constitutively expressing AAC1, CRD1, MIM1, OAC1, PSD1, or UPS2 from the GPD promoter were generated by integration of the expression cassette into yeast chr I (199456-199457) using standard techniques (Hughes and Gottschling, 2012). Plasmids for integration of the GPD-driven expression cassette are described in Table S3. Correct insertion of the expression cassette into chr I was confirmed by colony PCR across the chromosomal insertion site.
Wild-type yeast strains AHY12872 and AHY11976, which were rendered prototrophic with pHLUM (Mülleder et al., 2012) (see Table S4) to prevent complications caused by amino acid auxotrophies in the BY strain background, were used to quantify amino acid dependencies of MDC formation and for analysis of whole-cell lipid levels. Wild-type yeast strain AHY2318 was used for mating with the yeast deletion collection to create a new deletion library containing fluorescently tagged TOM70 and TIM50 for the MDC screen. AHY6760, AHY7019, AHY7021, AHY9543, AHY10568, AHY11351, AHY11353, AHY11357, AHY11359, AHY11367, AHY11435, AHY11438, AHY11442, AHY11533, AHY11634, AHY11766, AHY11768, AHY11966, AHY11990, AHY12011, AHY12518, AHY12555, AHY12775, AHY12803, AHY13656, AHY14155, AHY14157, AHY14226, AHY14228, AHY14230, AHY14232, AHY14234, AHY14261, AHY14740, AHY14832, AHY14835, AHY14838, AHY14911, AHY14945, AHY14947, AHY14949, AHY14953, AHY14955, AHY14963, AHY14993, AHY14995, AHY14997, and AHY14999 were used for quantification of MDC formation using widefield microscopy. AHY14041, AHY14045, AHY14138, AHY14142, AHY14742, AHY14744, AHY140789, and AHY14791 were used for obtaining microscopy images using super-resolution microscopy. AHY14923, AHY14925, AHY14927, AHY14929, and AHY14943 were used for yeast growth assays. Lipid analysis was performed in BY4741, AHY2677, AHY4230, AHY11362, AHY11370, AHY11440, AHY11533, AHY11636, AHY11833, AHY11874, AHY11964, AHY12516, AHY12736, and AHY12801. A complete list of all strains used in this manuscript can be found in Table S4.
Yeast cell culture and media
Yeast cells were grown exponentially for 15 h at 30°C to a density of 4–8 × 106 cells/ml before the start of all experiments described in the paper, including MDC assays and sample preparation for lipidomic analysis. This period of overnight log-phase growth was carried out to ensure mitochondrial uniformity across the cell population and is essential for consistent MDC activation. Unless otherwise indicated, cells were cultured in YPAD medium (1% yeast extract, 2% peptone, 0.005% adenine, and 2% glucose), which is the high amino acid media used for amino acid dependency experiments. For amino acid dependency experiments, SD medium (low amino acid medium) (0.67% yeast nitrogen base without amino acids, 2% glucose, supplemented nutrients 0.074 g/liter each adenine, alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, histidine, myo-inositol, isoleucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, uracil, valine, 0.369 g/liter leucine, 0.007 g/liter para-aminobenzoic acid) or minimal medium (no amino acid medium) (0.67% yeast nitrogen base without amino acids, 2% glucose) was used. For ethanolamine addition, ethanolamine was added to a final concentration of 5 mM. The pH of the media was adjusted to the value equal to before ethanolamine addition. Drugs were added to cultures at final concentrations of 500 nM for Concanamycin A, 10 μg/ml for Cycloheximide, 20 mg/ml for Doxycycline, and 200 nM for Rapamycin.
