


The E4 variant of APOE strongly predisposes individuals to late-onset Alzheimer’s disease. We demonstrate that in response to lipogenesis, apolipoprotein E (APOE) in astrocytes can avoid translocation into the endoplasmic reticulum (ER) lumen and traffic to lipid droplets (LDs) via membrane bridges at ER–LD contacts. APOE knockdown promotes fewer, larger LDs after a fatty acid pulse, which contain more unsaturated triglyceride after fatty acid pulse-chase. This LD size phenotype was rescued by chimeric APOE that targets only LDs. Like APOE depletion, APOE4-expressing astrocytes form a small number of large LDs enriched in unsaturated triglyceride. Additionally, the LDs in APOE4 cells exhibit impaired turnover and increased sensitivity to lipid peroxidation. Our data indicate that APOE plays a previously unrecognized role as an LD surface protein that regulates LD size and composition. APOE4 causes aberrant LD composition and morphology. Our study contributes to accumulating evidence that APOE4 astrocytes with large, unsaturated LDs are sensitized to lipid peroxidation, which could contribute to Alzheimer’s disease risk.
Introduction
Lipids comprise 60% of the brain’s dry mass (O’Brien and Sampson, 1965). Consequently, sophisticated molecular mechanisms evolved to manage the distribution and utilization of the diverse lipid species present in the brain. Astrocytes coordinate many aspects of brain lipid homeostasis (Pfrieger and Ungerer, 2011). They mediate lipid uptake from the blood, synthesize lipids for neurons such as cholesterol and polyunsaturated fatty acids, and take up peroxidated lipids from neurons for detoxification (Pfrieger and Barres, 1997; Mauch et al., 2001; Liu et al., 2015; Nakato et al., 2015; Ioannou et al., 2019). Cytoplasmic lipid droplets (LDs) are pivotal components of astrocyte lipid homeostasis (Ralhan et al., 2021). LDs store lipids including triglycerides (TGs) and cholesterol esters (CEs) in a neutral lipid core surrounded by an amphipathic phospholipid monolayer. Proteins that coat the surface of LDs regulate their biogenesis and turnover, as well as a medley of other cellular processes including cell signaling, protein homeostasis, and inflammation (Olzmann and Carvalho, 2019). LDs store energy in the form of fatty acids that can be beta-oxidized and buffer against lipotoxic stress by preventing the accumulation of harmful lipid intermediates (Olzmann and Carvalho, 2019). Therefore, processes that regulate LD biogenesis and turnover are critical to protecting cells from lipid-related insults. Notably, Alois Alzheimer first observed the accumulation of “adipose inclusions” in the glia of patient brain tissue in his foundational study, which also describes extracellular amyloid beta plaques and tau neurofibrillary tangles (Alzheimer, 1907; translated in Alzheimer et al. [1995]). More recent work has demonstrated that both astrocytes and microglia accumulate LDs during aging and in pathologies such as ischemia, neuroinflammation, and neurodegenerative diseases including Alzheimer’s disease (Hamilton et al., 2015; Farmer et al., 2020; Ralhan et al., 2021). However, it is unclear whether LD accumulation is a cause or consequence of pathology.
A key protein mediator of brain lipid homeostasis is apolipoprotein E (APOE), a 34-kD secreted protein expressed primarily by astrocytes and microglia as well as neurons under stress (Xu et al., 2006). APOE is a component of high-density lipoprotein particles that transport lipids between cells in the brain. Nascent APOE lipoproteins are assembled in the lumen of endoplasmic reticulum (ER) prior to secretion, although the mechanism for this stage of biogenesis is unknown (Windham and Cohen, 2023). After secretion, APOE-coated lipoproteins are lipidated via reverse cholesterol transport from the plasma membrane mediated by ABC transporters, primarily ABCA1 (Wahrle et al., 2004). APOE on lipoproteins binds to cell-surface lipoprotein receptors, including LDLR and LRP1, triggering lipoprotein uptake via receptor-mediated endocytosis (Bu, 2009). Astrocyte-derived lipoproteins supply key lipids, predominately cholesterol and phospholipids, to neurons for building their high surface area membranes and cholesterol-rich lipid nanodomains at synapses (van Deijk et al., 2017). Other studies demonstrated that lipid peroxides formed in neurons under oxidative or excitotoxic stress can be transported by APOE-lipoproteins to astrocytes; astrocytes store these neuron-derived lipids in LDs, which protect against neurotoxicity and may play a role in detoxifying these deleterious lipid species (Walker et al., 2006; Liu et al., 2015, 2017; Ioannou et al., 2019; Moulton et al., 2021). APOE is notably the strongest genetic risk factor for late-onset Alzheimer’s disease. The APOE4 variant is a cysteine to arginine substitution at residue 112, with individuals possessing the APOE4 variant significantly more likely to develop late-onset Alzheimer’s disease than those homozygous for the APOE3 variant in a dose-dependent manner (Corder et al., 1993). There is no consensus mechanism for how the E4 variant predisposes individuals to Alzheimer’s disease.
Although early work focused on the effects of APOE4 on the clearance of amyloid-β, many recent studies draw a compelling connection between APOE4 and glia-specific lipid dishomeostasis (Windham and Cohen, 2023). Multiple transcriptomic studies of APOE4-expressing iPSC-derived astrocytes and microglia demonstrate alterations in the expression of lipid metabolic genes as well as defects in cholesterol trafficking and metabolism (Lin et al., 2018; de Leeuw et al., 2022; Tcw et al., 2022). Another study demonstrated that iPSC-derived APOE4 astrocytes accumulate LDs rich in highly unsaturated TGs. Blocking fatty acid desaturation through inhibition of stearoyl-CoA desaturase 1, an enzyme elevated in Alzheimer's disease patient brains, or supplementing cells with choline reduced LD accumulation in APOE4 iPSCs (Astarita et al., 2011; Sienski et al., 2021). APOE4 iPSCs also exhibit defects in endocytosis (Narayan et al., 2020). Mouse astrocytes expressing human APOE4 have defects in LD metabolism as well as in fatty acid uptake and oxidation (Farmer et al., 2019; Qi et al., 2021). However, it is unclear whether these phenotypes are downstream of APOE-mediated lipid secretion and uptake or another function of APOE within the cell.
Hints that APOE may play roles beyond lipid secretion came from studies of the LD surface proteome. A proximity ligation strategy identified APOE as a protein on the cytoplasmic face of LDs in Huh7 hepatocarcinoma cells (Bersuker et al., 2018). To be a positive hit, the protein must be accessible to APEX2-tagged PLIN2 on the surface of LDs, significantly reducing the possibility that this was a false positive caused by contamination of ER membranes in the LD preparation. This suggested that in addition to being secreted, APOE also has the capacity to target LDs in the cytoplasm. APOE was identified in subsequent studies of the LD proteome in mouse liver and THP-1 macrophages (Krahmer et al., 2018; Mejhert et al., 2020). Studies on the trafficking of APOE have also suggested that the cleaved C-terminus of APOE can exist in the cytoplasm and associate with mitochondria in neurons (Chang et al., 2005). Other members of the exchangeable apolipoprotein family have also been found to associate with LDs. APOE and APOCIII were identified on nuclear LDs in HepG2 cells, and APOAV has previously been reported on the surface of cytoplasmic LDs in adipocytes (Shu et al., 2010; Gao et al., 2012; Sołtysik et al., 2019). A putative LD-associated pool of APOE within glial cells could play an important role in maintaining lipid homeostasis in the brain.
We sought to determine whether APOE is a bona fide LD protein in astrocytes. We observed that APOE localized to the cytoplasmic surface of LDs in astrocytes under conditions of neutral lipid synthesis. APOE is trafficked to LDs by avoiding translocation into the ER, targeting the cytoplasmic side of the ER before moving onto LDs at membrane bridges between the ER and LDs. By utilizing an oleic acid (OA) pulse-chase assay, we found that LD-associated APOE regulates the size of LDs. Knockdown of APOE caused a smaller number of large LDs to form during lipogenesis. Expression of an exclusively LD-targeted APOE chimeric construct rescued this phenotype, supporting a physiological role for APOE on the LD surface. Like APOE knockdown cells, APOE4-expressing cells had larger LDs than E3-expressing cells after OA pulse-chase, suggesting a lipid turnover defect. These lipolysis-resistant E4 LDs were dramatically enriched in unsaturated triglycerides and were more sensitive to lipid peroxidation than E3 LDs. Our results contribute to growing evidence that lipid homeostatic defects in E4 sensitize astrocytes to stress, which could promote a reactive, proinflammatory state that contributes to Alzheimer’s pathogenesis.
Results
APOE localizes to the cytoplasmic surface of LDs
LD accumulation in glial cells occurs in aging and Alzheimer's disease (Hamilton et al., 2015; Shimabukuro et al., 2016; Farmer et al., 2019; Sienski et al., 2021). Previous proteomics studies identified APOE as a putative LD protein in liver cells and macrophages (Bersuker et al., 2018; Krahmer et al., 2018; Mejhert et al., 2020). Therefore, we hypothesized that APOE could localize to LDs in astrocytes. However, APOE possesses a canonical N-terminal signal peptide that should target it for cotranslational translocation into the ER lumen (Zannis et al., 1984). Because of this, a cytoplasmic localization would be rather unprecedented. Thus, it was imperative to test the LD targeting of APOE by probing its endogenous localization before relying on overexpression, which could cause APOE to mislocalize.
To visualize endogenous APOE in astrocytes, we employed immortalized APOE-targeted replacement astrocytes. These cells are astrocytes isolated from APOE-targeted replacement mice—a model in which endogenous mouse coding exons 2–4 are replaced with the corresponding human APOE3 or APOE4 coding exons via homologous recombination—and subsequently immortalized via stable SV40 expression (Sullivan et al., 1997; Morikawa et al., 2005). The cells are termed TRAE3-H and TRAE4-H, which express human APOE3 or human APOE4, respectively. We chose this model because it provided an easily scalable, continuous cell with which we could readily study the endogenous localization of human APOE and also directly compare the differential effects of APOE variants.
We stained fixed TRAE3-H cells for human APOE, LDs, and GM-130, a marker of the Golgi. siRNA-mediated knockdown demonstrated the specificity of the APOE antibody (Fig. S1, A–C). At their baseline state, TRAE3-H cells contained few LDs, and APOE localized largely to the Golgi apparatus, as expected (Fig. 1, A–C). To model an LD-accumulating state, we loaded TRAE3-H cells with 400 µM OA for 5 h, which profoundly induced LD formation (Fig. 1, A–D). Surprisingly, APOE relocalized to the surface of cytoplasmic LDs in a majority of cells after OA induction (Fig. 1, A–C). The percentage of cells exhibiting rings of APOE on the surface of LDs by visual assessment increased from 0% to 61% ± 11.5 upon OA treatment (Fig. 1 E). This observation was corroborated by an unbiased, quantitative measurement of APOE protein enrichment on the LD surface (Fig. 1 F). By contrast, we observed a concomitant reduction in colocalization of APOE with the Golgi marker GM130 after OA treatment (Fig. 1, C and G).
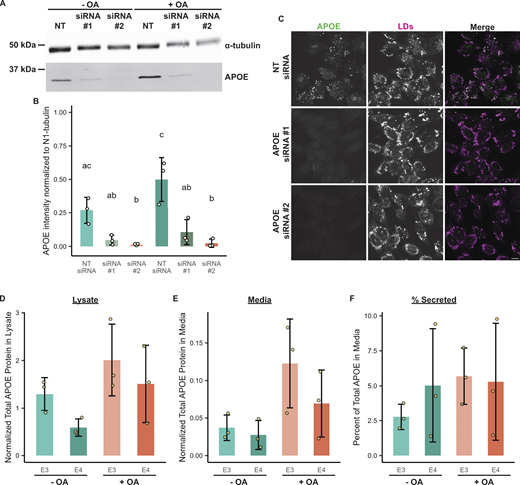
Validation of APOE antibody via siRNA-mediated knockdown and measurement of intracellular and secreted APOE ± OA. (A) Western blot of lysates of TRAE3-H cells transfected with a non-targeting siRNA or one of two different siRNAs against APOE and treated ± OA for 5 h. The antibody used to probe for endogenous APOE is the same one used for both the immunofluorescence and immunogold experiments. (B) Quantification of Western blots of APOE knockdown from three independent biological replicates. APOE siRNA #2 demonstrated ∼94% knockdown of APOE in both − and + OA conditions, and was used for subsequent loss of function studies in Figs. 5 and 6. (C) Representative confocal slices of TRAE3-H cells transfected with NT siRNA or one of two APOE siRNAs and treated with 400 µM OA for 5 h. Cells were fixed and stained for endogenous APOE with an anti-APOE antibody and for LDs with BODIPY 493/503. Little to no endogenous APOE signal was observed by immunofluorescence upon APOE knockdown. Scale bar, 10 µm. (D) Normalized total APOE protein present in TRAE3-H and TRAE4-H lysates ± 5-h OA treatment. Cells were lysed in 100 µl of lysis buffer, and APOE protein concentrations in µg/ml were measured by ELISA. The APOE concentration in µg/ml was multiplied by the total lysate volume of 0.1 ml to derive the total amount of APOE protein in the sample. These values were then normalized by dividing the corresponding total lysate protein concentration of each sample measured via Bradford assay to account for differences in cell number. N = 3 independent biological replicates. Data are expressed in bar graphs as means, and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. (E) Normalized total APOE protein present in TRAE3-H and TRAE4-H media ± 5-h OA treatment. Cells were grown in 1 ml of media, and APOE protein concentrations in µg/ml were measured by ELISA. The APOE concentration in µg/ml was multiplied by the total media volume of 1 ml to derive the total amount of APOE protein in the sample. These values were then normalized by dividing the total lysate protein concentration of the corresponding lysate sample measured via Bradford assay to account for variations in total material. N = 3 independent biological replicates. Data are expressed in bar graphs as means and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. (F) The percentage of APOE present in the media out of the total APOE protein present in the lysate + media. Around 5% of total APOE protein was secreted into the media after 5 h ± OA treatment in all conditions. N = 3 independent biological replicates. Data are expressed in bar graphs as means, and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. Source data are available for this figure: SourceData FS1.

