


Glycosylation is essential to facilitate cell–cell adhesion and differentiation. We determined the role of the dolichol phosphate mannosyltransferase (DPM) complex, a central regulator for glycosylation, for desmosomal adhesive function and epidermal differentiation. Deletion of the key molecule of the DPM complex, DPM1, in human keratinocytes resulted in weakened cell–cell adhesion, impaired localization of the desmosomal components desmoplakin and desmoglein-2, and led to cytoskeletal organization defects in human keratinocytes. In a 3D organotypic human epidermis model, loss of DPM1 caused impaired differentiation with abnormally increased cornification, reduced thickness of non-corneal layers, and formation of intercellular gaps in the epidermis. Using proteomic approaches, SERPINB5 was identified as a DPM1-dependent interaction partner of desmoplakin. Mechanistically, SERPINB5 reduced desmoplakin phosphorylation at serine 176, which was required for strong intercellular adhesion. These results uncover a novel role of the DPM complex in connecting desmosomal adhesion with epidermal differentiation.
Introduction
Desmosomes are vital mediators of intercellular adhesion and are mechanistically linked to several diseases, such as the blistering skin disease pemphigus, arrhythmogenic cardiomyopathy, and different cancer entities (Broussard et al., 2015; Najor, 2018; Yang et al., 2006). Desmosomes are highly ordered complexes comprised of adhesion molecules and plaque proteins. Desmogleins (DSG) 1–4 and desmocollins (DSC) 1–3 belong to the cadherin family of adhesion molecules, which interact in a calcium-dependent manner (Delva et al., 2009; Chitaev and Troyanovsky, 1997). While desmosomal cadherins mediate cell–cell contact by means of their extracellular domains, the intracellular domains interact with the armadillo proteins plakophilin 1–3 and plakoglobin (PG). These connect to desmoplakin (DSP), which links the entire complex to intermediate filaments of the cytoskeleton, thereby providing mechanical strength to cells and tissues (Kouklis et al., 1994). In addition to intermediate filaments, the actin cytoskeleton also has implications on desmosome assembly, where actomyosin contractility and the cortical actin network are required for membrane translocation and mobility of desmosomal proteins (Godsel et al., 2005). Cell–cell adhesion not only provides mechanical strength and tissue integrity but also influences differentiation processes. A well-coordinated balance of cell–cell adhesion via desmosomes, adherens junctions, and tight junctions, as well as differentiation of keratinocytes into the cornified layer, is important to maintain barrier functions of the epidermis (Candi et al., 2005). Alterations in several desmosomal components lead to barrier and differentiation defects of the epidermis, highlighting the importance of desmosomal cadherins for maintaining tissue integrity and mediating a proper differentiation process (Jonkman et al., 2005; Chidgey et al., 2001; Sumigray and Lechler, 2015; Koch et al., 1997; Spindler and Waschke, 2018). However, the precise underlying mechanisms are only partially understood.
Various posttranslational modifications (PTMs) play important roles in mediating the subcellular localization of desmosomal proteins, and glycosylation serves as an important PTM in this context. Studies have shown the impact of glycosylation in maintaining tissue homeostasis, protein trafficking, cell signaling, proliferation, differentiation, and cell adhesion (Ruoslahti, 1989; Ruoslahti and Yamaguchi, 1991). Glycosylation is a complex protein modification where glycans attached to proteins undergo processing and maturation with different end products (mannose rich, complex, and hybrid). In mammals, all glycans are made from nine monosaccharide building blocks: mannose, glucose, galactose, fucose, N-acetyl galactosamine, N-acetylglucosamine, xylose, glucoronic acid, and Neu5Ac (sialic acid) (Stanley et al., 2009). The dolichol phosphate mannosyltransferase (DPM) complex is a crucial mediator in the process as it is responsible for donating dolichol-phosphate-mannose (Dol-P-Man) residues that feed into N-glycosylation, C- and O-Man glycosylation, and GPI-anchor biosynthesis pathways (Maeda et al., 1998; Kornfeld and Kornfeld, 1985; Herscovics and Orlean, 1993). The DPM1 complex is made of three subunits, DPM1, DPM2, and DPM3. DPM1 is the catalytic subunit that synthesizes the Dol-P-Man donor substrate from GDP-mannose and dolichol-phosphate in the endoplasmic reticulum (ER) membrane (Colussi et al., 1997). DPM2 and DPM3 have regulatory functions, which facilitate DPM1 localization to ER membrane and enable DPM1 to exert its catalytic activity (Maeda et al., 2000). Defects in DPM1 lead to congenital glycosylation disorders and seizures (Kim et al., 2000). With regard to desmosome function, it has been shown that inhibiting N-linked glycosylation results in impaired desmosome formation and stability (Jin and Chung, 2018). It has also been demonstrated that N-linked glycosylation at multiple sites is responsible for the incorporation of DSC2 in the plasma membrane (Brodehl et al., 2019). Further, O-linked glycosylation of PG at the N-terminus protects it from proteolytic degradation and enhances cell–cell adhesion (Hatsell et al., 2003). Apart from its role in mediating cell–cell adhesion, glycosylation has also been studied in the context of epidermal differentiation. CRISPR/Cas9-based editing of several glycosylation genes in organotypic models of epidermis led to differentiation defects (Dabelsteen et al., 2020). Further, glycosylation was implicated in the process of desquamation and shedding of corneocytes from the surface (Walsh and Chapman, 1991). However, the molecular mechanisms of how glycosylation regulates cell–cell adhesion and subsequent effects on epidermal differentiation are not well understood.
Here, we study the contribution of the DPM complex to desmosomal adhesion and epidermal differentiation using keratinocytes and organotypic models of the human epidermis.
Results
DPM1 modulates cell–cell adhesion and differentiation in a human organotypic epidermis model
We first aimed to understand the role of the DPM group of glycosylation modulators for regulation of intercellular adhesion. We generated CRISPR/Cas9-mediated knockdown (KD) cell lines of the three DPM isoforms with respective non-targeting control lines (NT) in HaCaT keratinocytes. In line with the occurrence as a complex, loss of DPM2 or DPM3 led to loss of DPM1 (Fig. S1 a). Functionally, DPM1 KD reduced the amounts of various glycan structures (oligomannose and complex type), including highly diminished intensities of some oligomannose structures such as 5 mannose-2 N-acetylglucosamine (Man5), 7 mannose-2 N-acetylglucosamine (Man7), or 8 mannose-2 N-acetylglucosamine (Man8), as measured by mass-spectrometry-based glycomic analysis from cell lysates (Fig. S1 b). Dispase-based intercellular adhesion assays showed that loss of DPM1, and to milder extent loss of DPM2 and DPM3, impaired intercellular adhesion, indicated by an increased number of fragments after applying shear stress to a detached monolayer (Fig. 1, a and b). A KD cell line for the desmosomal protein DSG2 was used as a positive control for reduced cell–cell adhesion. CRISPR/Cas9-mediated KD of DPM1 in primary human keratinocytes also resulted in loss of cell–cell adhesion, supporting the relevance of DPM1 for keratinocyte intercellular adhesion (Fig. 1, c and d). Interestingly, biochemical characterization of desmosomal proteins in HaCaT keratinocytes showed no major changes in total protein content upon loss of the DPM complex (Fig. S1, c and d). Because DPM1 is the enzymatically active protein of the complex, we focused on DPM1 for further experiments. The human epidermis is a stratified epithelium that differentiates into several layers during its course of maturation to provide a fully functional epidermis (Fig. 1 e). Immunostaining in human foreskin tissue showed expression of DPM1 in all layers of the differentiated epidermis, suggesting a contribution of this protein to epidermal homeostasis (Fig. S1 e). We generated an organotypic 3D-reconstructed human epidermis model (3D-RHE), which reproduced the differentiation process of interfollicular epidermis as indicated by CK10 and filaggrin staining and which showed expression of DPM1 throughout all non-corneal layers (Fig. 1 f and Fig. S1, e and f). KD of DPM1 in 3D-RHEs caused defects in the differentiation process, indicated by thickening of the corneal layer and reduced thickness of the other layers, with the total epidermal thickness being unchanged (Fig. 1, f–i). These changes were unrelated to proliferation in the basal layer under DPM1 loss, as indicated by the proliferation marker Ki67 being similar in both sgNT1 and sgDPM1 conditions (Fig. 1 j). Further, DPM1 loss was accompanied by the presence of intercellular spaces within the viable layers of the epidermis, indicating perturbed cell–cell adhesion (Fig. 1 k).
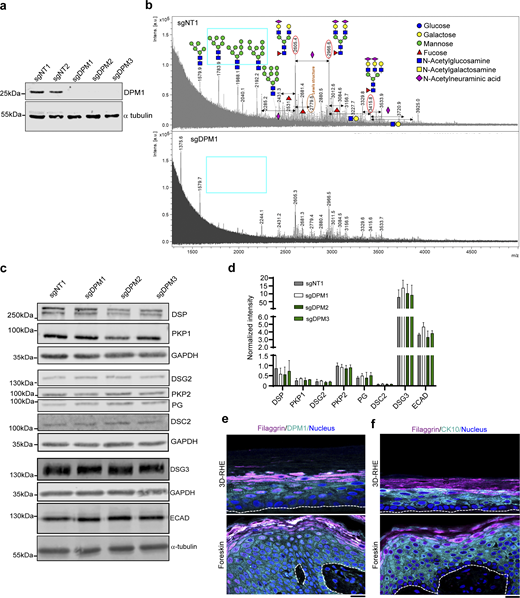
Loss of DPM1 leads to reduced oligomannose structures in keratinocytes. (a) Western blot showing DPM1 in sgDPM1, sgDPM2, and sgDPM3 HaCaT keratinocytes. α-Tubulin was used as internal loading control. (b) Mass-spectrometry analysis of N-glycome from sgNT1 and sgDPM1 cell lysates. X-axis denotes mass to charge (m/z) ratio and y-axis denotes the intensities of the corresponding m/z peaks (A.U.). The m/z peaks denote the respective glycan structures as depicted and the monosaccharide codes are provided in the color-coded legends. Cyan box marks oligomannose structures in sgNT1 conditions that are drastically reduced in sgDPM1. (c and d) Western blot images and quantifications of desmosomal proteins from sgDPM1, sgDPM2, and sgDPM3 HaCaT keratinocytes. Representative images of three biological replicates are shown. GAPDH was used as internal loading control (N = 3). (e) Immunofluorescence staining of DPM1 and filaggrin in 3D-RHE and human foreskin tissue, as indicated. Filaggrin was used as a differentiation marker. Dashed line indicates insert membrane/basement membrane. Scale bar: 10 µm. Panel shows representative of three biological replicates. (f) Immunostaining of CK10 and filaggrin in 3D-RHE and human foreskin tissue, as indicated. Dashed line indicates insert and basement membrane. Scale bar: 10 µm distance. Panel shows representative of three biological replicates. Source data are available for this figure: SourceData FS1.
Loss of DPM1 leads to reduced oligomannose structures in keratinocytes. (a) Western blot showing DPM1 in sgDPM1, sgDPM2, and sgDPM3 HaCaT keratinocytes. α-Tubulin was used as internal loading control. (b) Mass-spectrometry analysis of N-glycome from sgNT1 and sgDPM1 cell lysates. X-axis denotes mass to charge (m/z) ratio and y-axis denotes the intensities of the corresponding m/z peaks (A.U.). The m/z peaks denote the respective glycan structures as depicted and the monosaccharide codes are provided in the color-coded legends. Cyan box marks oligomannose structures in sgNT1 conditions that are drastically reduced in sgDPM1. (c and d) Western blot images and quantifications of desmosomal proteins from sgDPM1, sgDPM2, and sgDPM3 HaCaT keratinocytes. Representative images of three biological replicates are shown. GAPDH was used as internal loading control (N = 3). (e) Immunofluorescence staining of DPM1 and filaggrin in 3D-RHE and human foreskin tissue, as indicated. Filaggrin was used as a differentiation marker. Dashed line indicates insert membrane/basement membrane. Scale bar: 10 µm. Panel shows representative of three biological replicates. (f) Immunostaining of CK10 and filaggrin in 3D-RHE and human foreskin tissue, as indicated. Dashed line indicates insert and basement membrane. Scale bar: 10 µm distance. Panel shows representative of three biological replicates. Source data are available for this figure: SourceData FS1.