Plasmids
Plasmids used in this study are listed in the Table S3. pHLUM, a yeast plasmid expressing multiple auxotrophic marker genes from their endogenous promoters, was obtained from Addgene (#40276) (Mülleder et al., 2012). Plasmids for GPD-driven expression of AAC1, CRD1, MIM1, OAC1, PSD1, or UPS2 were generated by gateway-mediated transfer of the corresponding ORF (Harvard Institute of Proteomics) from pDONR201/221 into pAG306GPD-ccdB chr 1 (Hughes and Gottschling, 2012) using Gateway LR Clonase II Enzyme mix (Thermo Fisher Scientific) according to the manufacturer’s instructions, followed by sequencing to verify correct integrations. To integrate the resulting expression plasmid into yeast chr I (199456-199457), pAG306GPD-ORF chr 1 was digested with NotI-HF (#10133990; NEB) prior to transformation. All plasmids expressing alternative versions of Psd1-FLAG were generated through Gibson assembly into the yeast expression plasmid pRS416 (Gibson et al., 2009; Sikorski and Hieter, 1989). In brief, PSD1 was endogenously tagged with 5×FLAG by one-step PCR-mediated C-terminal endogenous epitope tagging using standard techniques and oligo pairs listed in Table S5 to create the yeast strain AHY11974, which contains PSD1-FLAG (see Table S4). Plasmid templates for FLAG epitope tagging were from the pFA6a-5FLAG-KanMX6 vector (Noguchi et al., 2008). Correct integrations were confirmed by colony PCR and the correct protein size by Western blot. To create a plasmid expressing Psd1-FLAG, the PSD1-5×FLAG ORF was amplified from AHY11974 genomic DNA by PCR and contained ∼500 bp upstream (PSD1 5′UTR) and the ADH1 terminator sequence and contained additional 20–25 bp flanking regions that were homologous to the DNA regions directly flanking the EcoRI recognition site found in the multiple cloning sites of pRS416. Alternatively, for plasmids expressing Psd1Mic60(1-57)-FLAG, Psd1Sec66(1-60)-FLAG, and Psd1Tom70(1-36)-FLAG, only PSD1 (codons 102-500)-5×FLAG and the ADH1 terminator sequence were PCR-amplified from AHY11974 genomic DNA, which were fused in frame with and behind PCR amplicons that contained either ∼500 bp of the MIC60 5′UTR and MIC60 ORF (codons 1-57), ∼500 bp of the SEC66 5′UTR and SEC66 ORF (codons 1-60), or ∼500 bp of the TOM70 5′UTR and TOM70 ORF (codons 1-36), respectively, which were further inserted into the EcoRI cut-site of pRS416 by tandem Gibson assembly respectively.
RNA isolation and RT-qPCR
Yeast RNA was isolated and the concentration was determined by RT-qPCR. For RNA isolation, 5 × 107 of overnight log-phase yeast cell cultures grown in high amino acid media were harvested and resuspended in Trizol reagent to a final density of 6 × 106 cells/ml. Cells were lysed with glass beads using an Omni Bead Ruptor 12 Homogenizer. Lysis was performed in six cycles of 20 s and was followed by centrifugation at 10,000 × g for 1 min at 4°C. Supernatants were transferred to a new tube and ethanol was added to a final concentration of 50%. RNA was isolated following the instruction of RNeasy Mini Kit (Qiagen) and the concentration was determined by Nanodrop. RNA samples were treated with TURBO DNase kit (AM2238; Invitrogen) to clear DNA contamination. High-capacity cDNA Reverse Transcription Kits (4368814; Applied Biosystems) were used for reverse transcription of RNA samples, followed by RT-qPCR using TaqMan Real-Time PCR Assays (Thermo Fisher Scientific). MRL1 was used as the internal control. Oligo pairs used for RT-qPCR are listed in Table S5.
Yeast growth assays
Fivefold serial dilutions of exponentially growing yeast cells were diluted in ddH2O. 3 μl of each dilution was spotted onto the agar media (3% agar) denoted in each figure legend. For ethanolamine addition, ethanolamine was added to the respective media to a final concentration of 5 mM, and the pH was adjusted prior to the addition of agar. The total cells plated in each dilution spot were 5,000; 1,000; 20; 40; and 8. Plates were cultured at 30°C for 24 h before obtaining images.