Validation of APOE antibody via siRNA-mediated knockdown and measurement of intracellular and secreted APOE ± OA. (A) Western blot of lysates of TRAE3-H cells transfected with a non-targeting siRNA or one of two different siRNAs against APOE and treated ± OA for 5 h. The antibody used to probe for endogenous APOE is the same one used for both the immunofluorescence and immunogold experiments. (B) Quantification of Western blots of APOE knockdown from three independent biological replicates. APOE siRNA #2 demonstrated ∼94% knockdown of APOE in both − and + OA conditions, and was used for subsequent loss of function studies in Figs. 5 and 6. (C) Representative confocal slices of TRAE3-H cells transfected with NT siRNA or one of two APOE siRNAs and treated with 400 µM OA for 5 h. Cells were fixed and stained for endogenous APOE with an anti-APOE antibody and for LDs with BODIPY 493/503. Little to no endogenous APOE signal was observed by immunofluorescence upon APOE knockdown. Scale bar, 10 µm. (D) Normalized total APOE protein present in TRAE3-H and TRAE4-H lysates ± 5-h OA treatment. Cells were lysed in 100 µl of lysis buffer, and APOE protein concentrations in µg/ml were measured by ELISA. The APOE concentration in µg/ml was multiplied by the total lysate volume of 0.1 ml to derive the total amount of APOE protein in the sample. These values were then normalized by dividing the corresponding total lysate protein concentration of each sample measured via Bradford assay to account for differences in cell number. N = 3 independent biological replicates. Data are expressed in bar graphs as means, and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. (E) Normalized total APOE protein present in TRAE3-H and TRAE4-H media ± 5-h OA treatment. Cells were grown in 1 ml of media, and APOE protein concentrations in µg/ml were measured by ELISA. The APOE concentration in µg/ml was multiplied by the total media volume of 1 ml to derive the total amount of APOE protein in the sample. These values were then normalized by dividing the total lysate protein concentration of the corresponding lysate sample measured via Bradford assay to account for variations in total material. N = 3 independent biological replicates. Data are expressed in bar graphs as means and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. (F) The percentage of APOE present in the media out of the total APOE protein present in the lysate + media. Around 5% of total APOE protein was secreted into the media after 5 h ± OA treatment in all conditions. N = 3 independent biological replicates. Data are expressed in bar graphs as means, and error bars represent ± standard deviation. P value calculated via Tukey’s HSD. All pairwise comparisons were statistically insignificant. Source data are available for this figure: SourceData FS1.

APOE localizes to cytoplasmic LDs in astrocytes during lipogenesis. (A) Representative fields of TRAE3-H cells untreated (− OA) or treated with 400 µM oleic acid for 5 h (+ OA). Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody, an anti-GM130 antibody to label the Golgi, and LDs with BODIPY 493/503. In the merged image, APOE is yellow, GM130/Golgi is cyan, and LDs are magenta. White dotted lines outline the plasma membranes of individual cells in the field. Scale bar, 50 µm. (B) Individual cells from A (labeled by white inset boxes) show the increase in LDs and the enrichment of APOE on the LD surface upon OA loading. Scale bar, 10 µm. (C) Inset of images from B. The top panels show the colocalization of APOE with the Golgi and the lack of LD-associated APOE in untreated cells. The bottom panels show APOE coating the surface of LDs and the concomitant reduction in Golgi colocalization upon OA treatment. Scale bar, 1 µm. (D) Quantification of the total area of BODIPY 493/503-labeled LDs per cell ± OA. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (E) Percentage of cells that have APOE on the surface of LDs ± OA. Each data point represents the percentage of 50 randomly selected cells from one independent experiment with APOE on the surface of LDs. 61.1% ± 11.5 of cells have APOE on the surface of LDs after 5 h 400 µM OA treatment. (F) Quantification of APOE enrichment on the surface of LDs in TRAE3-H cells ± OA. LD enrichment is equal to the mean intensity of APOE signal surrounding LDs divided by the mean intensity of the entire cell minus the LDs. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (G) Quantification of colocalization of APOE with the Golgi marker GM130 as measured by the Mander’s coefficient in TRAE3-H cells ± OA. The Mander’s coefficient was calculated by dividing the area of overlap between APOE and GM130 masks by the total area of the APOE mask. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (H) Immunogold electron micrographs of endogenous APOE in TRAE3-H cells treated with 400 µM OA for 5 h. The primary APOE antibody was included in images labeled “+ Primary,” and not included in the negative control images labeled “− Primary.” Silver-enhanced gold particles localize directly to the surface of LDs at the interface between the LD monolayer surface and the cytoplasm. Scale bars: 200 nm (left), 20 nm for zoom (right). P values for D, F, and G were calculated using a clustered Wilcoxon rank sum test via the Rosner-Glynn-Lee method. **** P < 0.0001. P value for E calculated via unpaired two-tailed t test. * P < 0.05.
APOE localizes to cytoplasmic LDs in astrocytes during lipogenesis. (A) Representative fields of TRAE3-H cells untreated (− OA) or treated with 400 µM oleic acid for 5 h (+ OA). Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody, an anti-GM130 antibody to label the Golgi, and LDs with BODIPY 493/503. In the merged image, APOE is yellow, GM130/Golgi is cyan, and LDs are magenta. White dotted lines outline the plasma membranes of individual cells in the field. Scale bar, 50 µm. (B) Individual cells from A (labeled by white inset boxes) show the increase in LDs and the enrichment of APOE on the LD surface upon OA loading. Scale bar, 10 µm. (C) Inset of images from B. The top panels show the colocalization of APOE with the Golgi and the lack of LD-associated APOE in untreated cells. The bottom panels show APOE coating the surface of LDs and the concomitant reduction in Golgi colocalization upon OA treatment. Scale bar, 1 µm. (D) Quantification of the total area of BODIPY 493/503-labeled LDs per cell ± OA. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (E) Percentage of cells that have APOE on the surface of LDs ± OA. Each data point represents the percentage of 50 randomly selected cells from one independent experiment with APOE on the surface of LDs. 61.1% ± 11.5 of cells have APOE on the surface of LDs after 5 h 400 µM OA treatment. (F) Quantification of APOE enrichment on the surface of LDs in TRAE3-H cells ± OA. LD enrichment is equal to the mean intensity of APOE signal surrounding LDs divided by the mean intensity of the entire cell minus the LDs. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (G) Quantification of colocalization of APOE with the Golgi marker GM130 as measured by the Mander’s coefficient in TRAE3-H cells ± OA. The Mander’s coefficient was calculated by dividing the area of overlap between APOE and GM130 masks by the total area of the APOE mask. N = 150 cells per condition, with 50 cells from each independent experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. (H) Immunogold electron micrographs of endogenous APOE in TRAE3-H cells treated with 400 µM OA for 5 h. The primary APOE antibody was included in images labeled “+ Primary,” and not included in the negative control images labeled “− Primary.” Silver-enhanced gold particles localize directly to the surface of LDs at the interface between the LD monolayer surface and the cytoplasm. Scale bars: 200 nm (left), 20 nm for zoom (right). P values for D, F, and G were calculated using a clustered Wilcoxon rank sum test via the Rosner-Glynn-Lee method. **** P < 0.0001. P value for E calculated via unpaired two-tailed t test. * P < 0.05.
The shift in APOE localization away from the Golgi and onto LDs upon OA treatment suggested that secretion of APOE may be reduced. To quantitatively compare intracellular and secreted APOE, we used ELISA to measure the amount of APOE in cell lysates (Fig. S1 D) versus the amount of APOE secreted into the media (Fig. S1 E) after 5 h in the absence or presence of OA. In the absence of OA, ∼3–5% of total measured APOE was secreted over a 5-h period (Fig. S1 F). OA loading did not significantly change the ratio of secreted to total APOE (Fig. S1 F), although both intracellular and secreted APOE trended higher in response to OA in TRAE3-H and TRAE4-H cells (Fig. S1, B, D, and E). Our interpretation of these data is that OA stimulates global APOE upregulation, with the subset of cells that route APOE to LDs accounting for increased intracellular APOE, while the subset of cells which lack LD-associated APOE account for the increase in APOE secretion.
To test if overexpressing APOE drives it to target LDs, we transfected TRAE3-H cells with human APOE3 tagged at the C-terminus with mEmerald (APOE3-mEm) and compared the localization of endogenous to overexpressed APOE. In the absence of OA, APOE3-mEm localized exclusively to the secretory pathway and never coated LDs, matching the endogenous localization pattern. Upon OA loading, APOE3-mEm localized to LDs in the same percentage of cells as endogenous APOE (Fig. S2, A and B). Therefore, the expression of exogenous APOE3-mEm is representative of the trafficking of the endogenous protein, and overexpression does not drive APOE to LDs.

Effect of APOE overexpression on LD targeting; targeting of APOE to LDs in iAstros and HMC3 cells and in response to multiple unsaturated fatty acids. (A) TRAE3-H cells transfected with APOE3-mEm, labeled for LDs with BODIPY 665/676, and treated ± OA. APOE3-mEm localizes to the secretory pathway in the absence of OA and shifts to LDs after OA treatment. Scale bar, 10 µm. (B) Percentage of cells that have either endogenous APOE or APOE3-mEm on the surface of LDs ± OA. Each data point represents the percentage of 35–50 randomly selected cells from one independent experiment with APOE on the surface of LDs. Data for endogenous APOE fractions are the same as in Fig. 1 E. There is no significant difference between endogenous and overexpressed APOE in the fraction of cells with APOE on LDs. (C) Representative confocal slices of induced pluripotent stem cell-derived astrocytes (iAstros) transfected with APOE3-mEm and labeled for LDs with BODIPY 665/676. Under baseline media conditions, ∼49.3% of iAstros have APOE on LDs, while 50.7% of iAstros do not have APOE on LDs. Scale bar, 10 µm. (D) Quantification of total LD area per cell in iAstros that do not exhibit APOE on LDs versus iAstros that have LD-associated APOE. LD localization of APOE correlates with LD abundance. N = 54–55 cells per condition. Data were collected and pooled from three biologically independent experiments. (E) Representative confocal slices of human microglial HMC3 cells transfected with APOE3-mEm and labeled for LDs with BODIPY 665/676. Under baseline media conditions, ∼41.6% of HMC3 cells have APOE on LDs, while 58.4% of HMC3 cells do not have APOE on LDs. Scale bar, 10 µm. (F) Quantification of total LD area per cell in HMC3 cells that do not exhibit APOE on LDs versus HMC3 cells that have LD-associated APOE. LD localization of APOE correlates with LD abundance. N = 44–54 cells per condition. Data were collected and pooled from three biologically independent experiments. (G) TRAE3-H cells loaded with oleic acid (OA), linoleic acid (LA), or arachidonic acid (ARA), fixed, and stained for endogenous APOE and LDs with BODIPY 493/503. Each fatty acid stimulated LD biogenesis and APOE trafficking to LDs in TRAE3-H cells. Scale bar, 20 µm (field), 10 µm (single cell). (H) Percentage of TRAE3-H cells loaded with OA, LA, or ARA with APOE on the surface of LDs. Each data point represents the percentage of 50 randomly selected cells from one independent experiment with APOE on the surface of LDs. There is no significant difference in APOE trafficking among the different fatty acid loading conditions. P values for B and P values for D and F were calculated using a Wilcox rank sum test. **** P <0.0001.