DPM1 loss impairs cell cohesion and epidermal differentiation. (a and b) Dispase-based dissociation assays to semiquantitatively assess cell–cell adhesion in HaCaT keratinocytes (N = 4). One-way ANOVA, Dunn’s multiple comparison test. Higher fragment numbers represent lower adhesive strength. (c and d) Dispase-based dissociation assays of primary human keratinocytes (N = 5). Unpaired Student’s t test. (e) Schematic of epidermis (top) and of 3D-RHE model design from primary human keratinocytes (bottom). (f) H&E staining and immunostainings for CK10/Filaggrin, DPM1, and Ki67 of control (sgNT1) and DPM1 KD (sgDPM1) 3D-RHEs 12 days after airlift. Insets (zoomed 2× of original image) and arrows denote interfollicular gaps in different layers of epidermal equivalents of sgDPM1. White dashed line indicates insert membrane. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. (g–i) Quantification of epidermal thickness parameters from four independent biological replicates, from two different donors. Each dot represents one biological replicate. Unpaired Student’s t test. (j) Analysis showing Ki67-positive nuclei normalized to total number of nuclei (N = 3). Unpaired Student’s t test. (k) Violin plot showing quantification of intercellular spaces within the epidermis in sgNT1 and sgDPM1 3D-RHE. The cutoff for defining intercellular spaces was set to be >50 µm2 to exclude shrinking artifacts. A minimum of 20 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test.
DPM1 loss impairs cell cohesion and epidermal differentiation. (a and b) Dispase-based dissociation assays to semiquantitatively assess cell–cell adhesion in HaCaT keratinocytes (N = 4). One-way ANOVA, Dunn’s multiple comparison test. Higher fragment numbers represent lower adhesive strength. (c and d) Dispase-based dissociation assays of primary human keratinocytes (N = 5). Unpaired Student’s t test. (e) Schematic of epidermis (top) and of 3D-RHE model design from primary human keratinocytes (bottom). (f) H&E staining and immunostainings for CK10/Filaggrin, DPM1, and Ki67 of control (sgNT1) and DPM1 KD (sgDPM1) 3D-RHEs 12 days after airlift. Insets (zoomed 2× of original image) and arrows denote interfollicular gaps in different layers of epidermal equivalents of sgDPM1. White dashed line indicates insert membrane. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. (g–i) Quantification of epidermal thickness parameters from four independent biological replicates, from two different donors. Each dot represents one biological replicate. Unpaired Student’s t test. (j) Analysis showing Ki67-positive nuclei normalized to total number of nuclei (N = 3). Unpaired Student’s t test. (k) Violin plot showing quantification of intercellular spaces within the epidermis in sgNT1 and sgDPM1 3D-RHE. The cutoff for defining intercellular spaces was set to be >50 µm2 to exclude shrinking artifacts. A minimum of 20 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test.
Loss of DPM1 impairs DSP surface localization and cytoskeletal arrangement in human keratinocytes
Based on the biochemical analysis, changes in the protein expression of desmosomal components and E-cadherin did not account for the reduced intercellular adhesion upon loss of DPM1. We thus investigated the localization of the essential desmosomal protein DSP in HaCaT and primary human keratinocytes. The amount of DSP puncta was significantly reduced at cell borders upon loss of DPM1, indicating reduced number of desmosomes (Fig. 2, a and b). DSG2, which is a desmosomal cadherin specifically clustering in desmosomes (Wanuske et al., 2021), was also reduced at the cell membrane under sgDPM1 conditions in HaCaT keratinocytes. In addition, DPM1 KD in primary human keratinocytes reproduced the findings from HaCaT keratinocytes (Fig. 2 c). Interestingly, DSP staining also appeared fragmented and more localized to the cytosol in sgDPM1 3D-RHEs compared with sgNT1 control (Fig. 2 d). With DSP serving as an anchor for keratin intermediate filaments, we also tested whether the reduced DSP membrane localization was associated with changes in keratin distribution. Pan-cytokeratin staining showed a redistribution away from cell–cell contact sites, creating a less dense keratin network at the cell cortex (Fig. 2, e–g). Depletion of DPM1 further led to redistribution of the actin cytoskeleton, with the cortical actin belt appearing more diffusely localized throughout the cytosol (Fig. 2, h–j). Changes in the cytoskeletal organizations affect the mechanical properties of the cell (Ramms et al., 2013). We used an atomic force microscopy (AFM)–based approach to quantify the elasticity of both control and DPM1 KD HaCaT keratinocytes. DPM1 KD cells showed significantly reduced cellular elasticity, quantified as Young’s modulus (Fig. 2 k). Further, to address whether the reduced expression of DSP on the surface in DPM1 KD conditions correlates with changes in the membrane stability of DSP, we performed fluorescence recovery after photobleaching (FRAP) experiments to assess the dynamics of DSP at cell–cell interfaces. FRAP showed a significant increase in the mobile fraction of DSP, indicating reduced DSP stability at cell junctions upon loss of DPM1 (Fig. 2, l and m). Together these results suggest a modulation of DSP localization and function by DPM1.

DPM1 modulates DSP localization and cytoskeletal organization. (a) Immunostaining of DSP and DSG2 in HaCaT keratinocytes. Merge indicates overlap between DSP, DSG2, and nuclei stained with DAPI. Scale bar: 10 µm distance. (b) Quantification of the number of DSP puncta over the respective length of cell membrane (µm) from individual cells (represented by individual dots), N = 3. Unpaired Student’s t test. (c) Immunostaining of DSP and DSG2 in primary human keratinocytes. Scale bar: 10 µm distance. Panel shows representatives of three biological replicates. (d) Immunostaining of DSP in 3D-RHE of control (sgNT1) and sgDPM1 conditions. White dashed line indicates insert membrane. Magenta dashed rectangles mark regions magnified on the right (zoomed 2× of original image). Representative of three biological replicates. Scale bar: 10 µm distance. (e–g) Keratin staining depicted by pan-cytokeratin in control and DPM1 KD HaCaT keratinocytes. Analysis done by quantifying the keratin staining intensity (A.U.) across cell borders spanning a distance of 10 µm. Peak width was calculated between the two highest points from the distribution plot profile graph. Scale bar: 10 µm distance. 30 individual cells were quantified from three independent biological replicates. Unpaired Student’s t test. (h–j) F-actin stained by phalloidin in control and DPM1 KD HaCaT keratinocytes. Analysis done by quantifying the F-actin staining intensity (A.U.) across cell borders spanning a distance of 10 µm. The width of the peaks was calculated between the baseline values from distribution plot profile graphs. Scale bar: 10 µm distance. 30 individual cells were analyzed from three independent biological replicates. Unpaired Student’s t test. (k) Schematic of the AFM setup to measure cellular elasticity. Graph shows cellular elasticity, indicated by Young’s modulus (kPa). Each dot represents single cells from three biological replicates. Unpaired Student’s t test. (l and m) Kymographs and mobile fraction analysis derived from FRAP assays used to measure DSP stability at cell–cell contact sites (N = 3). Scale bar on the y-axis = 1 µm. Each dot represents one biological replicate. Unpaired Student’s t test.
DPM1 modulates DSP localization and cytoskeletal organization. (a) Immunostaining of DSP and DSG2 in HaCaT keratinocytes. Merge indicates overlap between DSP, DSG2, and nuclei stained with DAPI. Scale bar: 10 µm distance. (b) Quantification of the number of DSP puncta over the respective length of cell membrane (µm) from individual cells (represented by individual dots), N = 3. Unpaired Student’s t test. (c) Immunostaining of DSP and DSG2 in primary human keratinocytes. Scale bar: 10 µm distance. Panel shows representatives of three biological replicates. (d) Immunostaining of DSP in 3D-RHE of control (sgNT1) and sgDPM1 conditions. White dashed line indicates insert membrane. Magenta dashed rectangles mark regions magnified on the right (zoomed 2× of original image). Representative of three biological replicates. Scale bar: 10 µm distance. (e–g) Keratin staining depicted by pan-cytokeratin in control and DPM1 KD HaCaT keratinocytes. Analysis done by quantifying the keratin staining intensity (A.U.) across cell borders spanning a distance of 10 µm. Peak width was calculated between the two highest points from the distribution plot profile graph. Scale bar: 10 µm distance. 30 individual cells were quantified from three independent biological replicates. Unpaired Student’s t test. (h–j) F-actin stained by phalloidin in control and DPM1 KD HaCaT keratinocytes. Analysis done by quantifying the F-actin staining intensity (A.U.) across cell borders spanning a distance of 10 µm. The width of the peaks was calculated between the baseline values from distribution plot profile graphs. Scale bar: 10 µm distance. 30 individual cells were analyzed from three independent biological replicates. Unpaired Student’s t test. (k) Schematic of the AFM setup to measure cellular elasticity. Graph shows cellular elasticity, indicated by Young’s modulus (kPa). Each dot represents single cells from three biological replicates. Unpaired Student’s t test. (l and m) Kymographs and mobile fraction analysis derived from FRAP assays used to measure DSP stability at cell–cell contact sites (N = 3). Scale bar on the y-axis = 1 µm. Each dot represents one biological replicate. Unpaired Student’s t test.
Proteome analysis suggests SERPINB5 as an interacting partner of DSP in keratinocytes
As the DPM complex mediates essential steps of glycosylation, we tested whether loss of DPM1 results in altered glycosylation patterns of DSG2 or DSP. PNGase F–based mobility shift assay showed that DSG2 migrates at a lower molecular weight upon treatment with PNGase F (which cleaves N-linked oligosaccharides in mammals), indicating that DSG2 is glycosylated. However, we did not observe differences in the migration pattern between sgNT1 and sgDPM1 cells. DSP showed no mobility shift upon PNGase F treatment (Fig. S2 a). This indicates that the altered localization of these molecules is independent of N-linked glycosylation. To gain more mechanistic insights into the regulatory roles of DPM1 on desmosomes, we used a global proteomics approach to identify differentially regulated proteins in DPM1 KD HaCaT keratinocytes. Here, various sets of proteins exhibited differential expression, indicating that loss of DPM1 has a profound impact on the proteome of HaCaT keratinocytes (Fig. S2, b and c). Glycoproteins, proteases, and several proteins of the SERPIN group were significantly downregulated in DPM1 KD conditions, while various keratins, RAB GTPase, and some Sec complex proteins were upregulated (Fig. 3 a). STRING (Search Tool for Retrieval of Interacting Genes/proteins)-based pathway analysis of differentially expressed proteins showed significant upregulation of pathways correlating with cornification, actin filament assembly, cytoskeletal reorganization, and protein folding upon loss of DPM1. Specifically, the changes in cornification and cytoskeletal organization pathways are in agreement with the structural alterations observed upon loss of DPM1. Vice versa, many metabolic pathways such as nitrogen metabolic process, carbohydrate-derived metabolic process, glycosyl compound metabolic pathway, catabolic processes, and purine ribonucleoside were downregulated upon loss of DPM1 (Fig. 3 b). In addition to the global changes observed, we focused on the interacting partners of DSP since we saw major impairments in DSP surface localization and stability. DSP immunoprecipitation-based (Fig. S2 d) proteomic analysis outlined binding partners of DSP in sgNT1 or sgDPM1 cells (Fig. 3 c). Interestingly, SERPINB5 appeared as the most differential hit, with an interaction with DSP being present in control cells only. This was further supported by proximity ligation assays, which showed a close localization of endogenous DSP and SERPINB5 in sgNT1 but not in sgDPM1 cells. (Fig. 3, d and e). Moreover, overexpression of SERPINB5-GFP and GFP-based pull-down of the fusion protein confirmed interaction with DSP (Fig. 3 f). To gain further insights into the interaction of DSP with SERPINB5, we used DSP truncation constructs with different domains missing, as indicated (Fig. S2 e). Normalized to the amount of DSP construct that was pulled down, coimmunoprecipitation of SERPINB5 was strongly reduced upon deletion of tail, rod, and some parts of head domain (DSP-1945 and DSP 584), although some interaction was retained. (Fig. S2, f and g). In line with these observations, we detected a reduced expression of SERPINB5 in DPM1 KD HaCaT keratinocytes by western blot and immunofluorescence-based assays, while SERPINB5 transcript levels were unchanged (Fig. S2, h–l), suggesting posttranslational regulation (Fig. S2 l). EGFR (epidermal growth factor receptor)-dependent signaling mediates SERPINB5 phosphorylation and modulates its localization in the cell (Tamazato Longhi and Cella, 2012; Tamazato Longhi et al., 2016). Interestingly, the membrane localization of EGFR in sgDPM1 cells was perturbed and EGFR showed reduced activity, as indicated by phosphorylation at Y1068 (Fig. 3, g–i). Importantly, the interaction of SERPINB5 with DSP depended on EGFR activity, as inhibition using the EGFR-specific tyrosine kinase inhibitor erlotinib resulted in reduced levels of DSP coprecipitated with SERPINB5-GFP (Fig. 3, j and k).