Yeast MDC assays
For yeast MDC assays, overnight log-phase cell cultures were directly harvested, or harvested after being grown in the presence of DMSO or the indicated drug(s) for 2 h. After incubation, cells were harvested by centrifugation, resuspended in an imaging buffer (5% wt/vol Glucose, 10 mM HEPES pH 7.6), and optical z-sections of live yeast cells were acquired with an AxioObserver (Carl Zeiss). The percentage of cells with MDCs was quantified from maximum intensity projected wide field images generated in ZEN (Carl Zeiss). Unless otherwise indicated, all quantifications show the mean ± SEM from three biological replicates with n = 100 cells per experiment. MDCs were identified as Tom70-positive Tim50-negative structures of varying size and shape. Unless specified, maximum-intensity projected images are displayed for all yeast images.
Yeast MDC screen
For the yeast MDC screen, a collection of yeast strains endogenously expressing Tom70-yeGFP and Tim50-mCherry were created by crossing a Tom70-yeGFP Tim50-mCherry query strain (AHY2318, see Table S4) to the yeast non-essential deletion collection (Giaever et al., 2002) using Synthetic Genetic Array (SGA) technology (Tong et al., 2001) with a ROTOR Robot (Singer) and standard techniques for high-throughput strain construction. The final library was contained within 96-well plates. The library was grown in YPAD (200 µl for each well) with constant agitation for over 24 h at 30°C to reach the stationary phase. The saturated library was then diluted and cultured in YPAD (1 ml for each well) in deep-well 96-well plates with constant agitation for overnight growth at 30°C. In this way, most mutants in the library reach log-phase at a cell density of 2–6 × 106 cells/ml. Strains with a low amount of cells for screening are noted in Table S1. The library was centrifuged in deep-well 96-well plates and resuspended in 1 ml of YPAD containing 200 nM Rap and cultured for 2 h at 30°C with constant agitation. For imaging, the library was harvested by centrifugation in deep-well 96-well plates, washed once with double-distilled water, and resuspended in 500 µl SD medium supplemented with 2 mg/ml cas-amino acids and 200 nM Rap. The supplementation of amino acids is to ensure consistent MDC formation. The library (100 µl for each well) was then added to glass-bottom 96-well plates precoated with 1 mg/ml Concanavalin A to allow the attachment of yeast cells to the bottom of wells. The surfaces of glass-bottom plates contacting the water immersion objective were coated with Sigmacote. Optical z-sections of the library were acquired with an AxioObserver (Carl Zeiss). The percentage of cells with MDCs was quantified from widefield images generated in ZEN (Carl Zeiss). At least n = 100 cells were assessed unless the total number of cells was <100 as noted in Table S1. Strains with a total number of cells fewer than five per well were noted as “low growth” in Fig. 1 B. MDCs were identified as Tom70-enriched, Tim50-negative structures of varying size and shape. Genetic ontology analysis was performed on gene deletions that lead to MDC formation in <20% or higher than 60% of cells using FunSpec (Robinson et al., 2002).