Effect of APOE overexpression on LD targeting; targeting of APOE to LDs in iAstros and HMC3 cells and in response to multiple unsaturated fatty acids. (A) TRAE3-H cells transfected with APOE3-mEm, labeled for LDs with BODIPY 665/676, and treated ± OA. APOE3-mEm localizes to the secretory pathway in the absence of OA and shifts to LDs after OA treatment. Scale bar, 10 µm. (B) Percentage of cells that have either endogenous APOE or APOE3-mEm on the surface of LDs ± OA. Each data point represents the percentage of 35–50 randomly selected cells from one independent experiment with APOE on the surface of LDs. Data for endogenous APOE fractions are the same as in Fig. 1 E. There is no significant difference between endogenous and overexpressed APOE in the fraction of cells with APOE on LDs. (C) Representative confocal slices of induced pluripotent stem cell-derived astrocytes (iAstros) transfected with APOE3-mEm and labeled for LDs with BODIPY 665/676. Under baseline media conditions, ∼49.3% of iAstros have APOE on LDs, while 50.7% of iAstros do not have APOE on LDs. Scale bar, 10 µm. (D) Quantification of total LD area per cell in iAstros that do not exhibit APOE on LDs versus iAstros that have LD-associated APOE. LD localization of APOE correlates with LD abundance. N = 54–55 cells per condition. Data were collected and pooled from three biologically independent experiments. (E) Representative confocal slices of human microglial HMC3 cells transfected with APOE3-mEm and labeled for LDs with BODIPY 665/676. Under baseline media conditions, ∼41.6% of HMC3 cells have APOE on LDs, while 58.4% of HMC3 cells do not have APOE on LDs. Scale bar, 10 µm. (F) Quantification of total LD area per cell in HMC3 cells that do not exhibit APOE on LDs versus HMC3 cells that have LD-associated APOE. LD localization of APOE correlates with LD abundance. N = 44–54 cells per condition. Data were collected and pooled from three biologically independent experiments. (G) TRAE3-H cells loaded with oleic acid (OA), linoleic acid (LA), or arachidonic acid (ARA), fixed, and stained for endogenous APOE and LDs with BODIPY 493/503. Each fatty acid stimulated LD biogenesis and APOE trafficking to LDs in TRAE3-H cells. Scale bar, 20 µm (field), 10 µm (single cell). (H) Percentage of TRAE3-H cells loaded with OA, LA, or ARA with APOE on the surface of LDs. Each data point represents the percentage of 50 randomly selected cells from one independent experiment with APOE on the surface of LDs. There is no significant difference in APOE trafficking among the different fatty acid loading conditions. P values for B and P values for D and F were calculated using a Wilcox rank sum test. **** P <0.0001.
One caveat of the TRAE3-H cells is that human APOE is expressed in a mouse cell background, and may therefore not faithfully recapitulate the behavior of human APOE in a human cell. To verify that human APOE also traffics to LDs in a genetically homologous system, we employed both induced pluripotent stem cell-derived astrocytes (iAstros) and the HMC3 human microglia cell line. In these cell types, APOE3-mEm constitutively localized to LDs in a subset of cells at baseline—∼49.3% of iAstros and 41.6% of HMC3 cells (Fig. S2, C and E). In both cell types, APOE localization to LDs correlated with LD abundance; cells with APOE-coating LDs contained an abundance of LDs, while cells with APOE in the secretory pathway had few or no LDs (Fig. S2, C–F). This is in contrast to the TRAE3-H cells, which, at baseline, exhibit few LDs and no LD-associated APOE (Fig. 1, D and E). For subsequent functional studies, we chose to employ the TRAE3-H cells because we could shift from a baseline state in which cells have few LDs and no LD-associated APOE to a state in which the majority of cells contain LD-associated APOE. This ability to easily shift between the two states allows us to better separate the functions of secreted APOE versus LD-associated APOE.
To test whether the shift of APOE onto LDs is specific to OA treatment, we loaded cells with the unsaturated fatty acids linoleic acid (LA; 18:2) or arachidonic acid (ARA; 20:4). Like OA (18:1), LA and ARA also induced LD formation and caused APOE to traffic to LDs. We observed no differences among the three fatty acid loading conditions in the percent of cells with LD-associated APOE (Fig. S2, G and H). This suggests that stimulating exogenous fatty acid-induced LD biogenesis drives APOE trafficking to LDs, irrespective of the type of fatty acid.
The ability of APOE to localize to cytoplasmic LDs is particularly striking, as APOE is normally a secreted protein and possesses a canonical N-terminal signal peptide that targets it for translocation into the ER lumen (Zannis et al., 1984). Prior studies have shown that proteins with LD binding moieties that are targeted to the ER lumen become enriched within the ER at ER–LD contact sites. Their localization pattern resembles LD surface proteins, but the protein is within the lumen of the ER (Mishra et al., 2016). Therefore, although APOE appeared enriched on the surface of LDs, it was not clear whether this signal represented APOE protein bound to the cytoplasmic LD surface or within the ER lumen at an ER–LD contact site. To distinguish between these two possibilities, we utilized immunogold electron microscopy, staining for APOE in TRAE3h cells. We observed gold particles dotting the cytoplasmic face of both LDs and the ER (Fig. 1 H). Gold was observed on the LD surface in the absence of any associated ER membrane, suggesting that APOE is a bona fide LD protein. Out of 71 fields of view analyzed, we observed gold particles on 78/191 LDs. We never observed gold within the lumen of the ER.
To further verify the topology of LD-associated APOE, we performed fluorescence protease protection assays (FPPs) (Fig. 2 A) (Lorenz et al., 2006). Primary cortical rat astrocytes expressing a fluorescent marker of the ER lumen and APOE-mEm or another fluorescently tagged LD protein were first treated with digitonin. Digitonin selectively permeabilizes the plasma membrane without permeabilizing intracellular membranes. After digitonin permeabilization, proteinase K was added to degrade cytoplasmic-facing fluorophores while leaving ER-luminal proteins intact. Upon addition of proteinase K, the signal of the ER luminal marker was retained, while LD-associated proteins, including APOE, were degraded (Fig. 2, B and C). These data strongly support the conclusion that APOE indeed coats the cytoplasmic-facing monolayer surface of LDs.

LD-associated APOE is exposed to the cytoplasm. (A) Cartoon schematic of the fluorescence protease protection (FPP) assay to test the topology of fluorescently tagged proteins in live cells. Cells are treated with 30 µM digitonin for 1 min, which selectively permeabilizes the plasma membrane but not the internal membranes. After permeabilization, cells are treated with 50 µg/ml proteinase K (PK), which enters the permeabilized plasma membrane and degrades all cytoplasmic-facing fluorophores (green). Because the ER membrane is not permeabilized, proteinase K does not enter into the ER lumen and ER lumen-facing fluorophores are retained (blue). (B) Representative confocal slices of FPP performed on primary cortical rat astrocytes (− OA) transiently transfected with APOE3-mEm, the ER marker TagBFP2-KDEL, and labeled for LDs with BODIPY 665/676. After digitonin permeabilization and PK treatment, APOE signal on the surface of LDs was lost, but the luminal ER marker fluorescence was retained. A Gaussian filter with a radius of 1 pixel was applied to all images to improve visibility for print. Scale bars: 10 µm (left), 5 µm for zoom (right). (C) Quantification of the FPP assay demonstrated in A and B. The fluorescence intensity of the indicated marker after PK treatment was divided by its fluorescence intensity just before PK treatment. For the “ER ratio,” the mean intensity of TagBFP2-KDEL within the entire cell was measured before and after PK treatment. For the “LD ratio,” the mean fluorescence intensity of the indicated LD protein (APOE, PLIN2, or LiveDrop) surrounding BODIPY 665/676-labeled LDs was measured before and after PK treatment. Ratios close to 1 indicate minimal loss of signal after proteinase K treatment, as observed with the ER marker TagBFP2-KDEL. Lower ratios indicate loss of fluorescence upon PK treatment. N = 8–18 cells per condition, collected from three independent experiments. *P < 0.05, **** P < 0.0001. Dig., 30 µM digitonin. PK, +50 µg/ml Proteinase K. P values were calculated via the Wilcoxon rank sum test and Bonferonni-corrected for multiple comparisons.
LD-associated APOE is exposed to the cytoplasm. (A) Cartoon schematic of the fluorescence protease protection (FPP) assay to test the topology of fluorescently tagged proteins in live cells. Cells are treated with 30 µM digitonin for 1 min, which selectively permeabilizes the plasma membrane but not the internal membranes. After permeabilization, cells are treated with 50 µg/ml proteinase K (PK), which enters the permeabilized plasma membrane and degrades all cytoplasmic-facing fluorophores (green). Because the ER membrane is not permeabilized, proteinase K does not enter into the ER lumen and ER lumen-facing fluorophores are retained (blue). (B) Representative confocal slices of FPP performed on primary cortical rat astrocytes (− OA) transiently transfected with APOE3-mEm, the ER marker TagBFP2-KDEL, and labeled for LDs with BODIPY 665/676. After digitonin permeabilization and PK treatment, APOE signal on the surface of LDs was lost, but the luminal ER marker fluorescence was retained. A Gaussian filter with a radius of 1 pixel was applied to all images to improve visibility for print. Scale bars: 10 µm (left), 5 µm for zoom (right). (C) Quantification of the FPP assay demonstrated in A and B. The fluorescence intensity of the indicated marker after PK treatment was divided by its fluorescence intensity just before PK treatment. For the “ER ratio,” the mean intensity of TagBFP2-KDEL within the entire cell was measured before and after PK treatment. For the “LD ratio,” the mean fluorescence intensity of the indicated LD protein (APOE, PLIN2, or LiveDrop) surrounding BODIPY 665/676-labeled LDs was measured before and after PK treatment. Ratios close to 1 indicate minimal loss of signal after proteinase K treatment, as observed with the ER marker TagBFP2-KDEL. Lower ratios indicate loss of fluorescence upon PK treatment. N = 8–18 cells per condition, collected from three independent experiments. *P < 0.05, **** P < 0.0001. Dig., 30 µM digitonin. PK, +50 µg/ml Proteinase K. P values were calculated via the Wilcoxon rank sum test and Bonferonni-corrected for multiple comparisons.
APOE moves onto cytoplasmic LDs via ER membrane bridges
We hypothesized two possible routes by which APOE could traffic to the LD surface. In the first route, APOE first fully translocates into the ER lumen. It is then exported from the ER lumen to the cytoplasmic compartment via retrotranslocation to reach LDs. It was previously suggested that APOB retrotranslocates from the ER to the surface of LDs at ER–LD contact sites and is subsequently degraded (Ohsaki et al., 2006; Suzuki et al., 2012). In the other possible route, APOE avoids translocation into the ER, instead re-routing onto the cytoplasmic surface of the ER before targeting LDs at ER–LD contact sites. This second possibility is similar to the trafficking of membrane proteins such as DGAT2 that traffic to LDs via ER membrane bridges using the ER to LD (ERTOLD) pathway (Wilfling et al., 2013; Song et al., 2022).
If APOE translocates into the ER before trafficking to LDs, we expect the APOE signal peptide to be cleaved. Therefore, we first examined whether the signal peptide of LD-associated APOE was cleaved by signal peptidase or retained. However, the signal peptide of APOE is quite small, with a molecular weight of ∼1 kD, and no corresponding mass shift of APOE was resolved via SDS-PAGE of OA-treated cell lysates (Fig. S1 A). We instead employed an epitope-tagging strategy wherein a FLAG tag was appended to the N-terminal end of the signal peptide. Cells transfected with this construct and treated with OA had an APOE signal that was positive for both FLAG and Emerald, indicating that the signal peptide was retained (Fig. S3 A). By contrast, APOE in the secretory pathway was Emerald but not FLAG positive, indicating the signal peptide was cleaved in the luminal pool. This suggests that LD-targeted APOE has not been exposed to the ER lumen.