SERPINB5 binds to DSP preferably at the rod and tail domains. (a) Western blot showing migration of DSG2 and DSP upon treatment with PNGaseF in sgNT1 versus sgDPM1 HaCaT keratinocytes. (b) Heat map showing differential protein expression profiles in sgNT1 and sgDPM1 HaCaT keratinocytes from three biological replicates. Color scale represents log2 fold change of intensity values. (c) Principal component (PC) analysis of the samples used in b. (d) Immunoprecipitation (I.P.) assay showing DSP pulldown in sgNT1 and sgDPM1 cells. IgG used as negative control. Panel shows representative of two biological replicates. (e) Schematic depicting the different truncated DSP constructs fused with GFP at the C-terminus. (f and g) Coimmunoprecipitation assay showing SERPINB5 binding to respective DSP truncated-GFP mutants expressed in HaCaT sgDSP cells. Quantification of SERPINB5 in I.P. samples normalized to the respective amounts of GFP immunoprecipitated. One-way ANOVA, Dunnett’s multiple comparison test, from three independent biological replicates. (h and i) Western blot and corresponding quantification of SERPINB5 expression in sgDPM1 HaCaT keratinocytes. N = 4, unpaired Student’s t test. (j and k) Images and quantification of SERPINB5 immunostainings in sgNT1 and sgDPM1 HaCaT keratinocytes. Scale bar: 10 µm distance. Panel represents three biological replicates. Unpaired Student’s t test (N = 3). (l) qPCR analysis of relative expression of SERPINB5 transcripts in sgNT1 and sgDPM1 cells. GAPDH used as housekeeping control and SERPINB5 transcript expression normalized to GAPDH. Each dot represents individual biological replicates, Unpaired Student’s t test. Source data are available for this figure: SourceData FS2.
SERPINB5 binds to DSP preferably at the rod and tail domains. (a) Western blot showing migration of DSG2 and DSP upon treatment with PNGaseF in sgNT1 versus sgDPM1 HaCaT keratinocytes. (b) Heat map showing differential protein expression profiles in sgNT1 and sgDPM1 HaCaT keratinocytes from three biological replicates. Color scale represents log2 fold change of intensity values. (c) Principal component (PC) analysis of the samples used in b. (d) Immunoprecipitation (I.P.) assay showing DSP pulldown in sgNT1 and sgDPM1 cells. IgG used as negative control. Panel shows representative of two biological replicates. (e) Schematic depicting the different truncated DSP constructs fused with GFP at the C-terminus. (f and g) Coimmunoprecipitation assay showing SERPINB5 binding to respective DSP truncated-GFP mutants expressed in HaCaT sgDSP cells. Quantification of SERPINB5 in I.P. samples normalized to the respective amounts of GFP immunoprecipitated. One-way ANOVA, Dunnett’s multiple comparison test, from three independent biological replicates. (h and i) Western blot and corresponding quantification of SERPINB5 expression in sgDPM1 HaCaT keratinocytes. N = 4, unpaired Student’s t test. (j and k) Images and quantification of SERPINB5 immunostainings in sgNT1 and sgDPM1 HaCaT keratinocytes. Scale bar: 10 µm distance. Panel represents three biological replicates. Unpaired Student’s t test (N = 3). (l) qPCR analysis of relative expression of SERPINB5 transcripts in sgNT1 and sgDPM1 cells. GAPDH used as housekeeping control and SERPINB5 transcript expression normalized to GAPDH. Each dot represents individual biological replicates, Unpaired Student’s t test. Source data are available for this figure: SourceData FS2.

Proteomic analysis identifies SERPINB5 as an interacting partner of DSP in keratinocytes. (a) Volcano plot showing differential protein expression in sgDPM1 with respect to control sgNT1 HaCaT keratinocytes from three biological replicates. −Log 10 P values above 1.3 (0.05) were considered significant (marked by gray line on plot). Orange dots indicate proteins which were significantly downregulated in DPM1 KD cells, whereas blue dots indicate proteins that were significantly upregulated in DPM1 KD cells. (b) STRING-based biological pathway analysis of significantly modulated proteins (orange: downregulated pathways and blue: upregulated pathways). X-axis denotes −log10 of FDR values, with a cutoff set to 0.05 for significance. (c) Heat map showing binding partners of DSP that were absent in either sgNT1 or sgDPM1 cells in at least two biological replicates (N = 3). (d and e) PLA assay showing SERPINB5 and DSP interaction as white dots. Nucleus stained with DAPI. Scale bar: 10 µm distance. Each dot represents individual biological replicates. Unpaired Student’s t test. (f) Co-immunoprecipitation assay showing DSP binding to SERPINB5-GFP expressed in HaCaT sgNT1 cells. Expression of GFP served as negative control. (g) Immunostaining of EGFR in sgNT1 and sgDPM1 HaCaT keratinocytes. White dashed boxes indicate areas zoomed in below. Scale bar: 10 µm distance. (h and i) Western blot and respective quantification of phosphorylated EGFR at Y1068 in sgNT1 and sgDPM1 HaCaT keratinocytes. Each dot represents individual biological replicates. Unpaired Student’s t test. (j and k) Co-immunoprecipitation assay showing DSP binding to SERPINB5-GFP expressed in HaCaT sgNT1 cells treated with DMSO and erlotinib, respectively. Quantitation of DSP co-immunoprecipitated signal intensity normalized to respective immunoprecipitated GFP intensity. Each dot represents individual biological replicates. Unpaired Student’s t test. Source data are available for this figure: SourceData F3.
Proteomic analysis identifies SERPINB5 as an interacting partner of DSP in keratinocytes. (a) Volcano plot showing differential protein expression in sgDPM1 with respect to control sgNT1 HaCaT keratinocytes from three biological replicates. −Log 10 P values above 1.3 (0.05) were considered significant (marked by gray line on plot). Orange dots indicate proteins which were significantly downregulated in DPM1 KD cells, whereas blue dots indicate proteins that were significantly upregulated in DPM1 KD cells. (b) STRING-based biological pathway analysis of significantly modulated proteins (orange: downregulated pathways and blue: upregulated pathways). X-axis denotes −log10 of FDR values, with a cutoff set to 0.05 for significance. (c) Heat map showing binding partners of DSP that were absent in either sgNT1 or sgDPM1 cells in at least two biological replicates (N = 3). (d and e) PLA assay showing SERPINB5 and DSP interaction as white dots. Nucleus stained with DAPI. Scale bar: 10 µm distance. Each dot represents individual biological replicates. Unpaired Student’s t test. (f) Co-immunoprecipitation assay showing DSP binding to SERPINB5-GFP expressed in HaCaT sgNT1 cells. Expression of GFP served as negative control. (g) Immunostaining of EGFR in sgNT1 and sgDPM1 HaCaT keratinocytes. White dashed boxes indicate areas zoomed in below. Scale bar: 10 µm distance. (h and i) Western blot and respective quantification of phosphorylated EGFR at Y1068 in sgNT1 and sgDPM1 HaCaT keratinocytes. Each dot represents individual biological replicates. Unpaired Student’s t test. (j and k) Co-immunoprecipitation assay showing DSP binding to SERPINB5-GFP expressed in HaCaT sgNT1 cells treated with DMSO and erlotinib, respectively. Quantitation of DSP co-immunoprecipitated signal intensity normalized to respective immunoprecipitated GFP intensity. Each dot represents individual biological replicates. Unpaired Student’s t test. Source data are available for this figure: SourceData F3.
These data identify SERPINB5 as an interaction partner of DSP dependent on the presence of DPM1 and EGFR signaling.
SERPINB5 overexpression rescues cell–cell adhesion, DSP localization, and cytoskeletal distribution
To characterize the functional effect of SERPINs on cell–cell adhesion, we expressed SERPINB5, SERPINB4, and the functionally related SERPINB3 in DPM1 KD HaCaT keratinocytes (Fig. S3 a). In adhesion assays, both SERPINB3 and SERPINB5 significantly rescued the loss of cell–cell adhesion in response to DPM1 loss, while SERPINB4 showed no protective effect (Fig. 4, a and b). In line with these results, we confirmed that expression of SERPINB5 also rescued loss of cell cohesion induced by sgDPM1 in primary human keratinocytes (Fig. S3, b and c). Thus, overexpression of SERBINB5 rescues the adhesive defect induced by DPM1 loss. In line with this observation, SERPINB5 overexpression in DPM1 KD background led to rescue of DSP localization at cell junctions, indicating enhanced number of desmosomes. SERPINB4, which showed no protective effect with regard to intercellular adhesion, did not ameliorate DSP membrane localization (Fig. 4, c and d). Similarly, SERPINB5 overexpression in primary human keratinocytes under DPM1 KD condition rescued DSP localization (Fig. S3 d). Consistent with these effects, SERPINB5 overexpression in DPM1 KD cells led to keratin organization similar to controls (Fig. 4, e–g), a more regular distribution of the cortical actin belt underneath the plasma membrane (Fig. 4, h–j) and more stable DSP localization at the membrane as indicated by FRAP (Fig. 4, k and l). These changes upon SERPINB5 rescue were also reflected in the mechanical properties of the cells, where SERPINB5 overexpression in HaCaT keratinocytes resulted in an increase in cell elasticity (Fig. 4 m). To address whether SERPINB5 modulates DSP without the background of DPM1 loss, we generated sgSERPINB5 cell lines from HaCaT background (Fig. S3 e). Adhesion assays revealed significant loss of cell–cell adhesion in sgSERPINB5 cells (Fig. 4, n and o), and loss of SERPINB5 led to significantly reduced DSP localization at the cell membrane (Fig. 4, p and q). Thus, SERPINB5 modulates DSP membrane localization, even though the total protein levels of DSP remained unaltered (Fig. S3, f and g). To understand the interdependency of SERPINB5 and DPM1, we checked DPM1 expression in SERPINB5 KD cells, where DPM1 levels were unaltered, suggesting DPM1 acts upstream of SERPINB5 (Fig. S3, h and i).

SERPINB5 rescues defects induced by DPM1 loss in primary keratinocytes. (a) Western blot showing expression of the respective SERPIN-GFP constructs in sgDPM1 HaCaT keratinocytes. GFP used as control. GAPDH used as loading control. Panel shows representative of three biological replicates. (b and c) Dispase-based dissociation assays to semiquantitatively assess cell–cell adhesion in primary human keratinocytes overexpressing SERPINB5-GFP in sgDPM1 background (N = 4). One-way ANOVA, Dunnett’s multiple comparison test used for statistics. (d) Images of DSP immunostainings in sgNT1-GFP, sgDPM1-GFP, and sgDPM1-SERPINB5 GFP in primary human keratinocytes. Scale bar: 10 µm distance. Panel represents three biological replicates. (e) Western blot showing SERPINB5 levels in sgNT1 and sgSERPINB5 HaCaT keratinocytes. Image represents three biological replicates. (f and g) Western blot images and quantifications of desmosomal proteins from sgNT1 and sgDPM1 HaCaT keratinocytes expressing the indicated SERPIN-GFP constructs. GAPDH used as internal loading control (N = 3). (h and i) Western blot images and quantifications of DPM1 from sgNT1 and sgSERPINB5 HaCaT keratinocytes. GAPDH used as internal loading control (N = 3). Unpaired Student’s t test. Source data are available for this figure: SourceData FS3.
SERPINB5 rescues defects induced by DPM1 loss in primary keratinocytes. (a) Western blot showing expression of the respective SERPIN-GFP constructs in sgDPM1 HaCaT keratinocytes. GFP used as control. GAPDH used as loading control. Panel shows representative of three biological replicates. (b and c) Dispase-based dissociation assays to semiquantitatively assess cell–cell adhesion in primary human keratinocytes overexpressing SERPINB5-GFP in sgDPM1 background (N = 4). One-way ANOVA, Dunnett’s multiple comparison test used for statistics. (d) Images of DSP immunostainings in sgNT1-GFP, sgDPM1-GFP, and sgDPM1-SERPINB5 GFP in primary human keratinocytes. Scale bar: 10 µm distance. Panel represents three biological replicates. (e) Western blot showing SERPINB5 levels in sgNT1 and sgSERPINB5 HaCaT keratinocytes. Image represents three biological replicates. (f and g) Western blot images and quantifications of desmosomal proteins from sgNT1 and sgDPM1 HaCaT keratinocytes expressing the indicated SERPIN-GFP constructs. GAPDH used as internal loading control (N = 3). (h and i) Western blot images and quantifications of DPM1 from sgNT1 and sgSERPINB5 HaCaT keratinocytes. GAPDH used as internal loading control (N = 3). Unpaired Student’s t test. Source data are available for this figure: SourceData FS3.