Microscopy and image analysis
Optical z-sections of live yeast cells were acquired with a Zeiss AxioObserver 7 equipped with a PCO Edge 4.2LT Monochrome, Air Cooled, USB 3 CCD camera with a Solid-State Colibri 7 LED illuminator and 63× oil-immersion objective (Plan Apochromat, NA 1.4), a Zeiss AxioImager M2 equipped with an Axiocam 506 monochromatic camera and 63× oil-immersion objective (Plan Apochromat, NA 1.4), or a Zeiss LSM800 equipped with an Airyscan detector, 63× oil-immersion objective (Plan Apochromat, NA 1.4) at room temperature. Yeast cells were resuspended in a buffer containing 10 mM HEPES, pH 7.6, and 5% glucose prior to imaging (Hughes et al., 2016). Optical z-sections of live yeast cell libraries for the MDC screen were acquired with a Zeiss AxioObserver 7 equipped with a PCO Edge 4.2LT Monochrome, Air Cooled, USB 3 CCD camera with a Solid-State Colibri 7 LED illuminator and 63× oil-immersion objective (Plan Apochromat, NA 1.4). Widefield images were acquired with ZEN (Carl Zeiss) and processed in Fiji (Schindelin et al., 2012). Super-resolution images were acquired with ZEN (Carl Zeiss) and processed using the automated Airyscan processing algorithm in ZEN (Carl Zeiss) and then further processed in Fiji (Schindelin et al., 2012). Fluorochromes are indicated in figure legends. Individual channels of all images were minimally adjusted in Fiji (Schindelin et al., 2012) to match the fluorescence intensities between channels for better visualization.
Yeast indirect immunofluorescence (IIF) staining
Yeast IIF detecting all versions of Psd1-FLAG was conducted as previously described, (Xiao et al., 2021; Shakya et al., 2021). Specifically, overnight log-phase psd1∆ mutants expressing different Psd1 constructs were grown in SD- Ura (SD medium that excludes uracil) to a final density of 4 × 106 cells/ml. Cells were harvested by centrifugation and fixed with 4% paraformaldehyde in SD- Ura for 1 h. Fixed yeast cells were washed twice and incubated with 10 mM DTT at room temperature for 10 min, and spheroplasted with 0.25 mg/ml Zymolyase at 30°C for 40 min. Spheroplasts were gently diluted in 1:20 and attached to glass slides pre-coated with 0.1% poly-L-Lysine (2 mg/ml). Samples were permeabilized in cold PBS containing 0.1% Triton X-100 for 10 min at 4°C, briefly dried, and blocked with 1% BSA at room temperature for 30 min. After blocking, samples were incubated with primary antibodies (Monoclonal ANTI-FLAG M2 antibody produced in mouse [Sigma-Aldrich], 1:300 diluted; Tom70 endogenous antibody produced in rabbit [Wiedemann and Pfanner, 2017], 1:250 diluted) for 1 h 30 min at room temperature and washed 10 times before incubated with secondary antibodies (Goat anti-Mouse IgG [H+L] Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, 1:300 diluted; Goat anti-Rabbit IgG [H+L] Cross-Adsorbed Secondary Antibody, Alexa Fluor 568, 1:300 diluted) for 45 min at room temperature. Samples were washed 10 times. Slides were washed twice before sealing and mounted with HardSet medium (ProLong Glass Antifade Mountant with NucBlue Stain [Invitrogen]) overnight. Wide-field images were acquired as described above.
Extraction of lipids from yeast whole cell lysates
For analysis of whole-cell lysate lipid levels, cells were grown exponentially in the indicated media for 15 h at 30°C to a density of 6–8 × 106 cells/ml or after being treated with the indicated chemicals for 2 h. A total of 5 × 107 yeast cells were harvested by centrifugation, washed twice with double-distilled water, and cell pellets were shock-frozen in liquid nitrogen.