LD-associated APOE retains its signal peptide and is not retrotranslocated from the ER. (A) Construct design and representative confocal slices of TRAE3-H cells transfected with APOE3-mEm or FLAG-SS-APOE3-mEm and treated with 400 µM OA for 5 h. Cells were then fixed and stained for the FLAG tag with an anti-FLAG antibody. Fluorescence signal on the surface of LDs is positive for both FLAG and mEmerald, indicating that LD-associated APOE retains its N-terminal signal peptide. By contrast, APOE in the secretory pathway is mEm positive but does not stain for FLAG, indicating that the pool of APOE in the secretory pathway is properly processed. Scale bar, 10 µm. (B) Representative confocal slices of TRAE3-H treated with 400 µM OA for 5 h together with 0.1% DMSO vehicle or 10 µM DBeQ. Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody and labeled for LDs with BODIPY 493/503. In the merged image, APOE is in green and LDs are in magenta. Scale bar, 20 µm. (C) The LD enrichment fraction from B was calculated as described in Fig. 1. There is no significant difference in LD enrichment upon DBeQ-treatment, indicating that p97-dependent retrotranslocation is not required for LD-targeting of APOE. (D) Quantification of the total area of BODIPY 493/503-labeled LDs per cell from B. There is no significant difference in the total LD area per cell after DBeQ treatment, indicating that DBeQ does not measurably impact OA-induced LD biogenesis. (E) Representative confocal slices of TRAE3-H slices treated with 400 µM OA for 5 h together with 0.1% MeOH vehicle or 100 µg/ml cycloheximide. Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody and labeled for LDs with BODIPY 493/503. In the merged image, APOE is in green, and LDs are in magenta. Scale bar, 10 µm. (F) The LD enrichment fraction from E was calculated as described in Fig. 1. There is a significant reduction in APOE on LDs upon cycloheximide treatment, suggesting that LD-associated APOE is newly translated. (G) Quantification of the total area of BODIPY 493/503-labeled LDs per cell from E. There is also no significant difference in the total LD area per cell after cycloheximide treatment, indicating that cycloheximide does not measurably impact OA-induced LD biogenesis. (B–G)N = 50 cells per condition. Data were collected and pooled from three biologically independent experiments. Scale bars, 10 µm. ns, P > 0.05, **** P < 0.0001. P values were calculated using a Wilcox rank sum test.

LD-associated APOE retains its signal peptide and is not retrotranslocated from the ER. (A) Construct design and representative confocal slices of TRAE3-H cells transfected with APOE3-mEm or FLAG-SS-APOE3-mEm and treated with 400 µM OA for 5 h. Cells were then fixed and stained for the FLAG tag with an anti-FLAG antibody. Fluorescence signal on the surface of LDs is positive for both FLAG and mEmerald, indicating that LD-associated APOE retains its N-terminal signal peptide. By contrast, APOE in the secretory pathway is mEm positive but does not stain for FLAG, indicating that the pool of APOE in the secretory pathway is properly processed. Scale bar, 10 µm. (B) Representative confocal slices of TRAE3-H treated with 400 µM OA for 5 h together with 0.1% DMSO vehicle or 10 µM DBeQ. Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody and labeled for LDs with BODIPY 493/503. In the merged image, APOE is in green and LDs are in magenta. Scale bar, 20 µm. (C) The LD enrichment fraction from B was calculated as described in Fig. 1. There is no significant difference in LD enrichment upon DBeQ-treatment, indicating that p97-dependent retrotranslocation is not required for LD-targeting of APOE. (D) Quantification of the total area of BODIPY 493/503-labeled LDs per cell from B. There is no significant difference in the total LD area per cell after DBeQ treatment, indicating that DBeQ does not measurably impact OA-induced LD biogenesis. (E) Representative confocal slices of TRAE3-H slices treated with 400 µM OA for 5 h together with 0.1% MeOH vehicle or 100 µg/ml cycloheximide. Cells were then fixed and stained for endogenous APOE with an anti-APOE antibody and labeled for LDs with BODIPY 493/503. In the merged image, APOE is in green, and LDs are in magenta. Scale bar, 10 µm. (F) The LD enrichment fraction from E was calculated as described in Fig. 1. There is a significant reduction in APOE on LDs upon cycloheximide treatment, suggesting that LD-associated APOE is newly translated. (G) Quantification of the total area of BODIPY 493/503-labeled LDs per cell from E. There is also no significant difference in the total LD area per cell after cycloheximide treatment, indicating that cycloheximide does not measurably impact OA-induced LD biogenesis. (B–G)N = 50 cells per condition. Data were collected and pooled from three biologically independent experiments. Scale bars, 10 µm. ns, P > 0.05, **** P < 0.0001. P values were calculated using a Wilcox rank sum test.
To test if APOE trafficking to LDs required retrotranslocation, we treated cells with the p97 inhibitor DBeQ concomitantly with OA treatment. No reduction in APOE signal on LDs was observed upon p97 inhibition (Fig. S3, B and C). By contrast, inhibiting the translation of new protein during OA treatment with cycloheximide significantly attenuated the LD-associated pool of APOE (Fig. S3, E and F). Neither drug affected OA-induced LD biogenesis (Fig. S3, D and G). These data are consistent with newly synthesized APOE being routed to LDs without p97-dependent retrotranslocation.
Since our results did not support the trafficking of APOE to LDs after retrotranslocation from the ER, we next explored the second possibility: subversion of translocation into the ER followed by trafficking from the ER to LDs via membrane bridges. To track the targeting of APOE to LDs over time, we performed Airyscan live-cell imaging of APOE3-mEm, LDs, and the ER during OA loading. We observed both half-circles and full rings of APOE forming around LDs at ER-LD contact sites (Fig. 3 A; and Videos 1 and 2). Half-circles of APOE colocalized with ER wrapping around the surface of LDs. By contrast, only segments of APOE rings colocalized with the ER at a contact site, while other segments localized to the LD in the absence of ER. Our immunogold EM data also revealed ER–LD contact sites where gold labeling was observed both on the cytoplasmic face of the ER and on the surface of LDs (Fig. 3 B). These data suggest a model in which APOE moves from the cytoplasmic face of the ER onto the surface of LDs via membrane bridges, as described for ERTOLD membrane proteins.

APOE targets LDs from the ER. (A) Representative frames from fast Airyscan movies showing the localization of LD-associated APOE relative to the ER after 4 h of treatment with 400 µM OA in TRAE3-H cells. Cells were transfected with APOE3-mEm and the ER marker TagBFP2-KDEL and labeled for LDs with BODIPY 665/676. In the merged images, the ER is in magenta and APOE is in green. The yellow lines across the merged images indicate the line of pixels used to create the linescan graphs to the right of the images. In the linescan graphs, the relative fluorescence intensity of BODIPY 665/676-labeled LDs is in cyan, APOE3-mEm is green, and the ER is magenta. Two different localization patterns were observed: “half rings,” in which APOE partially covers the LD surface and colocalizes with the ER, and “full rings,” where APOE fully encloses the surface of the LD and only partially colocalizes with the ER. Scale bars, 500 nm. (B) Immunogold electron micrographs of endogenous APOE localization at membrane contact sites between the ER and LDs in TRAE3-H cells treated with 400 µM OA for 5 h. The blue arrow points to a direct membrane contact between the ER and an LD. Yellow arrows mark APOE localized to the cytoplasmic face of the ER membrane. The red arrow marks APOE localized to the cytoplasmic surface of the LD. Scale bars, 200 nm. (C) Representative frames from confocal FRAP movies of APOE3-mEm on the surface of BODIPY 665/676-labeled LDs in primary rat cortical astrocytes during an OA pulse (200 µM OA for 4 h) or an OA pulse-chase (200 µM OA for 4 h followed by 2 h chase in complete media—OA). APOE fluorescence was bleached at the 0 s timepoint. Scale bar, 1 µm. (D) Normalized intensity of APOE signal within the bleach ROI over time, with t = 0 s denoting the time at which APOE was bleached. The bold center line is the mean normalized intensity, and the upper and lower bounds of the ribbon represent ± standard deviation (SD). N = 28 cells per condition, collected from three independent experiments. (E) Comparison of the rate constant of recovery k between OA pulse and pulse-chase conditions. The rate constant was derived by fitting each recovery curve to the equation y = C (1 - e−kt). N = 28 cells per condition, collected from three independent experiments. * P < 0.05. (F) Comparison of the mobile fraction between OA pulse and pulse-chase conditions. The mobile fraction was derived by fitting each recovery curve to the equation y = C (1 - e−kt), where C is equal to the asymptote of the curve i.e., the mobile fraction. N = 28 cells per condition, collected from three independent experiments. **** P < 0.0001. P values were calculated via the Wilcoxon rank-sum test. (G) Schematic illustrating interpretation of the results of the FRAP experiment. When APOE on the LD is bleached during the OA pulse, it recovers very rapidly with a high mobile fraction. This indicates that bleached APOE on the LD is rapidly exchanged for unbleached APOE. After a short washout, LD-associated APOE recovers slowly or not at all, indicating that unbleached APOE molecules are unable to replace bleached ones on the LD. We hypothesize LD-APOE exchanges with APOE on the cytoplasmic face of the ER via membrane bridges during OA loading. These bridges are reduced or lost after OA washout, preventing exchange of APOE between LDs and the ER.
APOE targets LDs from the ER. (A) Representative frames from fast Airyscan movies showing the localization of LD-associated APOE relative to the ER after 4 h of treatment with 400 µM OA in TRAE3-H cells. Cells were transfected with APOE3-mEm and the ER marker TagBFP2-KDEL and labeled for LDs with BODIPY 665/676. In the merged images, the ER is in magenta and APOE is in green. The yellow lines across the merged images indicate the line of pixels used to create the linescan graphs to the right of the images. In the linescan graphs, the relative fluorescence intensity of BODIPY 665/676-labeled LDs is in cyan, APOE3-mEm is green, and the ER is magenta. Two different localization patterns were observed: “half rings,” in which APOE partially covers the LD surface and colocalizes with the ER, and “full rings,” where APOE fully encloses the surface of the LD and only partially colocalizes with the ER. Scale bars, 500 nm. (B) Immunogold electron micrographs of endogenous APOE localization at membrane contact sites between the ER and LDs in TRAE3-H cells treated with 400 µM OA for 5 h. The blue arrow points to a direct membrane contact between the ER and an LD. Yellow arrows mark APOE localized to the cytoplasmic face of the ER membrane. The red arrow marks APOE localized to the cytoplasmic surface of the LD. Scale bars, 200 nm. (C) Representative frames from confocal FRAP movies of APOE3-mEm on the surface of BODIPY 665/676-labeled LDs in primary rat cortical astrocytes during an OA pulse (200 µM OA for 4 h) or an OA pulse-chase (200 µM OA for 4 h followed by 2 h chase in complete media—OA). APOE fluorescence was bleached at the 0 s timepoint. Scale bar, 1 µm. (D) Normalized intensity of APOE signal within the bleach ROI over time, with t = 0 s denoting the time at which APOE was bleached. The bold center line is the mean normalized intensity, and the upper and lower bounds of the ribbon represent ± standard deviation (SD). N = 28 cells per condition, collected from three independent experiments. (E) Comparison of the rate constant of recovery k between OA pulse and pulse-chase conditions. The rate constant was derived by fitting each recovery curve to the equation y = C (1 - e−kt). N = 28 cells per condition, collected from three independent experiments. * P < 0.05. (F) Comparison of the mobile fraction between OA pulse and pulse-chase conditions. The mobile fraction was derived by fitting each recovery curve to the equation y = C (1 - e−kt), where C is equal to the asymptote of the curve i.e., the mobile fraction. N = 28 cells per condition, collected from three independent experiments. **** P < 0.0001. P values were calculated via the Wilcoxon rank-sum test. (G) Schematic illustrating interpretation of the results of the FRAP experiment. When APOE on the LD is bleached during the OA pulse, it recovers very rapidly with a high mobile fraction. This indicates that bleached APOE on the LD is rapidly exchanged for unbleached APOE. After a short washout, LD-associated APOE recovers slowly or not at all, indicating that unbleached APOE molecules are unable to replace bleached ones on the LD. We hypothesize LD-APOE exchanges with APOE on the cytoplasmic face of the ER via membrane bridges during OA loading. These bridges are reduced or lost after OA washout, preventing exchange of APOE between LDs and the ER.
Partial and full rings of APOE surrounding LDs contacting the ER. Airyscan live-cell imaging of TRAE3-H cells transfected with the ER lumen marker TagBFP2-KDEL (cyan) and APOE3-mEm (yellow), stained for LDs (magenta) with BODIPY 665/676, and treated with 400 µM OA for 4 h. The video shows both full and half rings of APOE on the surface of LDs and at ER-LD contact sites. Scale bar: 2 µm. Corresponds to images shown in Fig. 3 A. Cells were imaged every 7.9 s for 80 frames. The video plays at 10 frames per second.
Partial and full rings of APOE surrounding LDs contacting the ER. Airyscan live-cell imaging of TRAE3-H cells transfected with the ER lumen marker TagBFP2-KDEL (cyan) and APOE3-mEm (yellow), stained for LDs (magenta) with BODIPY 665/676, and treated with 400 µM OA for 4 h. The video shows both full and half rings of APOE on the surface of LDs and at ER-LD contact sites. Scale bar: 2 µm. Corresponds to images shown in Fig. 3 A. Cells were imaged every 7.9 s for 80 frames. The video plays at 10 frames per second.
Full rings of APOE surrounding LDs contacting the ER. Airyscan live-cell imaging of TRAE3-H cells transfected with the ER lumen marker TagBFP2-KDEL (cyan) and APOE3-mEm (yellow), stained for LDs (magenta) with BODIPY 665/676, and treated with 400 µM OA for 4 h. The video shows full rings of APOE on the surface of LDs and at ER-LD contact sites. Scale bar: 1 µm. Corresponds to the images shown in Fig. 3 A. Cells were imaged every 4.9 s for 29 frames. The video plays at 10 frames per second.
Full rings of APOE surrounding LDs contacting the ER. Airyscan live-cell imaging of TRAE3-H cells transfected with the ER lumen marker TagBFP2-KDEL (cyan) and APOE3-mEm (yellow), stained for LDs (magenta) with BODIPY 665/676, and treated with 400 µM OA for 4 h. The video shows full rings of APOE on the surface of LDs and at ER-LD contact sites. Scale bar: 1 µm. Corresponds to the images shown in Fig. 3 A. Cells were imaged every 4.9 s for 29 frames. The video plays at 10 frames per second.
To further test our model, we performed fluorescence recovery after photobleaching (FRAP) experiments on LD-associated APOE. When APOE on LDs was bleached during loading with 200 µM OA, it rapidly recovered, nearly reaching its initial intensity. By contrast, LD-associated APOE recovery was profoundly attenuated after an OA pulse-chase in which cells were first loaded with 200 µM OA-supplemented media for 4 h and then chased in unsupplemented complete media for 2 h (Fig. 3, C and D; and Videos 3 and 4). During the OA pulse, APOE had both a significantly higher rate constant of recovery and mobile fraction (Fig. 3, E and F). These observations suggest that during the period of OA-loading, bleached APOE molecules are rapidly replaced with unbleached molecules from the ER. Nascent LDs that form in response to OA treatment remain attached to the ER by membrane bridges immediately following biogenesis, which would allow APOE molecules on the ER to exchange with the LD pool. However, if OA is washed out in a pulse-chase experiment, then these nascent LDs mature and detach from the ER. Therefore, after the chase, unbleached molecules are unable to move onto LDs and exchange with the bleached ones. These data support a model in which APOE moves onto LDs via the ER at membrane bridges between the ER and LDs (Fig. 3 G).
FRAP of LD-associated APOE during an OA pulse. Confocal FRAP movies of primary astrocytes transfected with APOE3-mEm (green), stained for LDs with BODIPY 665/676 (magenta), and treated with 200 µM OA for 4 h. LD-associated APOE rapidly recovers when bleached during an OA pulse. Scale bar: 1 µm. Corresponds to images shown in Fig. 3 C. Cells were imaged every second for 96 frames. The video plays at 5 frames per second.
FRAP of LD-associated APOE during an OA pulse. Confocal FRAP movies of primary astrocytes transfected with APOE3-mEm (green), stained for LDs with BODIPY 665/676 (magenta), and treated with 200 µM OA for 4 h. LD-associated APOE rapidly recovers when bleached during an OA pulse. Scale bar: 1 µm. Corresponds to images shown in Fig. 3 C. Cells were imaged every second for 96 frames. The video plays at 5 frames per second.
FRAP of LD-associated APOE after an OA pulse-chase. Confocal FRAP movies of primary astrocytes transfected with APOE3-mEm (green), stained for LDs with BODIPY 665/676 (magenta), and treated with 200 µM OA for 4 h followed by a 2-h chase in complete media. LD-associated APOE recovers very slowly when bleached after an OA pulse-chase. Scale bar: 1 µm. Corresponds to images shown in Fig. 3 C. Cells were imaged every second for 96 frames. The video plays at 5 frames per second.
FRAP of LD-associated APOE after an OA pulse-chase. Confocal FRAP movies of primary astrocytes transfected with APOE3-mEm (green), stained for LDs with BODIPY 665/676 (magenta), and treated with 200 µM OA for 4 h followed by a 2-h chase in complete media. LD-associated APOE recovers very slowly when bleached after an OA pulse-chase. Scale bar: 1 µm. Corresponds to images shown in Fig. 3 C. Cells were imaged every second for 96 frames. The video plays at 5 frames per second.
APOE targeting LDs requires its amphipathic C-terminal domain
To further dissect the mechanism of APOE targeting LDs, we expressed different truncated variants of APOE and measured their capacity to target the surface of LDs. APOE has two domains separated by a short hinge region. Its N-terminal domain forms a soluble four-helix bundle and contains a patch of basic residues required for APOE binding to lipoprotein receptors; the C-terminal domain is an amphipathic alpha-helix that mediates binding of APOE to the surface of lipoproteins (Westerlund and Weisgraber, 1993). To determine which domains are required for LD targeting, we made fluorescently tagged versions of both the N and C terminal domains (Fig. 4 A). Additionally, we created versions of full-length APOE and separate domains in which the N-terminal signal peptide was deleted (Δss). Deleting the signal peptide prevents ER translocation and causes APOE to be present only in the cytoplasm. Western blotting confirmed the expression of each truncation construct at the correct molecular weight (Fig. 4 B). As we previously observed, full-length APOE targeted LDs upon OA treatment. Full-length APOE Δss localized to the cytoplasm and was enriched on the surface of LDs (Fig. 4, C and D). However, not every LD was coated with APOE, suggesting heterogeneity in the binding affinity of APOE to LDs even when APOE is available for binding in the cytosol. The N-terminal domain with a signal sequence did not target LDs. The N-terminal domain Δss was soluble in the cytoplasm but did not exhibit LD surface enrichment (Fig. 4, C and D). The C-terminal domain of APOE bound to LDs with or without a signal peptide, although its binding to LDs was less efficient than the full-length protein (Fig. 4, C and D). We conclude that the C-terminal domain is necessary for binding of APOE to LDs, but inefficient for proper targeting as observed with the full-length protein.