SERPINB5 rescues cell–cell adhesion and cytoskeletal rearrangements caused by DPM1 loss. (a and b) Dispase-based dissociation assays with sgDPM1 HaCaT keratinocytes overexpressing SERPINB3, SERPINB4, and SERPINB5 (N = 3). Each dot represents one biological replicate. One-way ANOVA, Dunnett’s multiple comparison test. (c) Images of DSP immunostainings in sgDPM1 and SERPIN overexpressing HaCaT keratinocytes. Panel shows representative of three biological replicates. Scale bar: 10 µm distance. (d) Quantification of the number of DSP puncta on the cell membrane normalized to the length of plasma membrane (µm). Each dot represents individual cells analyzed from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (e–g) Keratin staining depicted by pan-cytokeratin in sgNT1, DPM1 KD, and DPM1 KD overexpressing SERPINB5-GFP in HaCaT keratinocytes. Analysis done by measuring the keratin intensity across cell junctions (A.U.) over a distance of 10 µm. Peak width calculated between the two highest points from the distribution plot profile graph. Scale bar: 10 µm distance. 30 individual cells were quantified from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (h–j) F-actin staining by phalloidin in sgNT1, DPM1 KD, and DPM1 KD overexpressing SERPINB5-GFP in HaCaT keratinocytes. Scale bar: 10 µm distance. Analysis done by measuring the phalloidin intensity across cell junctions (A.U.) over a distance of 10 µm. The width of the peaks was calculated at baseline levels from distribution plot profile graphs. 30 individual cells were quantified from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (k and l) Kymographs and mobile fraction analysis derived from FRAP assays used to measure DSP stability at cell–cell contact sites (N = 3) in sgDPM1 and sgDPM1 SERPINB5 overexpressing cells. Scale bar on the y-axis = 1 µm. Each dot represents individual contact sites from three biological replicates. Unpaired Student’s t test. (m) Graph shows cellular elasticity, indicated by Young’s modulus (kPa). Each dot represents single cells from three biological replicates. Unpaired Student’s t test. (n and o) Dispase-based intercellular adhesion assays showing reduced cell–cell adhesion upon KD of SERPINB5 in HaCaT keratinocytes (N = 4). Unpaired Student’s t test. (p and q) Images and analysis of DSP at the cell membrane upon loss of SERPINB5 (N = 3). Each dot represents single cells from three biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test.
SERPINB5 rescues cell–cell adhesion and cytoskeletal rearrangements caused by DPM1 loss. (a and b) Dispase-based dissociation assays with sgDPM1 HaCaT keratinocytes overexpressing SERPINB3, SERPINB4, and SERPINB5 (N = 3). Each dot represents one biological replicate. One-way ANOVA, Dunnett’s multiple comparison test. (c) Images of DSP immunostainings in sgDPM1 and SERPIN overexpressing HaCaT keratinocytes. Panel shows representative of three biological replicates. Scale bar: 10 µm distance. (d) Quantification of the number of DSP puncta on the cell membrane normalized to the length of plasma membrane (µm). Each dot represents individual cells analyzed from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (e–g) Keratin staining depicted by pan-cytokeratin in sgNT1, DPM1 KD, and DPM1 KD overexpressing SERPINB5-GFP in HaCaT keratinocytes. Analysis done by measuring the keratin intensity across cell junctions (A.U.) over a distance of 10 µm. Peak width calculated between the two highest points from the distribution plot profile graph. Scale bar: 10 µm distance. 30 individual cells were quantified from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (h–j) F-actin staining by phalloidin in sgNT1, DPM1 KD, and DPM1 KD overexpressing SERPINB5-GFP in HaCaT keratinocytes. Scale bar: 10 µm distance. Analysis done by measuring the phalloidin intensity across cell junctions (A.U.) over a distance of 10 µm. The width of the peaks was calculated at baseline levels from distribution plot profile graphs. 30 individual cells were quantified from three independent biological replicates. One-way ANOVA, Dunnett’s multiple comparison test. (k and l) Kymographs and mobile fraction analysis derived from FRAP assays used to measure DSP stability at cell–cell contact sites (N = 3) in sgDPM1 and sgDPM1 SERPINB5 overexpressing cells. Scale bar on the y-axis = 1 µm. Each dot represents individual contact sites from three biological replicates. Unpaired Student’s t test. (m) Graph shows cellular elasticity, indicated by Young’s modulus (kPa). Each dot represents single cells from three biological replicates. Unpaired Student’s t test. (n and o) Dispase-based intercellular adhesion assays showing reduced cell–cell adhesion upon KD of SERPINB5 in HaCaT keratinocytes (N = 4). Unpaired Student’s t test. (p and q) Images and analysis of DSP at the cell membrane upon loss of SERPINB5 (N = 3). Each dot represents single cells from three biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test.
SERPINB5 prevents DPM1-induced differentiation defects in 3D-RHEs
Given the role of SERPINB5 in positively regulating cell–cell adhesion and DSP membrane localization in HaCaT and primary human keratinocytes, we asked if this would be reflected by changes in differentiation. Primary human keratinocytes transduced with sgDPM1 and either GFP or SERPINB5-GFP (Fig. 5 a) were allowed to differentiate to 3D-RHEs for 12 days. Similar to Fig. 1 f, DPM1 KD 3D-RHEs expressing GFP showed a thickened cornified layer with intercellular gaps, indicating impairment of cell–cell adhesion. Upon SERPINB5 overexpression, the cornified layer was significantly thinner and comparable with the control conditions (Fig. 5, b and c), although the thickness of the viable layers remained unchanged, resulting in a reduced total thickness of the epidermis (Fig. 5, d and e). In line with increased intercellular adhesion in response to SERPINB5 expression, the occurrence of intercellular gaps was also significantly diminished in SERPINB5-GFP expressing 3D-RHE (Fig. 5 f), while membrane localization of DSP at the cell surface was increased (Fig. 5, g and h). Interestingly, KD of DSP alone in 3D-RHEs, similar to DPM1 silencing, led to reduced thickness of non-corneal layers (Fig. 5, k–m), massive split formation (Fig. 5 n), and a disrupted staining pattern of the differentiation markers CK10 and filaggrin (Fig. 5 j). These data suggest that DPM1 together with SERPINB5 and at least in part by modulating DSP localization is required for epidermal differentiation.

SERPINB5 rescues defects in epidermal differentiation in response to DPM1 loss. (a) Western blot showing SERPINB5-GFP expression in primary human keratinocytes that lack DPM1. GFP served as control. GAPDH used as internal loading control. (b) H&E staining of 3D-RHE from sgDPM1 expressing GFP control or SERPINB5-GFP 12 days after airlift. Immunostainings of CK10 and filaggrin were used as differentiation markers. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. Insets zoomed 2× of original image. White dashed line indicates insert membrane. Panel shows representatives from three biological replicates. (c–e) Quantification of corneal layer thickness, total epidermal thickness, and viable (non-corneal) epidermal layer thickness from three independent biological replicates. Student’s t test, unpaired. (f) Violin plot showing quantification of intercellular spaces within the epidermis in sgDPM1 GFP and sgDPM1 SERPINB5 GFP 3D-RHE. The area for defining intercellular spaces was set to be >50 µm2 to exclude shrinking artifacts. A minimum of 15 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test. (g and h) Immunostaining and analysis of DSP intensity of sgDPM1 GFP and sgDPM1 SERPINB5-GFP 3D-RHEs. Panel shows representative of four biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test used to determine statistical significance (N = 4). (i and j) H&E staining of sgDSP 3D-RHEs 12 days after airlift. DSP staining shows depletion of DSP in sgDSP 3D-RHE. Immunostainings for CK10 and filaggrin used as differentiation markers. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. White dashed line indicates insert membrane. Panel shows representatives from three biological replicates. (k–m) Quantification of total epidermal thickness, viable (non-corneal) epidermal layer thickness, and corneal layer thickness from three independent biological replicates. Student’s t test, unpaired. (n) Violin plot showing quantification of intercellular spaces within the epidermis in sgNT1 and sgDSP 3D-RHE. A minimum of 15 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test. Source data are available for this figure: SourceData F5.
SERPINB5 rescues defects in epidermal differentiation in response to DPM1 loss. (a) Western blot showing SERPINB5-GFP expression in primary human keratinocytes that lack DPM1. GFP served as control. GAPDH used as internal loading control. (b) H&E staining of 3D-RHE from sgDPM1 expressing GFP control or SERPINB5-GFP 12 days after airlift. Immunostainings of CK10 and filaggrin were used as differentiation markers. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. Insets zoomed 2× of original image. White dashed line indicates insert membrane. Panel shows representatives from three biological replicates. (c–e) Quantification of corneal layer thickness, total epidermal thickness, and viable (non-corneal) epidermal layer thickness from three independent biological replicates. Student’s t test, unpaired. (f) Violin plot showing quantification of intercellular spaces within the epidermis in sgDPM1 GFP and sgDPM1 SERPINB5 GFP 3D-RHE. The area for defining intercellular spaces was set to be >50 µm2 to exclude shrinking artifacts. A minimum of 15 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test. (g and h) Immunostaining and analysis of DSP intensity of sgDPM1 GFP and sgDPM1 SERPINB5-GFP 3D-RHEs. Panel shows representative of four biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test used to determine statistical significance (N = 4). (i and j) H&E staining of sgDSP 3D-RHEs 12 days after airlift. DSP staining shows depletion of DSP in sgDSP 3D-RHE. Immunostainings for CK10 and filaggrin used as differentiation markers. Scale bar: 50 µm distance on H&E images and 10 µm on immunostaining images. White dashed line indicates insert membrane. Panel shows representatives from three biological replicates. (k–m) Quantification of total epidermal thickness, viable (non-corneal) epidermal layer thickness, and corneal layer thickness from three independent biological replicates. Student’s t test, unpaired. (n) Violin plot showing quantification of intercellular spaces within the epidermis in sgNT1 and sgDSP 3D-RHE. A minimum of 15 individual fields of view were used for analysis from three independent biological replicates. Unpaired Student’s t test. Source data are available for this figure: SourceData F5.
SERPINB5 and DPM1 modulate DSP localization and cell–cell adhesion through S176 phosphorylation of DSP
It is established that the localization and cytoskeletal anchorage of DSP are regulated by phosphorylation (Godsel et al., 2005; Albrecht et al., 2015; Rietscher et al., 2022). Given the role of DPM1 for DSP localization and keratin organization, we asked whether these changes are regulated by differential DSP phosphorylation. Phospho-proteomics-based analysis of DSP revealed in total 18 sites spanning all domains of DSP to be phosphorylated in sgNT1 conditions (Fig. 6 a). Of these sites, only S176 and S2024 showed a significant and consistent change of phosphorylation in sgDPM1 cells with comparable total DSP levels (Fig. 6 a and Fig. S4 a). Interestingly, only the increase in S176 phosphorylation in sgDPM1 cells was prevented by SERPINB5 overexpression (Fig. 6 b), indicating SERPINB5-mediated modulation of this site. To elucidate the relevance of DSP phosphorylation at S176, we generated a phospho-deficient mutant (DSP-S176A) by exchange of serine to alanine (Fig. 6 c and Fig. S4 b). Interestingly, introduction of DSP-S176A in DPM1 KD cells led to significantly enhanced localization of DSP at the cell surface (Fig. 6, d and e), and the overall size of individual DSP complexes was enlarged in DSP-S176A cells. FRAP showed a decrease in the mobile fraction of DSP S176A mutant, indicating a rescue in DSP stability at cell junctions upon S176A mutation of DSP in DPM1 KD cells (Fig. 6, f and g). In line with enhanced stability at the membrane, a trend toward increased amounts of PG, another desmosomal adapter protein, was detectable by co-precipitation of DSP-S176A-GFP (Fig. S4, c and d). Further, while reconstitution of DSP-GFP in sgDPM1 cells was sufficient to improve cell–cell adhesion, this effect was stronger under expression of DSP-S176A (Fig. 6, h and i). These results suggest that SERPINB5 suppresses DSP phosphorylation at S176, which promotes DSP localization and intercellular adhesion at the cell membrane. To gain further insights into the regulatory mechanism for differential phosphorylation of DSP through SERPINB5, we performed a kinase activity analysis from the phospho-proteomics dataset and predicted putative kinases specific to S176 of DSP using the GPS 6.0 tool (Xue et al., 2005). The latter suggested casein kinases (CK1,2), MAP3K, and PDK (Fig. S4 e), while the former showed GRK3 and PLK2 kinase to be upregulated in DPM1 KD cells, which was reverted upon SERPINB5 overexpression. Based on these data, we selected CK1, CK2, PLK2, CAMK2, and PDK1 for screening of DSP phosphorylation using inhibitors. Since S176 resides in the head domain of DSP, we applied the DSP-584 GFP construct. Phos-tag western blots showed reduced phosphorylated forms of DSP-584 upon treatment with PLK2 inhibitor (ON123) and CK1 inhibitor (D466), indicated by lower migrating bands in the gel (Fig. S4 g, white box). This suggests that at least PLK2 and CK1 contribute to the DSP phosphorylation in the head domain (Fig. S4 g).