Extraction of lipids was carried out using a biphasic solvent system of cold methanol, methyl tert-butyl ether (MTBE), and water as described (Matyash et al., 2008) with some modifications. In a randomized sequence, yeast lipids were extracted in bead-mill tubes (glass 0.5 mm; Qiagen) containing a solution of 230 µl MeOH containing internal standards (Cholesterol-d7 [75 µg/ml], and FA 16:0-d31 [28.8 µg/ml] all at 10 µl per sample; Avanti SPLASH LipidoMix) and 250 µl ammonium bicarbonate. Samples were homogenized in one 30-s cycle, transferred to microcentrifuge tubes (polypropylene 1.7 ml; VWR) containing 750 µl MTBE, and rested on ice for 1 h with occasional vortexing. Samples were then centrifuged at 15,000 × g for 10 min at 4°C and the upper phases were collected. A 1-ml aliquot of the upper phase of MTBE/MeOH/water (10:3:2.5, vol/vol/vol) was added to the bottom aqueous layer followed by a brief vortex. Samples were then centrifuged at 15,000 × g for 10 min at 4°C and the upper phases were combined and evaporated to dryness under speedvac. Lipid extracts were reconstituted in 500 µl of mobile phase B and transferred to a liquid chromatography–mass spectrometry (LC–MS) vial for analysis. Concurrently, a process blank sample was prepared and then a pooled quality control (QC) sample was prepared by taking equal volumes (∼50 µl) from each sample after final resuspension.
Extraction of lipids from isolated yeast mitochondria
Yeast mitochondria were isolated as previously described (Schuler et al., 2016) with some modifications. Specifically, yeast cells were grown overnight in log-phase as described above, then treated with either DMSO or indicated drugs for 2 h to a density of 6–8 × 106 cells/ml. Cells were then reisolated by centrifugation, washed with distilled water, and harvested. The pellet weight was determined. Subsequently, cells were resuspended in dithiothreitol (DTT) buffer (0.1 M Tris, 10 mM DTT) for 2 ml/g of pellets and incubated for 10 min at 30°C under constant rotation. After reisolation by centrifugation at 2,000 × g, DTT-treated cells were washed once in 1.2 M sorbitol and resuspended in sorbitol phosphate buffer (1.2 M sorbitol, 20 mM K2HPO4, pH = 7.4 with HCl) for 2 ml per gram of pellets. Cell walls were digested in 6.67 ml of sorbitol phosphate buffer per gram of pellet containing 2 mg lyticase per gram of pellets (4 mg lyticase per gram of pellets was used when cells were cultured in no amino acid media to allow complete digestion) for 50 min at 30°C under constant rotation. After lyticase digestion, spheroplasts were isolated by centrifugation at 1,200 × g and lysed by mechanical disruption in 13.3 ml of homogenization buffer (0.6 M sorbitol, 10 mM Tris, pH = 7.4, 1 mM EDTA, pH = 8.0 with KOH, 0.2% BSA, 1 mM PMSF) per gram of pellet at 4°C. Cell debris was removed from the homogenate by centrifugation twice at 1,500 × g and 2,000 × g successively for 5 min at 4°C. Crude mitochondria were pelleted at 12,000 × g for 12 min at 4°C. The mitochondrial pellet was resuspended in 10 ml of SEM buffer (250 mM sucrose, 1 mM EDTA, pH = 8.0 with KOH, 10 mM 3-[N-morpholino]-propansulfonic acid, pH = 7.2), reisolated by differential centrifugation as described above, and resuspended in SEM buffer. The concentration of mitochondria prepared was determined by Bicinchoninic Acid Protein Assay. For each sample used for lipidomics analysis, 200 µg of mitochondria (by protein) were pelleted at 17,500 × g for 12 min at 4°C before being shock-frozen in liquid nitrogen and stored at −80°C.
Extraction of lipids was carried out using a biphasic solvent system of cold methanol, methyl tert-butyl ether (MTBE), and PBS/water (Matyash et al., 2008) with some modifications. In a randomized sequence, 225 µl MeOH with internal standards described above and 750 µl MTBE were added to each sample. Samples were sonicated for 60 s and then incubated on ice with occasional vortexing for 1 h. 188 µl of PBS was then added to induce phase separation and briefly vortexed. Samples were rested at room temperature for 15 min and then centrifuged at 15,000 × g for 10 min at 4°C. The organic (upper) layer was collected and the aqueous (lower) layer was re-extracted with 1 ml of 10:3:2.5 (vol/vol/vol) MTBE/MeOH/dd-H2O, briefly vortexed, incubated at room temperature, and centrifuged at 15,000 × g for 10 min at 4°C. Upper phases were combined and evaporated to dryness under a speed vac. Lipid extracts were reconstituted in 450 µl of 4:1:1 (vol/vol/vol) IPA/ACN/water and transferred to an LC-MS vial for analysis. Concurrently, a process blank sample was prepared, and pooled QC samples were prepared by taking equal volumes from each sample after final resuspension.