The C-terminal domain is required for LD targeting of APOE. (A) Schematic of the APOE truncation constructs used in this experiment. (B) Western blot of lysates from TRAE3-H cell transfected with the indicated APOE truncation construct. Each construct was expressed and appeared at the expected molecular weight. Predicted molecular weights for each construct are as follows: FL: 63.94 kD, ΔSS FL: 62.03 kD, N: 51.83 kD, ΔSS N: 49.92 kD, C: 39.28 kD, C ΔSS: 37.36 kD. (C) Representative confocal slices of TRAE3-H cells transfected with the indicated construct, stained for LDs with BODIPY 665/676, and treated with 400 µM OA for 5 h. “O” denotes no enrichment of signal on the LD surface, “+” indicates partial enrichment, and “++” indicates full enrichment. (D) Quantification of LD targeting of each construct. The LD intensity ratio was calculated by dividing the mean mEm fluorescence intensity on LDs by the mean mEm fluorescence intensity of the rest of the cell. Letters indicate pairwise significance groups. Conditions denoted with the same letter have no statistically significant difference. N = 40 cells per condition. Each data point represents one cell. Data were collected and pooled from three independent experiments. P-values were calculated via Dunn’s Test for pairwise multiple comparisons. FL, full-length APOE. N-term., N-terminal domain of APOE. C-term., C-terminal domain of APOE. ΔSS, construct has the N-terminal signal peptide deleted. Source data are available for this figure: SourceData F4.
The C-terminal domain is required for LD targeting of APOE. (A) Schematic of the APOE truncation constructs used in this experiment. (B) Western blot of lysates from TRAE3-H cell transfected with the indicated APOE truncation construct. Each construct was expressed and appeared at the expected molecular weight. Predicted molecular weights for each construct are as follows: FL: 63.94 kD, ΔSS FL: 62.03 kD, N: 51.83 kD, ΔSS N: 49.92 kD, C: 39.28 kD, C ΔSS: 37.36 kD. (C) Representative confocal slices of TRAE3-H cells transfected with the indicated construct, stained for LDs with BODIPY 665/676, and treated with 400 µM OA for 5 h. “O” denotes no enrichment of signal on the LD surface, “+” indicates partial enrichment, and “++” indicates full enrichment. (D) Quantification of LD targeting of each construct. The LD intensity ratio was calculated by dividing the mean mEm fluorescence intensity on LDs by the mean mEm fluorescence intensity of the rest of the cell. Letters indicate pairwise significance groups. Conditions denoted with the same letter have no statistically significant difference. N = 40 cells per condition. Each data point represents one cell. Data were collected and pooled from three independent experiments. P-values were calculated via Dunn’s Test for pairwise multiple comparisons. FL, full-length APOE. N-term., N-terminal domain of APOE. C-term., C-terminal domain of APOE. ΔSS, construct has the N-terminal signal peptide deleted. Source data are available for this figure: SourceData F4.
APOE regulates LD size
We next set out to identify the function of APOE on LDs. We hypothesized that APOE on LDs modulates LD metabolism. To test this, we performed OA pulse-chase assays in TRAE3-H cells transfected with an siRNA against APOE or a non-targeting control siRNA. We tested two different siRNAs against APOE and demonstrated that siRNA #2 achieved an ∼94% reduction of APOE protein. OA treatment had no effect of the efficiency of the knockdown of siRNA #2, and this siRNA was used for subsequent APOE knockdown studies (Fig. S1 A). A 5-h OA pulse was used to stimulate LD biogenesis and drive APOE to LDs (Fig. 5 A). OA-containing media was subsequently washed out and replaced with OA-free complete media for 18 h, during which LDs were catabolized (Fig. 5 A). BODIPY 493/503-stained LDs were imaged in live cells at the baseline, OA pulse, and pulse-chase timepoints. This design tests the effect of APOE knockdown on both the biogenesis and turnover of LDs.