DPM1 and SERPINB5 modulate phosphorylation of DSP at S176, which is essential for cell–cell adhesion (a) Graph showing differential phosphorylation sites of DSP in sgDPM1 HaCaT keratinocytes, presented as log2 fold change (FC) of respective values from sgNT1 control cells. Each dot represents one biological replicate. * indicates P < 0.05, Student’s paired t test. Red bars indicate significantly altered sites. Schematic of the representation of phospho-sites in DSP. (b) Graph showing S176 phosphorylation intensity in sgNT1, sgDPM1, and sgDPM1 SERPINB5-GFP cells. Each dot represents one biological replicate. One-way ANOVA, Dunnett’s multiple comparison test (N = 3). (c) Schematic of the point mutation of S176 to A176 in the head domain of DSP. (d and e) Images and analysis of DSP-GFP and DSP S176A-GFP expression at the cell membrane in sgDPM1 HaCaT keratinocytes. Insets zoomed 2× of original image. Each dot represents individual cells from three biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test (N = 3). (f and g) Kymographs and mobile fraction analysis derived from FRAP assays were used to measure DSP and DSP-S176A stability at cell–cell contact sites (N = 3). Scale bar on the y-axis = 1 µm. Each dot represents individual cell–cell contact from three biological replicates. Unpaired Student’s t test. (h and i) Dispase-based dissociation assays in sgDPM1 HaCaT keratinocytes upon reconstitution of DSP-GFP and DSP S176A GFP. Unpaired Student’s t test (N = 3). Each dot represents one biological replicate.
DPM1 and SERPINB5 modulate phosphorylation of DSP at S176, which is essential for cell–cell adhesion (a) Graph showing differential phosphorylation sites of DSP in sgDPM1 HaCaT keratinocytes, presented as log2 fold change (FC) of respective values from sgNT1 control cells. Each dot represents one biological replicate. * indicates P < 0.05, Student’s paired t test. Red bars indicate significantly altered sites. Schematic of the representation of phospho-sites in DSP. (b) Graph showing S176 phosphorylation intensity in sgNT1, sgDPM1, and sgDPM1 SERPINB5-GFP cells. Each dot represents one biological replicate. One-way ANOVA, Dunnett’s multiple comparison test (N = 3). (c) Schematic of the point mutation of S176 to A176 in the head domain of DSP. (d and e) Images and analysis of DSP-GFP and DSP S176A-GFP expression at the cell membrane in sgDPM1 HaCaT keratinocytes. Insets zoomed 2× of original image. Each dot represents individual cells from three biological replicates. Scale bar: 10 µm distance. Unpaired Student’s t test (N = 3). (f and g) Kymographs and mobile fraction analysis derived from FRAP assays were used to measure DSP and DSP-S176A stability at cell–cell contact sites (N = 3). Scale bar on the y-axis = 1 µm. Each dot represents individual cell–cell contact from three biological replicates. Unpaired Student’s t test. (h and i) Dispase-based dissociation assays in sgDPM1 HaCaT keratinocytes upon reconstitution of DSP-GFP and DSP S176A GFP. Unpaired Student’s t test (N = 3). Each dot represents one biological replicate.

PLK2 and CK-1 are candidate kinases to modulate DSP head domain phosphorylation. (a) Graph showing endogenous total DSP protein quantification from mass spectrometry data in sgNT1, sgDPM1, and sgDPM1 SERPINB5 overexpressing cell lines. Y-axis denoted Log2 fold change of intensity values. Dots represent individual biological replicates. (b) Sequencing data showing point mutation of serine at 176 position to alanine of Dsp (HUMDP1 = hDsp1 WT, as template). (c and d) Co-immunoprecipitation assay showing DSP-GFP and DSP S176A-GFP binding to PG. Quantification shows immunoprecipitated PG normalized to the respective amount of DSP pulled down. Each dot represents individual biological replicates, Unpaired Student’s t test. (e) Kinase prediction of DSP phosphorylation site on serine at position 176 (indicated by peptide sequence in the table) by online GPS 6.0 biocuckoo tool. (f) Kinase activity analysis from phospho-proteomics datasets in sgDPM1-sgNT1 and sgDPM1 SERPINB5-sgDPM1 comparison. X-axis denotes dominant enrichment scores and Y axis denotes −log10 of dominant P values. Red indicates upregulated activities in respective kinases and blue indicates downregulated activities in respective kinases. Black dots indicate non-significantly changed kinases. The data were analyzed as described in the publication (Johnson et al., 2023). (g) Phosphate-affinity western blot showing differential phosphorylation forms of DSP-584 GFP truncated protein, upon treatment with respective kinase inhibitors. White box shows lower migrating phosphorylated forms of DSP-584 upon treatment with ON12 (PLK2 inhibitor) and D446 (CK1 inhibitor). Inset marked with red box below shows a higher exposure for clarity. Source data are available for this figure: SourceData FS4.
PLK2 and CK-1 are candidate kinases to modulate DSP head domain phosphorylation. (a) Graph showing endogenous total DSP protein quantification from mass spectrometry data in sgNT1, sgDPM1, and sgDPM1 SERPINB5 overexpressing cell lines. Y-axis denoted Log2 fold change of intensity values. Dots represent individual biological replicates. (b) Sequencing data showing point mutation of serine at 176 position to alanine of Dsp (HUMDP1 = hDsp1 WT, as template). (c and d) Co-immunoprecipitation assay showing DSP-GFP and DSP S176A-GFP binding to PG. Quantification shows immunoprecipitated PG normalized to the respective amount of DSP pulled down. Each dot represents individual biological replicates, Unpaired Student’s t test. (e) Kinase prediction of DSP phosphorylation site on serine at position 176 (indicated by peptide sequence in the table) by online GPS 6.0 biocuckoo tool. (f) Kinase activity analysis from phospho-proteomics datasets in sgDPM1-sgNT1 and sgDPM1 SERPINB5-sgDPM1 comparison. X-axis denotes dominant enrichment scores and Y axis denotes −log10 of dominant P values. Red indicates upregulated activities in respective kinases and blue indicates downregulated activities in respective kinases. Black dots indicate non-significantly changed kinases. The data were analyzed as described in the publication (Johnson et al., 2023). (g) Phosphate-affinity western blot showing differential phosphorylation forms of DSP-584 GFP truncated protein, upon treatment with respective kinase inhibitors. White box shows lower migrating phosphorylated forms of DSP-584 upon treatment with ON12 (PLK2 inhibitor) and D446 (CK1 inhibitor). Inset marked with red box below shows a higher exposure for clarity. Source data are available for this figure: SourceData FS4.
Discussion
DPM1 and SERPINB5 may serve as a link between differentiation and adhesion
The importance of O-linked and N-linked glycosylation in modulating cell–cell adhesion, desmosomes, epidermal differentiation, and wound healing has been established in the past (Ruoslahti, 1989; Ruoslahti and Yamaguchi, 1991). Since mannose monosaccharide forms the basis for most glycosylation pathways, we here used the CRISPR/Cas9-based gene editing system to target DPM1, which functions as a mannose donor for the glycosylation processes. CRISPR/Cas9-based gene editing of DPM1 in a 3D organotypic model of human epidermis showed impaired stratification and differentiation, with reduced thickness of the viable epidermal layers and abnormally increased corneal layer thickness. Defects in differentiation were accompanied by intercellular gap formation in the epidermis, indicating loss of cell–cell adhesion. These results of DPM1 loss were prevented by overexpression of SERPINB5, suggesting that restoration of adhesion contributed to the rescue of differentiation defects. Indeed, despite the primary function in providing intercellular adhesion, there is evidence that desmosomal adhesion, epidermal differentiation, and signaling are connected. As examples, loss of DSC1 and DSG4 led to differentiation defects in interfollicular epidermis (Chidgey et al., 2001; Kljuic et al., 2003). Ectopic expression of DSG3 in murine epidermis induces cornification defects and trans-epidermal water loss (Elias et al., 2001), and DSG3 expression in suprabasal layers results in abnormal differentiation and hyperproliferation (Merritt et al., 2002). Further, the use of ectodomain-deleted DSG3 and inhibiting peptides led to impaired epidermal differentiation and epithelial morphogenesis (Allen et al., 1996; Runswick et al., 2001). Importantly, it was also shown that DSG1 regulates epidermal differentiation, which was mechanistically dependent on EGFR and ERK signaling (Getsios et al., 2009). It is tempting to speculate that altered EGFR signaling affects epidermal differentiation also on the level of modulating DSP/SERPINB5 interactions, as indicated by our results. However, a detailed analysis of the effect of EGFR in this context needs further evaluation as it was shown that EGFR inhibition can promote intercellular adhesion in cardiomyocytes (Shoykhet et al., 2023) and in the context of pemphigus (Walter et al., 2019; Sayar et al., 2014).
These data all support a role of desmosomal adhesion in contributing to correct differentiation of the epidermis. Of course, adhesion-independent regulation of differentiation by desmosomal molecules is plausible, and a precise understanding needs further investigation. However, the data are consistent with a contribution of DPM1, via SERPINB5, for epidermal homeostasis at least in part on the level of regulating intercellular adhesion. Although DPM1 is a crucial constituent of glycosylation pathways and a global reduction of diverse mannose structures was evident in the sgDPM1 cells, it is so far unclear whether the changes we observed are mediated by altered glycosylation of desmosomal molecules. Our data does not support a direct role of impaired glycosylation for membrane trafficking of adhesion molecules. However, indirect contributions, e.g., by altered glycosylation of other molecules which may be required for the stability and localization of desmosomal molecules, are likely. Further analyses will have to clarify the contribution of altered glycosylation in more detail.
Mechanisms of DPM1-mediated modulation of cell–cell adhesion and differentiation
We found perturbed keratin and cortical actin organizations in DPM1 KD cells indicating that DPM1 contributes to the complex interplay between the different components of the cytoskeleton and adhesion molecules. It is unclear in this context whether reduced DSP at the cell surface leads to cytoskeletal remodeling or vice versa unstable DSP at cell junctions is a consequence of cytoskeletal impairments. A DSP mutant with higher binding affinity to keratin filaments strengthens intercellular adhesion (Dehner et al., 2014), and targeting DSG3 by autoantibodies from pemphigus patients induces uncoupling of keratins from the desmosome (Spindler et al., 2018). Vice versa, it has been demonstrated that loss of keratins from keratinocytes confers reduced binding forces and cell–cell junction mobility of DSG3 (Vielmuth et al., 2018). Keratinocytes deficient for all keratins showed increased PKC-mediated phosphorylation of DSP, leading to destabilization of desmosomes (Kröger et al., 2013). The actin cytoskeleton, in contrast, appears to exert more indirect effects on desmosomal adhesion. It has been demonstrated that DSP assembly into desmosomes occurs in several stages with the last being actin dependent (Godsel et al., 2005). Further, the actin-binding protein adducin and the cortical actin network are required for DSG3 membrane incorporation and cell–cell adhesion (Rötzer et al., 2014; Hiermaier et al., 2021). However, it has also been shown that desmosomal proteins in turn regulate actin organization in the cells (Godsel et al., 2010; Todorović et al., 2010). Together, this profound interdependency may explain the affection of both adhesion and cytoskeletal organization by DPM1 loss. Proteomic analysis of DPM1 KD keratinocytes revealed that cornification and cytoskeleton reorganization pathways were significantly upregulated, which is in line with the observed results discussed above.
SERPINB5 mediates DSP localization and intercellular adhesion
In an attempt to dissect the mechanisms underlying altered cell–cell adhesion in keratinocytes, we identified SERPINB5 as an interaction partner of DSP. Further analysis suggested the interaction to be dependent on the DSP head domain, although some interaction still remained in the rod and tail domain deletion mutants, leaving the possibility of some non-specificity. Nevertheless, BioID screening of the DSP interactome has shown the existence of SERPINB5 within the desmosomal plaque, supporting our findings (Badu-Nkansah and Lechler, 2020). The SERPIN class of proteins are classical serine peptidase inhibitors, however, SERPINB5 (MASPIN) is an exception. SERPINB5 does not have peptidase inhibitor activity (Pemberton et al., 1995), and its detailed functions are largely unclear. It was studied majorly in the context of breast cancer where it modulates cell adhesion, migration, and apoptosis of tumor cells (Maass et al., 2001; Jiang et al., 2002; Gao et al., 2004; Qin and Zhang, 2010). SERPINB5 localization was shown to be regulated by EGFR and through cell density (Tamazato Longhi et al., 2016). Indeed, the interaction of DSP and SERPINB5 was dependent on EGFR activity and the presence of DPM1.
We also show that deletion of SERPINB5 from HaCaT WT keratinocytes resulted in impaired DSP localization at the cell surface and loss of cell–cell adhesion, confirming that SERPINB5 is a regulator of DSP in keratinocytes. Moreover, overexpressing SERPINB5 rescued the effects of DPM1 loss. Corneal layer thickening in response to DPM1 loss was also ameliorated by ectopic expression of SERPINB5, although the thickness of the non-corneal layers was not affected. Nevertheless, this demonstrates an important contribution of SERPINB5 to the DPM1-dependent modulation of cornification. Along similar lines, SERPINB7 was recently shown to modulate epidermal differentiation, and its loss led to psoriasiform lesions in mice (Zheng et al., 2022). These roles of the SERPINB family members in epidermal differentiation may warrant a more detailed analysis also with regard to conditions such as wound healing.