LC-MS analysis (QTOF)
Lipid extracts were separated on an Acquity UPLC CSH C18 column (2.1 × 100 mm; 1.7 µm) coupled to an Acquity UPLC CSH C18 VanGuard precolumn (5 × 2.1 mm; 1.7 µm) (Waters) maintained at 65°C connected to an Agilent HiP 1290 Sampler, Agilent 1290 Infinity pump, and Agilent 6545 Accurate Mass Q-TOF dual AJS-ESI mass spectrometer (Agilent Technologies). Samples were analyzed in a randomized order in both positive and negative ionization modes in separate experiments acquiring with the scan range m/z 100–1700. For positive mode, the source gas temperature was set to 225°C, with a drying gas flow of 11 liters/min, nebulizer pressure of 40 psig, sheath gas temp of 350°C, and sheath gas flow of 11 l/min. VCap voltage is set at 3500 V, nozzle voltage 500 V, fragmentor at 110 V, skimmer at 85 V, and octopole RF peak at 750 V. For negative mode, the source gas temperature was set to 300°C, with a drying gas flow of 11 l/min, a nebulizer pressure of 30 psig, sheath gas temp of 350°C, and sheath gas flow 11 l/min. VCap voltage was set at 3,500 V, nozzle voltage 75 V, fragmentor at 175 V, skimmer at 75 V, and octopole RF peak at 750 V. Mobile phase A consisted of ACN:H2O (60:40, vol/vol) in 10 mM ammonium formate and 0.1% formic acid, and mobile phase B consisted of IPA:ACN:H2O (90:9:1, vol/vol/vol) in 10 mM ammonium formate and 0.1% formic acid. For negative mode analysis, the modifiers were changed to 10 mM ammonium acetate. The chromatography gradient for both positive and negative modes started at 15% mobile phase B then increased to 30% B over 2.4 min, it then increased to 48% B from 2.4 to 3.0 min, then increased to 82% B from 3 to 13.2 min, then increased to 99% B from 13.2 to 13.8 min where it is held until 16.7 min and then returned to the initial conditions and equilibrated for 5 min. The flow was 0.4 ml/min throughout, with injection volumes of 5 µl for positive and 10 µl negative mode. Tandem mass spectrometry was conducted using iterative exclusion, the same LC gradient at collision energies of 20 and 27.5 V in positive and negative modes, respectively.
LC-MS data processing
For data processing, Agilent MassHunter (MH) Workstation and software packages MH Qualitiative and MH Quantitative were used. The pooled QC (n = 8) and process blank (n = 4) were injected throughout the sample queue to ensure reliability of the acquired lipidomics data. For lipid annotation, accurate mass and MS/MS matching were used with the Agilent Lipid Annotator library and LipidMatch (Koelmel et al., 2017). Results from the positive and negative ionization modes from the Lipid Annotator were merged based on the class of lipid identified. Data exported from MH Quantitative were evaluated using Excel where initial lipid targets are parsed based on the following criteria. Only lipids with relative standard deviations (RSD) <30% in QC samples are used for data analysis. Additionally, only lipids with background AUC counts in process blanks that are <30% of QC are used for data analysis. The parsed Excel data tables are normalized based on the ratio to class-specific internal standards.