APOE modulates LD size distribution and triglyceride saturation. (A) Cartoon outlining the OA pulse-chase assay used in this figure, as well as Fig. 7. TRAE3-H cells are treated with 400 µM OA for 5 h to induce LD biogenesis. This is followed by a chase in unsupplemented complete media (CM) for 18 h, during which LDs are catabolized. Imaging and untargeted lipidomics were performed on cells at the baseline, OA pulse, and pulse-chase timepoints. (B) Representative TRAE3-H cells transfected with non-targeting (NT) or APOE (KD) siRNA, stained for LDs with BODIPY 493/503, and imaged live at each timepoint of the OA pulse-chase assay. Scale bar, 10 µm. (C–E) Quantification of LD parameters in NT or APOE KD cells at each timepoint of the OA pulse-chase assay. (C) Total LD area was measured as the area of the entire LD mask per cell in µm2. (D) Average LD size was calculated as the mean LD area per cell in µm2. (E) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 90 cells per genotype, timepoint, and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method and Bonferonni-corrected for multiple comparisons. (F) Comparison of total triglyceride between NT and APOE KD cells. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. There is no significant difference in the abundance of triglyceride between NT and APOE KD at any timepoint. ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. (G) Heatmap showing the relative abundance of measured lipid classes at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were then grouped by lipid class and Z-score normalized. ChE, cholesterol ester; PC, phosphatidyl choline; PI, phosphatidylinositol; PE, phosphatidyl ethanolamine; PG, phosphatidylglycerol; TG, triacylglycerol; DG, diacylglycerol. (H) Heatmap of the abundance of triglyceride species separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The green box frames lipid species enriched in APOE KD.
APOE modulates LD size distribution and triglyceride saturation. (A) Cartoon outlining the OA pulse-chase assay used in this figure, as well as Fig. 7. TRAE3-H cells are treated with 400 µM OA for 5 h to induce LD biogenesis. This is followed by a chase in unsupplemented complete media (CM) for 18 h, during which LDs are catabolized. Imaging and untargeted lipidomics were performed on cells at the baseline, OA pulse, and pulse-chase timepoints. (B) Representative TRAE3-H cells transfected with non-targeting (NT) or APOE (KD) siRNA, stained for LDs with BODIPY 493/503, and imaged live at each timepoint of the OA pulse-chase assay. Scale bar, 10 µm. (C–E) Quantification of LD parameters in NT or APOE KD cells at each timepoint of the OA pulse-chase assay. (C) Total LD area was measured as the area of the entire LD mask per cell in µm2. (D) Average LD size was calculated as the mean LD area per cell in µm2. (E) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 90 cells per genotype, timepoint, and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method and Bonferonni-corrected for multiple comparisons. (F) Comparison of total triglyceride between NT and APOE KD cells. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. There is no significant difference in the abundance of triglyceride between NT and APOE KD at any timepoint. ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. (G) Heatmap showing the relative abundance of measured lipid classes at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were then grouped by lipid class and Z-score normalized. ChE, cholesterol ester; PC, phosphatidyl choline; PI, phosphatidylinositol; PE, phosphatidyl ethanolamine; PG, phosphatidylglycerol; TG, triacylglycerol; DG, diacylglycerol. (H) Heatmap of the abundance of triglyceride species separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The green box frames lipid species enriched in APOE KD.
At each time point, there was no difference in the area of LDs per cell, suggesting that APOE knockdown does not affect the total amount of neutral lipid in the cell. However, we observed a marked shift in the size distribution of LDs in APOE knockdown cells compared with control cells (Fig. 5, B–E). After the OA pulse, we observed larger but fewer LDs in APOE knockdown cells. This suggests that, although the same total amount of neutral lipids is present in these cells, LD biogenesis is altered, causing neutral lipids to concentrate in a smaller number of larger LDs. The altered size distribution was maintained after the pulse chase.
The distribution of lipids in fewer, larger LDs after an OA pulse suggests APOE knockdown causes defects in the formation and growth of LDs. One mechanism that affects LD size is their composition. We hypothesized that APOE knockdown could impact the composition of LDs and have subsequent downstream effects on their size distribution. We performed untargeted whole-cell lipidomics on TRAE3-H cells transfected with non-targeting siRNA (NT) or APOE siRNA (KD) at each time point of the OA pulse-chase assay. As expected, we observed a pronounced increase in the total abundance of triglyceride (TG) and diglyceride (DG) after an OA pulse, and a subsequent decrease in TG and DG after the chase (Fig. 5, F and G). At each timepoint, there were no statistically significant differences in total TG or the abundance of any other measured lipid classes between control and APOE knockdown cells (Fig. 5, F and G; and Fig. S5, A and B). This matches our previous microscopy data showing that the total LD area is unchanged upon APOE depletion. However, APOE knockdown cells were markedly enriched in highly unsaturated TG species (Fig. 5 H). Thus, while APOE depletion does not significantly alter the total amount of cellular TG, it does result in the selective enrichment of polyunsaturated TG species.
APOE knockdown alters the composition and size distribution of LDs during lipogenesis and subsequent lipolysis (Fig. 5 H). However, global depletion of APOE via siRNA abrogates both LD-associated APOE and APOE in the secretory pathway, which is still observed in a subset of cells upon OA treatment (Fig. 1 C). It is possible that the LD phenotypes we observed upon APOE knockdown are in part due to the indirect effects of disrupting the balance of lipoprotein-based lipid secretion and uptake. To dissect the function of specifically LD-associated APOE, we performed rescue experiments with full-length APOE or a chimeric version of APOE that targets only LDs. We made two rescue constructs, one in which synonymous mutations were introduced into the APOE sequence to impart RNAi resistance and an “LD-only” chimeric construct in which the signal peptide of APOE was replaced with the LD-targeting hairpin domain of GPAT4 (Fig. 6 A). The siRNA used in the knockdown studies targets the region of the APOE mRNA that encodes the signal peptide, and, therefore, removing the signal peptide makes this construct also resistant to knockdown. Both rescue constructs were tagged with HA at the C-terminus. The inclusion of a C-terminal tag prevented the APOE antibody from binding to its epitope, allowing us to distinguish between endogenous and exogenous HA-tagged APOE using anti-APOE or anti-HA antibodies (Fig. 6 B). After APOE depletion, cells were transduced with lentivirus carrying either the full-length RNAi-resistant APOE or LD-only APOE and subjected to an OA pulse-chase. Western blot confirmed the depletion of endogenous APOE and expression of both HA-tagged rescue constructs (Fig. 6 B). Expression of either the RNAi-resistant full-length or the LD-only version of APOE fully rescued the LD size phenotype observed upon APOE knockdown (Fig. 6, C–F). This supports a role for LD-associated APOE in modulating LD metabolism.

LD-associated APOE modulates LD size. (A) Cartoon illustrating the conditions used in the APOE rescue experiment. Endogenous APOE3 protein is present in cells transfected with a non-targeting siRNA. APOE protein is depleted upon APOE knockdown. The siRNA used to knock down APOE targets the mRNA sequence encoding the N-terminal signal peptide. The RNAi-resistant full-length APOE (RR FL) rescue construct consists of APOE3 with synonymous mutations in the signal peptide that impart resistance to the APOE siRNA. The LD-only APOE construct has the signal sequence removed, making it insensitive to the APOE siRNA, and replaced with the hairpin domain of the LD protein GPAT4. This version of APOE only targets LDs and never enters the ER lumen. (B) Western blot of lysates of TRAE3-H cells transfected with the indicated siRNA and transduced with the indicated lentivirus. The same samples were run on two separate SDS-PAGE gels, with 10 µg of total protein loaded into each well. Gels were transferred and then blotted with anti-HA or an anti-APOE antibody together with an anti-tubulin antibody. Both the RR FL and LD-only APOE constructs were expressed in an endogenous APOE knockdown background. Moreover, the HA tag obstructs the epitope of the APOE antibody, allowing endogenous APOE and exogenous, HA-tagged APOE to be distinguished. (C) Representative confocal slices of cells transfected with non-targeting siRNA or APOE siRNA and transduced with an empty vector control, RR FL APOE, or LD-only APOE. Cells were subjected to an OA pulse-chase as described in Fig. 5A, fixed, and stained for LDs with BODIPY 493/503. Scale bar, 10 µm. (D–F) Quantification of LD parameters for the conditions described in A after an OA pulse-chase assay. (D) Total LD area was measured as the area of the entire LD mask per cell in µm2. (E) Average LD size was calculated as the mean LD area per cell in µm2. (F) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 60 cells per condition and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method and Bonferonni-corrected for multiple comparisons. Source data are available for this figure: SourceData F6.
LD-associated APOE modulates LD size. (A) Cartoon illustrating the conditions used in the APOE rescue experiment. Endogenous APOE3 protein is present in cells transfected with a non-targeting siRNA. APOE protein is depleted upon APOE knockdown. The siRNA used to knock down APOE targets the mRNA sequence encoding the N-terminal signal peptide. The RNAi-resistant full-length APOE (RR FL) rescue construct consists of APOE3 with synonymous mutations in the signal peptide that impart resistance to the APOE siRNA. The LD-only APOE construct has the signal sequence removed, making it insensitive to the APOE siRNA, and replaced with the hairpin domain of the LD protein GPAT4. This version of APOE only targets LDs and never enters the ER lumen. (B) Western blot of lysates of TRAE3-H cells transfected with the indicated siRNA and transduced with the indicated lentivirus. The same samples were run on two separate SDS-PAGE gels, with 10 µg of total protein loaded into each well. Gels were transferred and then blotted with anti-HA or an anti-APOE antibody together with an anti-tubulin antibody. Both the RR FL and LD-only APOE constructs were expressed in an endogenous APOE knockdown background. Moreover, the HA tag obstructs the epitope of the APOE antibody, allowing endogenous APOE and exogenous, HA-tagged APOE to be distinguished. (C) Representative confocal slices of cells transfected with non-targeting siRNA or APOE siRNA and transduced with an empty vector control, RR FL APOE, or LD-only APOE. Cells were subjected to an OA pulse-chase as described in Fig. 5A, fixed, and stained for LDs with BODIPY 493/503. Scale bar, 10 µm. (D–F) Quantification of LD parameters for the conditions described in A after an OA pulse-chase assay. (D) Total LD area was measured as the area of the entire LD mask per cell in µm2. (E) Average LD size was calculated as the mean LD area per cell in µm2. (F) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 60 cells per condition and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method and Bonferonni-corrected for multiple comparisons. Source data are available for this figure: SourceData F6.
APOE4 expression results in large LDs with impaired turnover
We repeated the OA pulse-chase assay in targeted replacement astrocytes expressing human APOE3 (TRAE3-H) or APOE4 (TRAE4-H) to test the effect of APOE4 on LD biogenesis and turnover. APOE4 targeted LDs upon OA treatment to the same degree as APOE3, indicating that the E4 mutation does not compromise LD targeting (Fig. S4, A–C). There were no significant differences in total LD area or LD number between TRAE3-H and TRAE4-H astrocytes at baseline, although E4 LDs were slightly larger (Fig. 7, A–D). After OA pulse, E4 LDs were larger and fewer than E3 LDs, yet the total LD area remained the same, similar to the knockdown phenotype (Fig. 7, A–D). However, after OA pulse-chase, E4 cells had a greater total LD area and larger LDs than E3, but no difference in LD number. This suggests that during LD biogenesis, neutral lipids are partitioned into a smaller number of larger LDs in E4, similar to the phenotype we observed upon APOE knockdown. However, unlike the knockdown, the large LDs in APOE4 cells displayed impaired turnover, as the total LD area was greater in APOE4 cells after pulse chase.

Targeting to LDs is unaffected in APOE4. (A) Representative confocal images of TRAE3-H cells or TRAE4-H cells with or without 5 h of OA loading. Cells were fixed and stained for endogenous APOE with anti-APOE antibody and for LDs with BODIPY 493/503. In merged images, APOE is in green, and LDs are in magenta. Scale bars, 10 µm. CM, complete media. +OA, 400 µM OA for 5 h. (B) Percentage of TRAE3-H or TRAE4-H cells with APOE on the surface of LDs after 5 h of OA. Each data point is the percentage of cells from 10 random fields of view with APOE on LDs in one experiment. APOE localization to LDs was determined qualitatively. N = 3 biologically independent experiments with 50 cells per experiment. ns, P >0.05. P value was calculated using an unpaired, two-sample t test. (C) LD enrichment fraction of TRAE3-H or TRAE4-H cells treated with OA for 5 h. LD enrichment fraction was calculated as described in Fig. 1. N = 50 cells per condition and experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. These are the same cells used in B, but LD enrichment was measured using an unbiased quantitative method rather than being assessed qualitatively. ns, P > 0.05. P value was calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method.

Targeting to LDs is unaffected in APOE4. (A) Representative confocal images of TRAE3-H cells or TRAE4-H cells with or without 5 h of OA loading. Cells were fixed and stained for endogenous APOE with anti-APOE antibody and for LDs with BODIPY 493/503. In merged images, APOE is in green, and LDs are in magenta. Scale bars, 10 µm. CM, complete media. +OA, 400 µM OA for 5 h. (B) Percentage of TRAE3-H or TRAE4-H cells with APOE on the surface of LDs after 5 h of OA. Each data point is the percentage of cells from 10 random fields of view with APOE on LDs in one experiment. APOE localization to LDs was determined qualitatively. N = 3 biologically independent experiments with 50 cells per experiment. ns, P >0.05. P value was calculated using an unpaired, two-sample t test. (C) LD enrichment fraction of TRAE3-H or TRAE4-H cells treated with OA for 5 h. LD enrichment fraction was calculated as described in Fig. 1. N = 50 cells per condition and experiment. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. These are the same cells used in B, but LD enrichment was measured using an unbiased quantitative method rather than being assessed qualitatively. ns, P > 0.05. P value was calculated using a clustered Wilcox rank sum test via the Rosner–Glynn–Lee method.