DPM1 and SERPINB5 mediated alterations in DSP localization are partly modulated by phosphorylation of DSP
DSP localization at the cell membrane and association with keratin intermediate filament (IF) is known to be regulated by DSP phosphorylation. As an example, dephosphorylation of S2849 in the tail domain of DSP leads to enhanced DSP-IF associations and strengthened cell–cell adhesion (Albrecht et al., 2015). DSP phosphorylation at S2849 is regulated by a balance between GSK3 kinase and PP2A-B55a phosphatase, which modulates DSP localization and desmosome assembly in keratinocytes (Albrecht et al., 2015; Perl et al., 2022, Preprint). Our phospho-proteomics screen confirmed several known (Rietscher et al., 2022) and identified some new phosphorylation sites in DSP, of which the phosphorylation of S176 was significantly increased in DPM1 KD cells and reduced again upon SERPINB5 overexpression. Interestingly, dephosphorylation of an adjacent serine phosphorylation site, S165/S166, was recently shown to increase intercellular adhesion and keratin network integrity along with reduced phosphorylation of S2849 (Rietscher et al., 2022). As these sites were not significantly altered in DPM1 KD cells, it appears that, specifically, S176 modulation is dependent on DPM1. As our data demonstrate that SERPINB5 directly interacts with DSP, it is possible that this interaction protects phosphorylation at S176. Initial approaches to identify the kinases responsible for S176 phosphorylation suggested CK1 and PLK2 as potential candidates. A full understanding of the mechanisms mediating this phosphorylation may add valuable insights into the understanding of the role of DSP phosphorylation for cell–cell adhesion and epidermal differentiation.
Taken together, we here showed that DPM1, at least in part by modulating SERPINB5–DSP interactions, contributes to intercellular adhesion and epidermal differentiation. These interactions were dependent on EGFR activity. In this context, we identified a novel phosphorylation site of DSP (S176), which is modulated by SERPINB5 and suppressed under conditions of strong intercellular adhesion and correct DSP membrane localization (Fig. 7).

Schematic showing DPM1 regulates DSP expression on the cell membrane through SERPINB5, which can directly interact with DSP in keratinocytes, and this interaction is dependent on EGFR activity. DPM1 loss has functional consequences on cell–cell adhesion, cytoskeletal organization, and epidermal differentiation, which can be partly restored by rescue of SERPINB5 expression. Further, DSP localization on the surface is negatively regulated by phosphorylation of DSP on the S176 position, which is significantly enhanced in DPM1 KD cells, and interestingly SERPINB5 rescues the phosphorylation of DSP at S176 in DPM1 KD background.
Schematic showing DPM1 regulates DSP expression on the cell membrane through SERPINB5, which can directly interact with DSP in keratinocytes, and this interaction is dependent on EGFR activity. DPM1 loss has functional consequences on cell–cell adhesion, cytoskeletal organization, and epidermal differentiation, which can be partly restored by rescue of SERPINB5 expression. Further, DSP localization on the surface is negatively regulated by phosphorylation of DSP on the S176 position, which is significantly enhanced in DPM1 KD cells, and interestingly SERPINB5 rescues the phosphorylation of DSP at S176 in DPM1 KD background.
Materials and methods
Cell culture and cloning
HaCaT keratinocytes (Boukamp et al., 1988) were cultivated at 5% CO2 and 37°C in Dulbecco’s Modified Eagle Medium (Sigma-Aldrich) containing 1.8 mM calcium and complemented with 10% fetal bovine serum (Merck), 50 U/ml penicillin, 50 µg/ml streptomycin (both AppliChem), and 4 mM L-glutamine (Sigma-Aldrich). Primary human keratinocytes were cultured in EpiLife medium (#11684842; Thermo Fisher Scientific) containing 0.06 mM CaCl2 (Gibco), supplemented with 1% Human Keratinocyte Growth Supplement (#10761364; Thermo Fisher Scientific) and 1% Pen/Strep. Erlotinib (#sc-202154; Santa Cruz Biotechnology) was added at a concentration of 1.5 µM for 16 h.
For cloning, oligos of sgNT1, sgDPM1, sgDPM2, sgDPM3, sgDSG2, sgSERPINB5, and sgDSP were synthesized by microsynth (primer sequences mentioned in Table S1 and gRNA in Table S2), phosphorylated using T4 PNK enzyme (#0201S; NEB), and annealed and ligated into Esp3I-digested (#R0734L; NEB) lentiCRISPR_v2 (#52961; Addgene) vector using T4 ligase (#M0202S; NEB). Ligated product was transformed in competent DH5α E. coli strain and plated on ampicillin-containing (100 μg/ml) agar plates. Colonies were amplified in 3 ml medium containing ampicillin (100 μg/ml). Plasmids were isolated via miniprep (#300287; Machery Nagel) and sequenced with U6_forward primer (5′-GAGGGCCTATTTCCCATGATT-3′).
For cloning SERPINB3, SERPINB4, and SERPINB5 overexpression constructs, PCR of HaCaT cDNA was performed using Platinum SuperFi 2 DNA Polymerase (#16410771; Thermo Fisher Scientific) according to the manufacturer’s protocol and primers listed in Table S1. The annealing temperature for the primers was set to 56°C. The PCR product was purified with PCR and gel purification kit (#28506; Qiagen). The PCR product and the plasmid pLenti-C-mGFP (#PS100071; OriGene Technologies) were digested with Asc1 (#R0558L; Bioconcept) and Xho1 (#300366; NEB) for 3 h at 37°C. The digested product was loaded on a 1% agarose gel and purified from the gel with PCR and gel purification kit (#28506; Qiagen). The insert was ligated into the vector with a ratio of 3:1 overnight at 16°C using T4 Ligase (#300361; Bioconcept). Positive clones were sequenced with forward and reverse primers (for 5′-TAATACGACTCACTATAGGG-3′; rev 5′-CTTGATCTCCAGCTTGCCGT-3′ for SERPINB4; 5′-TCAGTGAAGCCAACACCAAGT-3′ for SERPINB3 and 5′-GAAAAGGAGCCACTGGGCAA-3′ for SERPINB5).
For generating DSP S176A mutant, a Q5 Site-Directed Mutagenesis kit (#E0554S; Bioconcept) was used and mutagenesis was performed as per standard kit instructions by the manufacturer. DSP-GFP (Wanuske et al., 2021) plasmid was used as a template. Annealing temperature was 63°C and extension time used was 7 min (primer sequence in Table S1). T7 forward primer (5′-TAATACGACTCACTATAGGG-3′) was used for sequencing.
Generation of lentiviral constructs and stable cell lines
Lentiviral particles were generated according to standard procedures. HEK293T cells (between passages 9 and 11) were transfected with lentiviral packaging vector psPAX2 (#12259; Addgene), the envelope vector pMD2.G (#12260; Addgene), and the respective construct plasmid using TurboFect (Thermo Fisher Scientific). 48 h after transfection, virus-containing supernatant was collected and concentrated using Lenti-Concentrator (OriGene) for minimum 2 h at 4°C. Cells were transduced with the respective virus particles in an equal ratio using 5 µg/ml polybrene (Sigma-Aldrich) according to the manufacturer’s instructions. 24 h after transduction for HaCaT keratinocytes and 8 h later for primary human keratinocytes, medium was exchanged and puromycin selection was added. Cells were cultivated for at least 1 wk under selection before starting with the respective experiments. Expression of the respective construct was confirmed via western blot analysis.
Isolation of primary human keratinocytes
Foreskin tissue was obtained during routine circumcisions under informed consent according to the local ethical committee (EKNZ; date of approval: 11.06.2018, Project-ID: 2018-00963). Skin samples were washed three times in PBS containing 300 U/ml penicillin (#A1837; AppliChem), 300 U/ml streptomycin sulfate (#A1852; AppliChem), and 7.5 µg/ml amphotericin B (#A2942; Sigma-Aldrich). Excess tissue, blood vessels, and parts of the dermis were removed and skin was cut into pieces of 0.5 × 1 cm size. To separate dermis and epidermis, skin samples were digested at 4°C overnight in 5 mg/ml dispase II solution (#D4693; Sigma-Aldrich) in HBSS (#H8264; Sigma-Aldrich) containing 300 U/ml penicillin, 300 U/ml streptomycin sulfate, and 2.5 µg/ml amphotericin B. Epidermis was peeled off and washed once in PBS and digested in 0.25% trypsin and 1 mmol/l EDTA containing 100 U/ml penicillin and 100 U/ml streptomycin sulfate at 37°C for 20 min. Trypsin activity was stopped by diluting 1 + 1 with a 1 mg/ml solution of soy bean trypsin inhibitor (#10684033; Gibco) in PBS. Keratinocytes were isolated by scratching the epidermis fragments on the dish bottom and through a 70-µm cell strainer (#431751; Corning). The isolated normal human epidermal keratinocytes (NHEK) were then seeded at a density of ∼8 × 104 cells/cm2 in EpiLife medium containing 60 µmol/l CaCl2 (#MEPI500CA; Gibco) and 1% Human Keratinocyte Growth Supplement (#S0015; Gibco), 1% Pen/Strep, and 2.5 µg/ml amphotericin B. After 3 days, the medium was exchanged, and from there on amphotericin B was omitted. For experiments, 40,000 cells were seeded in a 24-well plate and grown until they reached confluency. Subsequently, differentiation was induced by adding 1.2 mmol/l CaCl2 for 24 h.
3D-RHE
3D-RHE generation was performed according to the protocol of CellnTec (CellnTec). In brief, NHEK cells were cultivated in CnT-PR medium (#CNT-PR CellnTec) to ∼50–70% confluency and transduced with respective constructs, as described above. Cells were selected in puromycin (Thermo Fisher Scientific) (2 µg/ml). The cells were then seeded (1.7 × 105 cells per insert) on 0.4 µm polycarbonate 24-well inserts (#10387523; Thermo Fisher Scientific), which were coated with 15 µg/cm2 rat-tail collagen I (#50201-IBIDI; Gräfelfing) in 0.02 M acetic acid at 37°C for 1 h. NHEKs were cultivated in CnT-PR medium. 24 h after seeding, the medium inside the insert and six wells was exchanged with CnT-PR-3D medium (#CnT-PR-3D; CellnTec). After 16-8 h, inserts were lifted to an air–liquid interface by aspirating the media inside the insert and adding CnT-PR-3D in the six wells up to the level of the insert membrane (1.3 ml); the CnT-PR-3D medium was exchanged every other day. The 3D organotypic cultures were harvested and analyzed 12 days after airlifting by fixing in 2% paraformaldehyde at 4°C for 4 h.
Western blot and phos-tag western blot
Confluent cell monolayers were lysed with SDS lysis buffer (25 mM HEPES, 2 mM EDTA, 25 mM NaF, 1% SDS, pH 7.6) supplemented with an equal volume of a protease inhibitor cocktail (cOmplete; Roche Diagnostics) by using a cell scraper. Lysates were sonicated and the total protein amount was determined with a BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The proteins were denatured by heating in Laemmli buffer for 10 min at 95°C. Membranes were blocked in odyssey blocking buffer (Li-Cor; Lincoln) for 1 h at room temperature. The following primary antibodies were diluted with odyssey blocking buffer in tris-buffered-saline containing 0.1% Tween20 (Thermo Fisher Scientific) and incubated overnight at 4°C, with rotation: mouse Dsc2 mAb (#60239-1-Ig; Proteintech), mouse Dsg2 mAb (clone 10G11, #BM5016; Acris), rabbit Dsg3 pAb (EAP3816; Elabscience), mouse Pkp1 mAb (clone 10B2, #sc-33636; Santa Cruz), mouse Pkp2 mAb (#651101; Progen), mouse PG mAb (clone PG5.1, #61005; Progen), mouse DSP mAb (#sc-390975; Santa Cruz), mouse E-Cad mAb (clone 36, #610181; BD Biosciences), rabbit DSC1 mAb (#ab150382; Abcam), mouse DSG1 (#651111; Progen), rabbit keratin 10 (#905403; BioLegend), mouse keratin 14 (ab7800; Abcam), rabbit DPM1 (#12403-2-AP; Proteintech), rabbit SERPINB5 (MASPIN #ab182785; Abcam), rabbit mGFP (#TA150122; Thermo Fisher Scientific), mouse GAPDH mAb (clone 0411, #sc-47724; Santa Cruz), and mouse α-tubulin (clone 10D8, #627901; BioLegend). Goat anti-mouse 800CW and goat anti-rabbit 680RD (#925-32210 and #925-68071; both Li-Cor) were used as secondary antibodies, incubated for 1 h at room temperature. Odyssey FC imaging system was used for imaging the blots and band density was quantified with Image Studio (both Li-Cor). Phosphate-trap SDS-PAGE was performed using Phos-tag (#300-93523v; Fujifilm WaKD Pure Chemical Industries), according to the manufacturer’s instructions. 50 μM of phos-tag acrylamide was used along with magnesium chloride in a 6% wt/vol polyacrylamide gel. A polyvinylidene difluoride membrane was used for transfer of proteins.