The number of replicates, what n represents, and dispersion and precision measures are indicated in the figure legends. All experiments were repeated at least three times. All attempts at replication were successful. Sample sizes were as large as possible to be representative but of a manageable size for quantifications. Specifically, for yeast MDC assays, N = three replicates, with n = 100 cells for each replicate. For lipidomic analysis, N = three to four biological replicates were analyzed in the same LC-MS run. All statistical analysis was performed in Prism (GraphPad), and the statistical test used is indicated in the corresponding figure legend. For statistical analysis of lipidomics experiments, two-way ANOVA with Holm-Šídák test was used as indicated in figure legends. No data were excluded from the analyses, with the exception of some lipidomic samples that did not meet the QC cutoff. In the latter case, all samples of the affected biological replicate were excluded from any further analysis. Data distribution was assumed to be normal, but this was not formally tested. No randomization or blinding was used as all experiments were performed with defined laboratory reagents and yeast strains of known genotypes.
Online supplemental material
Fig. S1 shows that depletion of CL levels prevents MDC biogenesis independently of affecting mitochondrial fission machinery. Fig. S2 shows that reducing mitochondrial PE levels activates MDC biogenesis. Fig. S3 contains data suggesting PE generated by mitochondrial-localized Psd1 is required for suppressing MDC biogenesis and shows that PSD1 deletion bypasses the requirement for ERMES components in MDC formation. Fig. S4 shows the PE level changes in the indicated mutant cells in response to the indicated treatments, demonstrates PE decline is downstream of amino acid overabundance stress, and shows Mim1 is required for MDC biogenesis. Table S1 lists the quantification results of the MDC screen and gene ontology analysis. Table S2 summarizes a complete profile of all major PL species, including their amount relative to total lipids and absolute abundance detected by mass spectrometry-based lipidomic analysis for all lipidomics results in the manuscript. Table S3 lists bacterial strains, chemicals, antibodies, plasmids, and software used in this study. Table S4 lists the yeast strains used in this study. Table S5 lists the oligonucleotides used in this study.
Data availability
All reagents used in this study are available upon request. All other data reported in this paper will be shared by the lead contact upon request. This paper does not report the original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank members of the A.L. Hughes laboratory for discussion and manuscript comments. We thank members of the Janet M. Shaw (J.M. Shaw) laboratory (Department of Biochemistry, University of Utah School of Medicine, Salt Lake City, UT, USA) for providing reagents, antibodies, and technical assistance. We thank Jenna M. Goodrum for helping construct the yeast deletion collection used for screening. We thank J.M. Shaw for contributing stipend support for T. Xiao. We thank Dan Cuthbertson of Agilent Technologies for assistance with tandem mass spectrometry.
Metabolomics analysis was performed at the Metabolomics Core Facility at the University of Utah. Mass spectrometry equipment was obtained through the NCRR Shared Instrumentation Grant 1S10OD016232-01, 1S10OD018210-01A1, and 1S10OD021505-01. Research was supported by National Institutes of Health (NIH) grants GM119694 and AG061376 (to A.L. Hughes); NIH T32GM007464 (to A.M. English); an American Heart Association Postdoctoral Fellowship, 20POST35200110 (to Z.N. Wilson); an American Cancer Society–Give Mas, Live Mas Southern Multifoods Postdoctoral Fellowship, PF-20-018-01-CCG (to Z.N. Wilson); and a University of Utah Graduate Research Fellowship (to T. Xiao).
Author contributions: conceptualization, T. Xiao and A.L. Hughes; methodology and formal analysis, T. Xiao, A.M. English, and J.A. Maschek; investigation, T. Xiao, A.M. English, Z.N. Wilson, and J.A. Maschek; visualization, T. Xiao and A.L. Hughes; funding acquisition, T. Xiao, A.M. English, Z.N. Wilson, A.L. Hughes, J.E. Cox, and J.M. Shaw; supervision, J.E Cox and A.L. Hughes; project administration, A.L. Hughes; writing—original draft, T. Xiao and A.L. Hughes; writing—review and editing, T. Xiao, A.M. English, Z.N. Wilson, J.A. Maschek, and A.L. Hughes.