APOE4 promotes large LDs with highly unsaturated triglyceride and impaired turnover. (A) Representative confocal slices of TRAE3-H or TRAE4-H cells labeled for LDs with BODIPY 493/503 and imaged live at each timepoint of the OA pulse-chase assay described in Fig. 5 A. Scale bar, 10 µm. (B–D) Quantification of LD parameters in TRAE3-H or TRAE4-H cells at each timepoint of the OA pulse-chase assay. (B) Total LD area was measured as the area of the entire LD mask per cell in µm2. (C) Average LD size was calculated as the mean LD area per cell in µm2. (D) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 90 cells per genotype, timepoint, and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner-Glynn-Lee method and Bonferonni-corrected for multiple comparisons. (E) Comparison of total triglyceride between TRAE3-H and TRAE4-H cells. Lipidomics data were collected from two independently performed experiments, which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors (purple and yellow) indicate data collected from the same independently performed experiment. There is significantly more total triglyceride in E4 cells at the pulse-chase timepoint. * P < 0.05, ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. (F) Heatmap showing the relative abundance of measured lipid classes at each timepoint of the assay. Heatmap values are derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were then grouped by lipid class and Z-score normalized. The green box frame lipid species that are enriched in E4, while the red box frames lipid species enriched in E3. PG, phosphatidylglycerol; ChE, cholesterol ester; PI, phosphatidylinositol; PC, phosphatidyl choline; PE, phosphatidyl ethanolamine; DG, diacylglycerol; TG, triacylglycerol. (G) Heatmap of the abundance of triglyceride species separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The green box frames lipid species enriched in E4.
APOE4 promotes large LDs with highly unsaturated triglyceride and impaired turnover. (A) Representative confocal slices of TRAE3-H or TRAE4-H cells labeled for LDs with BODIPY 493/503 and imaged live at each timepoint of the OA pulse-chase assay described in Fig. 5 A. Scale bar, 10 µm. (B–D) Quantification of LD parameters in TRAE3-H or TRAE4-H cells at each timepoint of the OA pulse-chase assay. (B) Total LD area was measured as the area of the entire LD mask per cell in µm2. (C) Average LD size was calculated as the mean LD area per cell in µm2. (D) Number of LDs per cell. Each data point represents one cell, and each color represents data collected from a separate, independent experiment. N = 90 cells per genotype, timepoint, and independent experiment. ns P > 0.05, **** P < 0.0001. P values were calculated using a clustered Wilcox rank sum test via the Rosner-Glynn-Lee method and Bonferonni-corrected for multiple comparisons. (E) Comparison of total triglyceride between TRAE3-H and TRAE4-H cells. Lipidomics data were collected from two independently performed experiments, which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors (purple and yellow) indicate data collected from the same independently performed experiment. There is significantly more total triglyceride in E4 cells at the pulse-chase timepoint. * P < 0.05, ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. (F) Heatmap showing the relative abundance of measured lipid classes at each timepoint of the assay. Heatmap values are derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were then grouped by lipid class and Z-score normalized. The green box frame lipid species that are enriched in E4, while the red box frames lipid species enriched in E3. PG, phosphatidylglycerol; ChE, cholesterol ester; PI, phosphatidylinositol; PC, phosphatidyl choline; PE, phosphatidyl ethanolamine; DG, diacylglycerol; TG, triacylglycerol. (G) Heatmap of the abundance of triglyceride species separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The green box frames lipid species enriched in E4.
To test how APOE4 expression alters cellular lipid metabolism, we performed whole-cell lipidomics on TRAE3-H or TRAE4-H cells at each timepoint of the OA pulse-chase assay. Again, we observed the expected increase in TG and DG after the OA pulse and subsequent decrease following the chase (Fig. 7, E and F). Notably, however, there was significantly more TG in E4 cells after the chase than in E3 cells, corroborating our microscopy data (Fig. 7, E and F). Similar to APOE-depleted cells, we also observed a particularly striking enrichment of unsaturated TG species after the chase in E4 cells (Fig. 7 G). Although there were no statistically significant differences in the abundance of any lipid classes other than TG, we observed that total cellular PC and PE were modestly decreased in E4 cells at the pulse-chase timepoint (Fig. 7 F). Examining the abundance of PE species by saturation revealed that E4 cells have markedly fewer PE species with 3, 4, or 5 double bonds (Fig. S5 D). With respect to LDs, the APOE4 phenotype is in the same direction as APOE knockdown, yet, more severe. Like APOE-depleted cells, E4 cells form a smaller number of large LDs that contain more unsaturated triglycerides. However, E4 LDs additionally exhibit reduced turnover.

Untargeted lipidomics data of other lipid species from APOE knockdown and APOE3 versus APOE4 experiments. (A) Comparison of the total abundance of other major lipid classes between non-targeting (NT) and APOE knockdown (KD) cells at each timepoint. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. There is no significant difference in the total abundance of any other major lipid classes between NT and APOE KD at any timepoint. ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. ChE, cholesterol ester; DG, diacylglycerol; PC, phosphatidyl choline; PE, phosphatidyl ethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol. (B) Heatmap of the abundance of phosphatidylethanolamine (PE) species in NT versus APOE KD cells separated by their degree of unsaturation at each timepoint of the assay. Heatmap values are derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. (C) Comparison of the total abundance of other major lipid classes between APOE3 and APOE4 cells at each timepoint. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. Other than TG, there is no significant difference in the total abundance of any major classes between E3 and E4 at any timepoint. (D) Heatmap of the abundance of PE species in E3 versus E4 cells separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The red box frames lipid species enriched in E3.

Untargeted lipidomics data of other lipid species from APOE knockdown and APOE3 versus APOE4 experiments. (A) Comparison of the total abundance of other major lipid classes between non-targeting (NT) and APOE knockdown (KD) cells at each timepoint. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. There is no significant difference in the total abundance of any other major lipid classes between NT and APOE KD at any timepoint. ns P > 0.05. P values were calculated using the Wilcox rank sum test and Bonferonni-corrected for multiple comparisons. ChE, cholesterol ester; DG, diacylglycerol; PC, phosphatidyl choline; PE, phosphatidyl ethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol. (B) Heatmap of the abundance of phosphatidylethanolamine (PE) species in NT versus APOE KD cells separated by their degree of unsaturation at each timepoint of the assay. Heatmap values are derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. (C) Comparison of the total abundance of other major lipid classes between APOE3 and APOE4 cells at each timepoint. Lipidomics data were collected from two independently performed experiments which each used three separate plates of cells as technical replicates. Each data point denotes a single technical replicate, and the dot colors indicate data collected from the same independently performed experiment. Other than TG, there is no significant difference in the total abundance of any major classes between E3 and E4 at any timepoint. (D) Heatmap of the abundance of PE species in E3 versus E4 cells separated by their degree of unsaturation at each timepoint of the assay. Heatmap values were derived from the means of three technical replicates from two independently performed experiments (shown as separate columns) for each condition. Means were grouped by lipid class and Z-score normalized. The red box frames lipid species enriched in E3.
APOE4 LDs are more sensitive to lipid peroxidation
The physiological consequence of accumulating unsaturated TG in large LDs as observed in APOE4 cells is unclear. Previous studies of LDs reconstituted in vitro demonstrated that having more unsaturated TG promotes larger droplets, similar to the phenotype we observed in cells (Lange et al., 2021). Moreover, these unsaturated LDs exhibit increased sensitivity to lipid peroxidation in vitro. We hypothesized that the enrichment of unsaturated TG in E4 LDs makes them more sensitive to lipid peroxidation. To test this hypothesis, we first performed the OA pulse-chase treatment in APOE3 or APOE4 cells as previously described. We then loaded cells with BODIPY C11—a fluorescent lipid peroxidation sensor (Fig. 8 A). BODIPY C11 contains an 11-carbon fatty acid moiety that allows it to incorporate into lipid membranes (Drummen et al., 2002). BODIPY C11 shifts its fluorescence emission wavelength from red to green when oxidized in the presence of lipid peroxides, and the ratio of green to red BODIPY C11 fluorescence acts as a readout of lipid peroxidation in cell membranes. We found that after loading BODIPY C11 into cells for 30 min followed by a 2-h chase in Hanks’ balanced salt solution (HBSS), the probe accumulated in LDs. The bright BODIPY C11 signal of LDs was easily segmented from the rest of the cell, allowing for the measurement of peroxidation specifically in LDs. To test whether APOE4 increases sensitivity to lipid peroxidation, we subjected TRAE3-H or TRAE4-H cells to an OA pulse-chase, loaded LDs with BODIPY C11, and treated cells with vehicle (EtOH) or cumene hydroperoxide (CHP), a drug that causes lipid peroxidation. When treated with EtOH, both genotypes exhibited minimal lipid peroxidation with no significant difference between E3 and E4. However, LDs in APOE4 cells treated with CHP showed a much greater ratio of green to red fluorescence than LDs in CHP-treated APOE3 cells (Fig. 8, B–D). This suggests that the larger, unsaturated LDs of APOE4 cells are more sensitive to lipid peroxidation than LDs in APOE3 cells.

APOE4 LDs are more sensitive to lipid peroxidation. (A) Schematic of a BODIPY C11-based assay for measuring the lipid peroxidation sensitivity of LDs. Cells are first subjected to an OA pulse-chase as described in Fig. 5 A, washed with HBSS, and then loaded with 2 µM BODIPY C11 in HBSS for 30 min. After 30 min, BODIPY C11-containing HBSS is replaced with C11-free HBSS, and cells are incubated for 2 h, during which time BODIPY C11 incorporates into LDs. Cells are then treated with 0.2% ethanol vehicle (EtOH) or 200 µM cumene hydroperoxide (CHP) for 2 h and subsequently imaged. (B) Representative confocal slices showing TRAE3-H or TRAE4-H cells labeled with BODIPY C11 and treated with either 0.2% EtOH or 200 µM cumene hydroperoxide as described in A. The magenta channel shows reduced BODIPY C11 fluorescence, and the green channel shows the fluorescence of BODIPY C11 oxidized by lipid peroxides. Scale bar, 10 µm. (C) Inset of B shows the difference in peroxidation of LDs between E3 and E4 cells. Scale bar, 1 µm. (D) Quantification of peroxidation in LDs in E3 or E4 cells treated with EtOH or CHP. The ratio was calculated by dividing the mean fluorescence intensity of green (oxidized) BODIPY C11 fluorescence in the LD mask divided by the mean intensity of red (reduced) BODIPY C11 fluorescence. ns P > 0.05, **** P < 0.0001. N = 50 cells per condition. Each data point represents one cell. Data were collected and pooled from three independent experiments. P values were calculated via the Wilcoxon rank-sum test and Bonferonni-corrected for multiple comparisons.
APOE4 LDs are more sensitive to lipid peroxidation. (A) Schematic of a BODIPY C11-based assay for measuring the lipid peroxidation sensitivity of LDs. Cells are first subjected to an OA pulse-chase as described in Fig. 5 A, washed with HBSS, and then loaded with 2 µM BODIPY C11 in HBSS for 30 min. After 30 min, BODIPY C11-containing HBSS is replaced with C11-free HBSS, and cells are incubated for 2 h, during which time BODIPY C11 incorporates into LDs. Cells are then treated with 0.2% ethanol vehicle (EtOH) or 200 µM cumene hydroperoxide (CHP) for 2 h and subsequently imaged. (B) Representative confocal slices showing TRAE3-H or TRAE4-H cells labeled with BODIPY C11 and treated with either 0.2% EtOH or 200 µM cumene hydroperoxide as described in A. The magenta channel shows reduced BODIPY C11 fluorescence, and the green channel shows the fluorescence of BODIPY C11 oxidized by lipid peroxides. Scale bar, 10 µm. (C) Inset of B shows the difference in peroxidation of LDs between E3 and E4 cells. Scale bar, 1 µm. (D) Quantification of peroxidation in LDs in E3 or E4 cells treated with EtOH or CHP. The ratio was calculated by dividing the mean fluorescence intensity of green (oxidized) BODIPY C11 fluorescence in the LD mask divided by the mean intensity of red (reduced) BODIPY C11 fluorescence. ns P > 0.05, **** P < 0.0001. N = 50 cells per condition. Each data point represents one cell. Data were collected and pooled from three independent experiments. P values were calculated via the Wilcoxon rank-sum test and Bonferonni-corrected for multiple comparisons.
Discussion
Despite being the greatest genetic risk factor for Alzheimer’s disease, the detailed trafficking mechanisms of APOE in astrocytes have not previously been studied. Here, we show that APOE can divert from secretion and instead target LDs, where it modulates LD size and triglyceride saturation. LD-associated APOE coats the cytoplasmic-facing monolayer surface of LDs and targets LDs from the cytoplasmic face of the ER membrane at ER–LD membrane bridges (Fig. 9 A). LD-associated APOE is required during lipogenesis for maintaining LD size, while the loss of APOE, or expression of APOE4, causes the accumulation of polyunsaturated TG molecules within LDs. Importantly, the large unsaturated LDs in APOE4-expressing cells exhibit increased sensitivity to lipid peroxidation (Fig. 9 B). The discovery that APOE traffics to LDs opens up many new avenues of investigation. What is the mechanism by which APOE is able to divert secretion and instead target LDs? How does LD-associated APOE regulate LD physiology? How do the LD defects observed in APOE4 glial cells contribute to Alzheimer’s pathology?