Quantitative real-time PCR (qRT-PCR)
Equal concentrations of RNA from all samples were reverse transcribed using SuperScript III Reverse Transcriptase (#300510; Invitrogen) and Oligo-dT-Primers (#300446; Thermo Fisher Scientific) according to the manufacturer's protocol. Power SYBR Green Master Mix (2x, #300427; Thermo Fisher Scientific) was used for qPCR, using 1:50 diluted cDNA and 0.5 μM primers (forward and reverse, Table S1). qRT-PCR was performed using StepOne Real-Time PCR System (#4376357; Applied Biosystems). GAPDH was used as housekeeping control.
Immunoprecipitation
Confluent cell monolayers were washed twice with ice-cold PBS and incubated with modified RIPA (radioimmunoprecipitation assay) buffer plus protease inhibitors (10 mM Na2HPO4, 150 mM NaCl, 1% Triton X-100, 0.25% SDS, 1% sodium deoxycholate, pH 7.3) for 30 min on ice. Cells were then scraped and homogenized on ice by passing 10 times through a 20 G and 25 G injection needle. Cell debris was removed by centrifugation at 7,000 g, 5 min, 4°C. Protein concentration was determined by BCA (Thermo Fisher Scientific) and equal amounts of protein were diluted in 1 ml of RIPA buffer. GFP trap magnetic agarose beads (gtma-20; ChromoTek) were washed three times with 1 ml RIPA buffer and 25 μl of beads were added to diluted lysates and incubated for 1.5 h at 4°C. Beads were washed 12 times with wash buffers and afterward mixed with Laemmli buffer and denaturized at 95°C for 10 min. The following samples from immunoprecipitation were subjected to western blotting as described above. Rabbit mGFP (#TA150122; Thermo Fisher Scientific) primary antibody was used to confirm immunoprecipitation and mouse DSP mAb (#sc-390975; Santa Cruz) antibody was used to detect DSP co-immunoprecipitation.
Dispase-based dissociation assay
HaCaT keratinocytes expressing various sgRNA and overexpression constructs or primary human keratinocytes were seeded in equal numbers in 24-well plates. After reaching confluency, cells were washed with prewarmed PBS and incubated with 250 μl dispase II (50 mg/10 ml in HBSS D4693; Sigma-Aldrich) for 20 min (HaCaTs) and 45 min for primary human keratinocytes at 37°C to detach the intact cell sheet from the well bottom. 150 μl HBSS was added to these wells and a constant mechanical shear stress was applied using an electrical pipette (Eppendorf) for 10 times each well. Wells were finally imaged with a binocular microscope (Olympus) and SLR camera (Canon). The number of fragments generated were a direct measure of cell cohesion, which is inversely proportional to adhesive strength.
Immunostaining
Cells were grown on 13-mm glass coverslips and fixed with either 2% PFA (Thermo Fisher Scientific) in PBS at room temperature or ice-cold methanol (Merck Millipore) for 10 min on ice. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 3% BSA and 1% normal goat serum in PBS for 1 h in a humidified chamber. The following primary antibodies were incubated overnight at 4°C: mouse Dsg2 mAb (clone 10G11, #BM5016; Acris), rabbit Dsg2 pAb (#610121; Progen), mouse Dsg3 mAb (clone 5G11, #326300; Invitrogen), rabbit DSP pAb (NW39), mouse DSP mAb (1G4) (both kind gifts from Kathleen Green, Northwestern University, Chicago, IL, USA), Phalloidin CruzFluor 488 (#sc-363791; Santa Cruz), and pan-cytokeratin (AE1/AE3) efluor 570 (#41-9003-80; eBioscience). Following primary antibody incubation, cells were washed three times with PBS and AlexaFluor (AF488, AF568) conjugated anti-rabbit or anti-mouse antibodies (#A-11008, A-11004; Thermo Fisher Scientific) were added and incubated for 1 h at room temperature. DAPI (Sigma-Aldrich) was added for 10 min to counterstain nuclei. Cells were washed three times with PBS and mounted with Prolong Diamond Antifade (Thermo Fisher Scientific). Image acquisitions were done using Stellaris 8 Falcon confocal microscope (Leica) with an HC PL APO CS2 63×/1.40 oil objective. Image analysis was done with ImageJ software for analyzing plot profiles and counting DSP puncta as described later in the data analysis section.
For staining of paraffin sections, tissues embedded in paraffin were cut into 5-µm-thick sections with an automated microtome (HM355S; Thermo Fisher Scientific). After deparaffinization, temperature-mediated antigen retrieval was performed in citrate buffer (10 mM citric acid monohydrate [20276.235; VWR], pH 6, 0.1% Triton X-100) for 20 min at 95°C. Tissue was permeabilized in 0.1% Triton X-100 in PBS for 5 min and blocked with 3% BSA/0.12% normal goat serum in PBS for 1 h. The following primary antibodies were incubated in PBS at 4°C overnight: mouse DSP (#61003; Progen), rabbit DSG2 RB5 (#610120; Progen Biotechnik), Filaggrin (#ab218395; Abcam), Keratin 10 (#905403; BioLegend), Ki67 B56 (#550609; BD Bioscience), rabbit SERPINB5 (MASPIN #ab182785; Abcam), and rabbit DPM1 (#12403-2-AP; Proteintech). Respective secondary goat anti-rabbit or goat anti-mouse antibodies coupled to Alexa Fluor 488, Alexa Fluor 568 (both Thermo Fisher Scientific), or cy5 (Dianova) were incubated for 1 h at room temperature and DAPI (Sigma-Aldrich) was added for 10 min to counterstain nuclei. Finally, samples were mounted with fluor mount Aqueous Mounting Medium (Sigma-Aldrich).
Histology
Fixed 3D-RHEs were removed from the cell culture insert using an 8-mm biopsy punch (#600213; Stiefel), cut into half, and embedded in histogel (#HG-4000-012; Epredia). After polymerization of the histogel, samples were paraffin-embedded using the TPC 15 Tissue Processor (Medite Medizintechnik): (1) 70% ethanol, 37°C, 45 min; (2) 80% ethanol, 37°C, 45 min; (3) 96% ethanol, 37°C, 30 min; (4) 96% ethanol, 37°C, 45 min; (5) 100% ethanol, 37°C, 30 min; (6) 100% ethanol, 37°C, 60 min; (7) 100% ethanol, 37°C, 60 min; (8) Xylene, 37°C, 30 min; (9) Xylene, 37°C, 45 min; 10) Xylene, 37°C, 60 min; (11) paraffin, 62°C, 45 min; (12) paraffin, 62°C, 60 min; and (13) paraffin, 62°C, 60 min. TES Valida embedding station (Medite Medizintechnik) was used to embed processed tissue into paraffin blocks and cut into 5-µm sections using an automated microtome (HM355S; Thermo Fisher Scientific). Haematoxylin and Eosin (H&E) staining was performed according to standard procedures. In brief, sections were stained with Mayer’s hemalum solution (#1.09249.1022; Sigma-Aldrich) for 5 min, washed, dehydrated in an increasing ethanol series, and stained with 0.5% (wt/vol) eosin solution for 5 min. After washing steps in ethanol and methyl salicylate, sections were mounted with DPX mounting media (#06522; Sigma-Aldrich).
AFM
A Nanowizard IV atomic force microscope (JPK Instruments) mounted on an inverted fluorescence microscope (IX83; Olympus) was used for cell stiffness measurements. Experiments were performed in culture medium at 37°C using Si3N4 AFM cantilevers (pyramid-shaped D-tip, MLCT cantilever, 4; Bruker) with a spring constant of 0.03 N/m. Spring constant was calibrated for each cantilever at 37°C applying the thermal noise method. Force-displacement curves were obtained in force spectroscopy mode using the following settings: relative setpoint 0.4 nN, z-length 5 μm, extend delay 0 s, pulling speed 2 μm/s, and recorded with the SPM (Scanning Probe Microscopy) control v.4 software 15 (JPK Instruments). Cells were seeded on glass coverslips. All measurements were performed on the cell center and conducted within 1 h. Force distance curves were analyzed using JPK SPM Data Processing software (version 6, JPK Instruments) and then fitted with the Hertz model for Young’s modulus calculation. The tip-half opening angle was 17.5°, the Poisson’s ratio was set as 0.5, and 300 nm of indentation depth was used.
FRAP
For FRAP measurements, HaCaT cells (sgNT1) and sgDPM1 were transduced with DSP-GFP (Wanuske et al., 2021) as described above and seeded in 8-well imaging chambers (IBIDI). After the formation of visible junctions, FRAP measurements were performed on a Stellaris 8 Falcon confocal microscope (Leica) with an HC PL APO CS2 63×/1.40 oil objective, at 37°C with 5% CO2 and constant humidity. The measurements were carried out and analyzed with the FRAP wizard software tool (Leica). Regions of interest were defined along cell–cell junctions containing a desmosome between two neighboring mGFP-positive cells. After five frames of recording the prebleach intensity, mGFP signal was bleached shortly for five frames, using the 488-nm laser line at 80% transmission on FRAP booster mode and the fluorescence recovery was recorded over 239 s with 100 frames for the initial 26 s and 45 frames for the remaining time. The fraction of mobile molecules was determined by the formula: Mobile fraction = Ie – Io/Ii – Io, where Ie is the intensity reached after recovery time, Io is the minimal intensity that was achieved right after bleaching, and Ii is the average prebleach intensity value.
Proteomics
Sample preparation for enrichment analysis
Samples were adjusted to the 5% of SDS, 100 mM TEAB (tetraethylammonium bromide), and 10 mM TCEP (Tris-(2 carboxyethly) phosphine), and subsequently reduced for 10 min at 95°C. Samples were then cooled down to room temperature and 0.5 μl of 1 M iodoacetamide was added to the samples. Cysteine residues were alkylated for 30 min at 25°C in the dark. Digestion and peptide purification were performed using S-trap technology (Protifi) according to the manufacturer’s instructions. In brief, samples were acidified by addition of 2.5 μl of 12% phosphoric acid (FA) (1:10) and then 165 μl of S-trap buffer (90% methanol, 100 mM TEAB, pH 7.1) was added to the samples (6:1). Samples were briefly vortexed and loaded onto S-trap micro spin-columns (Protifi) and centrifuged for 1 min at 4,000 g. Flow-through was discarded and spin-columns were then washed three times with 150 μl of S-trap buffer (each time samples were centrifuged for 1 min at 4,000 g and flow-through was removed). S-trap columns were then moved to the clean tubes and 20 μl of digestion buffer (50 mM TEAB, pH 8.0) and trypsin (at 1:25 enzyme to protein ratio) were added to the samples. Digestion was allowed to proceed for 1 h at 47°C. After, 40 μl of digestion buffer was added to the samples and the peptides were collected by centrifugation at 4,000 g for 1 min. To increase the recovery, S-trap columns were washed with 40 μl of 0.2% formic acid in water (400 g, 1 min) and 35 μl of 0.2% formic acid in 50% acetonitrile. Eluted peptides were dried under vacuum and stored at −20°C until further analysis.
Data acquisition for enrichment analysis
Dried peptides were resuspended 0.1% FA and 0.2 μg of peptides were subjected to LC–MS/MS (liquid chromatography mass spectrometry/mass spectrometry) analysis using a Q Exactive Plus Mass Spectrometer fitted with an EASY-nLC 1000 (both Thermo Fisher Scientific) and a custom-made column heater set to 60°C. Peptides were resolved using an RP-HPLC (Reversed Phase–High Performance Liquid Chromatography) column (75 μm × 30 cm) packed in-house with C18 resin (ReproSil-Pur C18–AQ, 1.9 μm resin; Dr. Maisch GmbH) at a flow rate of 0.2 ml/min-1. A linear gradient ranging from 5% buffer B to 45% buffer B over 60 min was used for peptide separation. Buffer A was 0.1% formic acid in water and buffer B was 80% acetonitrile, 0.1% formic acid in water. The mass spectrometer was operated in DDA (Data Dependent Acquisition) mode with a total cycle time of approximately 1 s. Each MS1 scan was followed by high-collision-dissociation (HCD) of the 20 most abundant precursor ions with dynamic exclusion set to 5 s. For MS1, 3e6 ions were accumulated in the Orbitrap over a maximum time of 25 ms and scanned at a resolution of 70,000 FWHM (full width at half maxima) (at 200 m/z). MS2 scans were acquired at a target setting of 1e5 ions, a maximum accumulation time of 110 ms, and a resolution of 35,000 FWHM (at 200 m/z). Singly charged ions, ions with charge state ≥6, and ions with unassigned charge state were excluded from triggering MS2 events. The normalized collision energy was set to 27%, the mass isolation window was set to 1.4 m/z, and one microscan was acquired for each spectrum.
Sample preparation for total proteomics
Cells were lysed in 50 μl of lysis buffer (1% sodium deoxycholate, 10 mM TCEP, 100 mM Tris, pH = 8.5) using 20 cycles of sonication (30 s on, 30 s off per cycle) on a Bioruptor (Dianode). Following sonication, proteins in the bacterial lysate were reduced by TCEP at 95°C for 10 min. Proteins were alkylated using 15 mM chloroacetamide at 37 °C for 30 min and further digested using sequencing-grade modified trypsin (1/50 wt/wt, ratio trypsin/protein; Promega) at 37°C for 12 h. After digestion, the samples were acidified using TFA (trifluoroacetic acid) (final 1%). Peptide desalting was performed using iST cartridges (PreOmics) following the manufacturer’s instructions. After drying the samples under vacuum, peptides were stored at −20°C and dissolved in 0.1% aqueous formic acid solution at a concentration of 0.5 mg/ml upon use.
Data acquisition for total proteomics
Dried peptides were resuspended 0.1% FA and 0.2 μg of peptides were subjected to LC–MS/MS analysis using an Orbitrap Fusion Lumos Mass Spectrometer fitted with an EASY-nLC 1200 (both Thermo Fisher Scientific) and a custom-made column heater set to 60°C. Peptides were resolved using a RP-HPLC column (75 μm × 36 cm) packed in-house with C18 resin (ReproSil-Pur C18–AQ, 1.9 μm resin; Dr. Maisch GmbH) at a flow rate of 0.2 μl/min-1. The following gradient was used for peptide separation: from 5% B to 12% B over 5 min to 35% B over 65 min to 50% B over 20 min to 95% B over 2 min followed by 18 min at 95% B. Buffer A was 0.1% formic acid in water and buffer B was 80% acetonitrile, 0.1% formic acid in water. The mass spectrometer was operated in DDA mode with a cycle time of 3 s between master scans. Each master scan was acquired in the Orbitrap at a resolution of 120,000 FWHM (at 200 m/z) and a scan range from 375 to 1,500 m/z followed by MS2 scans of the most intense precursors in the linear ion trap at “Rapid” scan rate with isolation width of the quadrupole set to 1.4 m/z. Maximum ion injection time was set to 50 ms (MS1) and 35 ms (MS2) with an automatic gain control (AGC) target set to 1e6 and 1e4, respectively. Only peptides with charge state 2–5 were included in the analysis. Monoisotopic precursor selection (MIPS) was set to Peptide, and the Intensity Threshold was set to 5e3. Peptides were fragmented by HCD with collision energy set to 35%, and one microscan was acquired for each spectrum. The dynamic exclusion duration was set to 30 s.
Phosphoproteomics
Cells were lysed in 2 M guanidium hydrochloride, 100 mM ammonium bicarbonate, 5 mM TCEP, phosphatase inhibitors (P5726&P0044; Sigma-Aldrich) by sonication (Bioruptor, 10 cycles, 30 s on/off; Diagenode). Proteins were subsequently reduced by 10 min incubation at 95°C and alkylated with 10 mM chloroacetamide for 30 min at 37°C. Samples were then diluted with 100 mM ammonium bicarbonate to a final guanidium hydrochloride concentration of 0.5 M. Proteins were digested by incubation with sequencing-grade modified trypsin (1/50, wt/wt; Promega) for 12 h at 37°C. After digestion, samples were acidified with 5% TFA and peptides were purified using C18 reverse-phase spin columns (Macrospin; Harvard Apparatus) according to the manufacturer’s instructions, dried under vacuum, and stored at −20°C until further use. Peptide samples were enriched for phosphorylated peptides using Fe (III)-IMAC cartridges on an Assay MAP Bravo platform. Phospho-enriched peptides were resuspended in 0.1% aqueous formic acid and subjected to LC–MS/MS analysis using an Orbitrap Fusion Lumos Mass Spectrometer fitted with an EASY-nLC 1200 (both Thermo Fisher Scientific) and a custom-made column heater set to 60°C. Peptides were resolved using a RP-HPLC column (75 μm × 36 cm) packed in-house with C18 resin (ReproSil-Pur C18–AQ, 1.9 μm resin; Dr. Maisch GmbH) at a flow rate of 0.2 μl min-1. The following gradient was used for peptide separation: from 5% B to 8% B over 5 min to 20% B over 45 min to 25% B over 15 min to 30% B over 10 min to 35% B over 7 min to 42% B over 5 min to 50% B over 3 min to 95% B over 2 min followed by 18 min at 95% B. Buffer A was 0.1% formic acid in water and buffer B was 80% acetonitrile, 0.1% formic acid in water. The mass spectrometer was operated in DDA mode with a cycle time of 3 s between master scans. Each master scan was acquired in the Orbitrap at a resolution of 120,000 FWHM (at 200 m/z) and a scan range from 375 to 16,00 m/z followed by MS2 scans of the most intense precursors in the Orbitrap at a resolution of 30,000 FWHM (at 200 m/z) with isolation width of the quadrupole set to 1.4 m/z. Maximum ion injection time was set to 50 ms (MS1) and 54 ms (MS2) with an AGC target set to 250% and “Standard,” respectively. Only peptides with charge state 2–5 were included in the analysis. MIPS was set to Peptide, and the Intensity Threshold was set to 2.5e4. Peptides were fragmented by HCD with collision energy set to 30%, and one microscan was acquired for each spectrum. The dynamic exclusion duration was 30 s.
Glycomics
10 million cells were resuspended in 300 μl of 7 M urea, 2 M thiourea, 40 mM Tris, 2% CHAPS, 10 mM DTT, and 1% protease inhibitor. The cell membranes were disrupted by 10 times 10 s sonication with 16 amplitudes and 1 min on ice in between, and subsequent shaking at 4°C overnight. 300 μl of 25 mM iodoacetamide was added for alkylation during 1 h in the dark. The peptides were generated using 0.1 mg of trypsin in 25 mM ABC (ammonium bicarbonate) buffer for 7 h. Following digestion, PNGase F was added at 37°C for overnight. PNGase F was further added for 6 h the next day. The glycan and peptide were separated by Sep-Pak C18 and PGC cartridge, Glycan from the PGC cartridge was washed with 2% ACN and eluted by 25% ACN. De-N-Glycopeptides were eluted by 20% isopropanol with 5% acetic acid and 40% isopropanol with 5% acetic acid and dried by SpeedVac. 1/20 samples were mixed with 1 μl of 10 mg/ml 2,4 Dihydroxybenzoic acid matrix and analyzed by Bruker RapiFlex MALDI-TOF-TOF 3/3. Permethylation of the N glycans was performed as described previously (Zhou et al., 2017).
Proximity ligation assay (PLA)
PLA was conducted as per manufacturer’s protocol (#DUO92008; Sigma-Aldrich). Shortly, cells were rinsed and fixed with ice-cold methanol at 4 °C for 10 min. Primary antibodies were added and incubated overnight at 4°C. The cells were then incubated with PLA secondary antibodies conjugated to DNA oligonucleotides for 60 min at 37°C, followed by a 30-min incubation at 37°C for ligation of nucleotides, and a 100-min incubation at 37°C for rolling circle polymerization.
Image processing and statistics
Figures were compiled with Photoshop CC and Illustrator CC (Adobe). Statistical analysis was performed using GraphPad Prism 8 with a two-tailed student’s t test for the comparison of two data sets and one-way or two-way ANOVA corrected by either Dunnett’s, Dunn’s, or Tukey’s test for more than two data sets. Statistical significance was determined at P <0.05. The data sets were first tested for normal distribution using Shapiro–Wilk normality test, and respective statistics were applied based on the distribution. Welch correction for unequal variances was applied where applicable. The error bars shown in all graphs were depicted as ± SD. A minimum of three biological replicates was used in each experiment.
Epidermal thickness analysis
Corneal layer and non-corneal layer (viable) thickness were normalized to total epidermal thickness to account for variances in overall thickness across biological replicates.
Ki67 analysis of 3D-RHE
Proliferation of 3D-RHE was analyzed on Ki67 staining from tissue sections, using QuPath software (Bankhead et al., 2017). In brief, three to five images per sample were analyzed in QuPath and nuclei were masked by detecting DAPI signal. The total numbers of nuclei per field were quantified. From these masks generated, Ki67 positive nuclei were counted and the ratio of Ki67 positive nuclei to total nuclei was calculated to determine the percentage of Ki67 positive cells.
ImageJ analysis
Image J was used to generate kymographs of FRAP recovery data for the DSP GFP signal over dynamic time scales before and after bleaching.
Keratin distribution and cortical actin distribution were analyzed by drawing a region of interest (ROI) rectangle of fixed size at the cell–cell contact sites (10 micron) and measuring the mean plot profile intensities along this area for individual cells. GraphPad PRISM software was used to create graphs and statistical analysis.
DSP dots were quantified by applying a threshold to the DSP staining on images, such that all the DSP puncta were accounted for. The membrane length for individual cells was measured by drawing ROI around the cell membranes, and the respective number of DSP puncta over this cell was counted using particle analysis in ImageJ.
Proteomic and glycomics data analysis
The raw data were analyzed using MaxQuant (v1.6.17.0) (Cox and Mann, 2008) with the default setting. In brief, the spectra were searched against a human database (protein sequences downloaded from https://www.uniprot.org on 2020/04/17) and commonly observed contaminants by the Andromeda search engine (Cox et al., 2011), using the following search criteria: full tryptic specificity was required (cleavage after lysine or arginine residues, unless followed by proline); three missed cleavages were allowed; carbamidomethylation (C) was set as fixed modification; oxidation (M) and acetylation (Protein N-term) were applied as variable modifications; mass tolerance of 20 ppm (precursor) and 20 ppm/0.6 Da (fragments) for QE-plus/Lumos. Label-free and iBAQ quantification as well as much between runs were enabled. The database search results were filtered to a false discovery rate (FDR) to 1% on the peptide and protein level and specified a minimum length of seven amino acids for peptides. Quantitative analysis results from label-free quantification were processed using the MS stats R package v4.0.1 (Choi et al., 2014).
Heat maps were generated by using ClustVis software freely available as an online tool. STRING database was used to identify molecular and biological pathways associated with the significantly modulated proteins across the samples.
Online supplemental material
Fig. S1 shows validation of DPM1 KD and its effect on the N-Glycome and desmosomal protein expression by western blots. The figure also includes validation of the 3D-RHE model. Fig. S2 includes heat maps and PCA analysis for differential protein expression in sgDPM1 HaCaT keratinocytes. Further, the figure includes verification of SERPINB5 downregulation in sgDPM1 cells and interaction of SERPINB5 to different domains of DSP. Fig. S3 shows validation of SERPINB5-mediated effects in primary human keratinocytes and biochemical characterization of desmosomal protein upon SERPINB5 overexpression in sgDPM1 HaCaT keratinocytes. Fig. S4 includes the sequencing confirmation of S176A mutation. Further, the figure shows binding of DSP-S176A mutant to PG and the analysis of putative kinases for differential phosphorylation of S176 in sgDPM1 versus sgDPM1-SERPINB5 overexpressing cells. Table S1 shows the list of primers used in the study. Table S2 includes guide RNA sequences and target exons.
Data availability
The data will be made available upon request.
Acknowledgments
The authors would like to acknowledge Dr. Diego Calabrese (Histology Core Facility) and Drs. Michael Abanto and Pascal Lorentz (Microscopy Core Facility), University of Basel, Switzerland; Dr. Florian Geier (Bioinformatics Facility), University of Basel; Dr. Chia-Wei Lin (Functional Genomics Centre), ETH Zurich, Zurich, Switzerland, for glycomics analysis; Prof. Kathleen Green, Northwestern University, Evanston, IL, USA, for providing DSP antibodies and Addgene plasmid #32227; and the Department of Urology (University Hospital, Basel, Switzerland) for providing human foreskin tissues.
The study was supported by the Swiss National Science Foundation (#197764 to V. Spindler), Swiss Heart Foundation (FF21098 to C. Schinner and V. Spindler), the Olga Mayenfisch Stiftung, and the Novartis Foundation for Medical-Biological Research (#22B086, both to C. Schinner) for funding.
Author contributions: Conceptualization: V. Spindler and M. Rathod; Data acquisition: M. Rathod, H. Franz, V. Beyersdorfer, M.T. Wanuske, K. Leal-Fischer, P. Hanns, C. Stüdle, A. Zimmermann, and K. Buczak; Data Analysis: M. Rathod, V. Beyersdorfer, M.T. Wanuske, K. Leal-Fischer, and K. Buczak; Funding Acquisition: V. Spindler and C. Schinner; Project Administration: V. Spindler and M. Rathod; Resources: V. Spindler and C. Schinner; Supervision: V. Spindler; Writing Original Draft Preparation: M. Rathod and V. Spindler; Writing - Review and Editing: V. Spindler, M. Rathod, H. Franz, V. Beyersdorfer, M.T. Wanuske, K. Leal-Fischer, P. Hanns, C. Stüdle, A. Zimmermann, and C. Schinner.