APOE targets cytoplasmic LDs via the ER and modulates LD composition and size. Schematic of our model of LD-associated APOE trafficking and function. (A) Normally, APOE is translocated into the lumen of the ER, where it assembles with nascent lipoprotein particles and is secreted. Under conditions that stimulate LD biogenesis, APOE subverts translocation via an unknown mechanism (retaining its signal peptide) and localizes to the cytoplasmic face of the ER membrane. It then moves onto LDs via membrane bridges between LDs and the ER. (B) On LDs, APOE is required to maintain triglyceride saturation and a dispersed LD size phenotype. In APOE4 cells, LDs are larger, accumulate highly unsaturated triglycerides, have impaired turnover, and are more sensitive to lipid peroxidation. We hypothesize that defects in the function of LD-associated APOE4 promote lipid dishomeostasis and sensitize astrocytes to stress, which could facilitate the progression of Alzheimer’s pathology.
APOE targets cytoplasmic LDs via the ER and modulates LD composition and size. Schematic of our model of LD-associated APOE trafficking and function. (A) Normally, APOE is translocated into the lumen of the ER, where it assembles with nascent lipoprotein particles and is secreted. Under conditions that stimulate LD biogenesis, APOE subverts translocation via an unknown mechanism (retaining its signal peptide) and localizes to the cytoplasmic face of the ER membrane. It then moves onto LDs via membrane bridges between LDs and the ER. (B) On LDs, APOE is required to maintain triglyceride saturation and a dispersed LD size phenotype. In APOE4 cells, LDs are larger, accumulate highly unsaturated triglycerides, have impaired turnover, and are more sensitive to lipid peroxidation. We hypothesize that defects in the function of LD-associated APOE4 promote lipid dishomeostasis and sensitize astrocytes to stress, which could facilitate the progression of Alzheimer’s pathology.
The molecular mechanism by which APOE diverts from translocation into the ER lumen and instead targets LDs is unclear. Several other secreted proteins with lipid-binding moieties can reroute to LDs. APOV and APOCIII have been shown to go to LDs (Shu et al., 2010; Gao et al., 2012; Sołtysik et al., 2019). One study demonstrated that the innate immune protein cathelicidin reroutes to LDs in response to OA loading or lipopolysaccharide treatment, retaining its N-terminal signal peptide (Bosch et al., 2020). Another apolipoprotein, APOB, possesses pause transfer sequences that cause it to stall during in vitro translocation, and parts of its nascent polypeptide chain become exposed to the solution (Chuck et al., 1990; Kivlen et al., 1997). Pausing during translocation could act as a checkpoint for topological decision-making during translocation. The ER membrane complex (EMC) is involved in the topological mediation of integral ER proteins (Hegde 2022). Proteins with uncleaved signal anchors can adopt orientations in which their N-terminus faces the cytoplasm or the ER lumen, a process regulated by EMC (Wu and Hegde 2023). EMC has also been implicated in topological decision-making of ER hairpin proteins that target LDs (Leznicki et al., 2022). Given the vast heterogeneity of targeting mechanisms during translocation, it is possible that some normally secreted proteins such as APOE utilize an uncharacterized conserved pathway to undertake alternative topological fates.
After avoiding translocation, APOE traffics to LDs from the cytoplasmic surface of the ER via membrane bridges. A recent study of the mechanism of protein trafficking from the ER to LDs identified both “early” and “late” ERTOLD pathways (Song et al., 2022). Given that we observe APOE coating LDs several hours into OA treatment, we hypothesize that it may move onto LDs via the late pathway. However, unlike most ERTOLD proteins, LD binding of APOE is mediated by an amphipathic alpha helix rather than a hairpin domain. Deletion of the C-terminal amphipathic helical region of APOE ablates LD targeting. However, the C-terminus of APOE alone is less efficient at LD targeting than the full-length protein, suggesting a potential complementary role of the N-terminus. The N-terminal domain consists of four amphipathic alpha helices that form a bundle in solution (Chen et al., 2021). The N-terminal domain of APOE bound to the ER or LDs could retain this bundled fold or adopt an unfolded conformation, with the N-terminal amphipathic helices also mediating membrane association. This could explain the inefficient LD targeting observed in the C-terminal-only construct.
APOE depletion and APOE4 expression both caused a smaller number of large LDs to form upon OA loading. We speculate that this is caused by the coalescence of smaller LDs into larger ones at the early stages of LD biogenesis in APOE knockdown and APOE4 cells. These large LDs are also more enriched in unsaturated TG species, while E4 cells have a reduction in unsaturated PE species. One possibility is that, in E4 cells, polyunsaturated fatty acids cleaved from PE via phospholipase A2 are re-esterified to triglycerides and stored in LDs, a mechanism that has previously been shown to protect against lipotoxic stress (Jarc et al., 2018). However, the precise role of LD-associated APOE in controlling TG saturation and LD size is unclear. One possibility is that APOE itself stabilizes LDs and prevents their coalescence. The amphipathic alpha helices of APOE’s C-terminal domain could bind to monolayer packing defects and reduce surface tension. In the absence of APOE, higher surface tension would make lowering the surface area to volume ratio of LDs via Oswalt ripening more thermodynamically favorable (Thiam et al., 2013). The increase in unsaturated TG observed upon APOE knockdown could be a downstream consequence of a change in LD size distribution, potentially due to impaired turnover of unsaturated TGs in larger droplets. Another possibility is that APOE acts as a scaffold, and the effects of APOE on LDs are due to a protein–protein interaction. The N-terminal domain of APOE forms a four-helix bundle that is homologous to the four-helix bundle domain of the perilipin family of LD proteins (Hickenbottom et al., 2004). In perilipins, this domain is involved in recruiting adipose triglyceride lipase to the surface of LDs. APOE has a patch of basic residues in helix 4 of its N-terminal domain that has previously been shown to mediate interactions between APOE and lipoprotein receptors. APOE may interact with other proteins on the surface of LDs, and this interaction could mediate the downstream effects of APOE on LD composition and size.
There is no consensus model regarding how APOE4 increases an individual’s risk for developing Alzheimer’s disease. Prior work on APOE has focused on resolving how secreted APOE4 could lead to neurodegeneration (Martens et al., 2022). In this paper, we show that another pool of APOE exists on LDs that could also drive pathology. Further work is necessary to integrate the contributions of both pools of APOE to lipid homeostasis and Alzheimer’s disease. APOE4 promotes the accumulation of unsaturated triglycerides in LDs. The large, unsaturated droplets in APOE4 cells have impaired turnover and increased sensitivity to lipid peroxidation. Ischemia is correlated with both increased lipid peroxidation and neutral lipid accumulation in the brain (Yoshida et al., 1982; Gasparovic et al., 2001; Ioannou et al., 2019). Instances of ischemia are a major risk factor for Alzheimer's disease, and increased sensitivity to hypoxic insult may play a role in driving pathology in APOE4 (Elman-Shina and Efrati 2022). An accumulation of large LDs that are difficult to turnover and accumulate lipid peroxides could stimulate astrocytes to adopt a proinflammatory reactive state that precedes neurodegeneration. Lipid accumulation in other brain cell types has also been linked to neurodegeneration. LDs rich in cholesterol esters drive the accumulation of phosphorylated tau in iPSC-derived neurons (van der Kant et al., 2019). Aged microglia accumulate LDs and adopt a proinflammatory state (Marschallinger et al., 2020). The presence of these lipid-laden microglia correlates with an impaired response to stroke in aged mice (Arbaizar-Rovirosa et al., 2023). More recent work has also shown that microglia near amyloid beta plaques accumulate LDs in vivo (Haney et al., 2023, Preprint; Prakash et al., 2023, Preprint). Since our results in HMC3 microglial cells suggest that APOE can traffic to LDs in microglia, we speculate that LD-associated APOE4 in microglia could also play a key role in Alzheimer's disease pathology. This work adds to the body of evidence that is beginning to uncover key connections between APOE and intracellular glial lipid metabolism in driving Alzheimer’s disease.
Materials and methods
Antibodies and chemicals
The following antibodies were used in this study. Dilutions used for Western blot (WB), immunofluorescence (IF), and immunogold (IG) are given in parenthesis along with the manufacturer, catalog number, and RRID.
Primary antibodies: recombinant rabbit anti-APOE mAb (1:300 IF; 1:50 IG; 1:500 WB; Cat# ab52607, RRID:AB_867704; Abcam), mouse anti-HA-Tag mAb 6E2 (1:250 IF; 1:1,000 WB; Cat# 2367, RRID:AB_10691311; Cell Signaling Technology), mouse anti- DYKDDDDK Tag mAb 9A3 (1:200 IF; Cat# 8146, RRID:AB_10950495; Cell Signaling Technology), mouse anti-GM130 mAb (1:250 IF; Cat# 610822, RRID:AB_398141; BD Biosciences), mouse anti-Tubulin, and alpha mAb DM1A (1:10,000 WB; Cat# ab7291, RRID:AB_2241126; Abcam).
Secondary antibodies: donkey anti-rabbit IgG (H+L) Alexa Fluor 568 (1:500 IF; Cat# A10042, RRID:AB_2534017; Thermo Fisher Scientific), donkey anti-mouse IgG (H+L) Alexa Fluor Plus 647 (1:500 IF; Cat# A32787, RRID:AB_2762830; Thermo Fisher Scientific), donkey anti-rabbit IgG (H+L) IRDye 680RD (1:15,000 WB; Cat# 926-68073, RRID:AB_10954442; LI-COR Biosciences), donkey anti-mouse IgG (H+L) (H+L) IRDye 800CW (1:15,000 WB; Cat# 926-32212, RRID:AB_621847; LI-COR Biosciences), goat anti-rabbit IgG (H+L) Ultrasmall EM grade 0.8 nm colloidal gold (1:100 IG; Aurion, Cat# 25101; Electron Microscopy Sciences).
The following chemicals were used in this work: BODIPY 493/503 (Cat# D3922; Thermo Fisher Scientific), BODIPY 665/676 (Cat# B3932; Thermo Fisher Scientific), cycloheximide (Cat# C7698; Sigma-Aldrich), DBeQ (Cat# SML0031; Sigma-Aldrich), digitonin (Cat# D141; Sigma-Aldrich), fibronectin (Cat# F4759; Sigma-Aldrich), Image-iT Lipid Peroxidation Kit (Cat# C10445; Thermo Fisher Scientific), normal donkey serum (Cat# S30; Sigma-Aldrich), 16% PFA solution (EM grade) (Cat# 15710; EMS), polybrene (Cat# TR-1003-G; Sigma-Aldrich), poly-D lysine (1.0 mg/ml) (Cat# A-003-E; Sigma-Aldrich), protease inhibitor cocktail (Cat# P8340; Sigma-Aldrich), proteinase K (Cat# P8107S; NEB), saponin (Cat# AAA1882014; Fisher), sodium arachidonate (Cat# 10006607; Cayman), sodium linoleate (Cat# 21909; Cayman), and sodium oleate (Cat# O7501; Sigma-Aldrich).
Plasmids
mEmerald-N1 (_53976; Addgene) and mApple-SiT (_54948; Addgene) were kind gifts from Dr. Michael Davidson (Florida State University, Tallahassee, FL, USA). GFP-Plin2 was a kind gift from Dr. Alan Kimmel (described in Hsieh et al. [2012]; National Institute of Diabetes and Digestive and Kidney Disease, Bethesda, MD, USA). The lentiviral vector pTK881 was a kind gift from Dr. Tal Kafri (UNC-Chapel Hill, Chapel Hill, NC, USA). TagBFP2-KDEL (_49150; Addgene) was a kind gift from Dr. Gia Voeltz (University of Colorado Boulder, Boulder, CO, USA). LiveDrop-mEmerald was generated in our lab (Miner et al. 2022).
For plasmids constructed for this study, all PCRs were performed using Q5 High Fidelity DNA polymerase (M0419; New England Biolabs). A plasmid containing human APOE3-TurboGFP was purchased from Origene (Cat# RG200395). The APOE3 ORF was amplified from APOE3-TurboGFP and subcloned into an mEmerald-N1 backbone via Gibson assembly using HiFi DNA Assembly Master mix (E2621; New England Biolabs) to make APOE3-mEm. Only APOE3-mEm, not APOE3-TurboGFP, was used for experiments in this study. APOE truncation mutants (APOE3-mEm ΔSS, APOE3-N-mEm (1–191), APOE3-N-mEm ΔSS (19–191), APOE3-C-mEm, APOE3-C-mEm ΔSS were created from APOE3-mEm using Q5 Site-Directed Mutagenesis Kit (E0554S; New England Biolabs). The N-terminal domain constructs consist of residues 19–209 with or without residues 1–18 of the N-terminal signal peptide. C-terminal domain constructs consist of residues 234–317 with or without residues 1–18 of the N-terminal signal peptide. FLAG-ss-APOE3-mEm was created by adding an N-terminal FLAG tag to APOE3-mEm using Q5 site-directed mutagenesis. The web tool “Synonymous Mutation Generator” was used to determine an optimal RNAi-resistant APOE sequence (Ong, 2021, Preprint). Synonymous mutations were then introduced into APOE3-mEm using Q5 site-directed mutagenesis kit. For lentiviral production, the RNAi-resistant APOE3 ORF was subcloned into pTK881 using Gibson assembly with a C-terminal HA tag added with PCR. HP-APOE3-mEm was created by amplifying the hairpin domain of GPAT4 (152–208) from LiveDrop-mEm and adding it to the N-terminus of APOE3-mEm ΔSS via Gibson Assembly. For lentiviral production, HP-APOE3 ORF was subcloned into pTK881 via Gibson Assembly with a C-terminal HA tag added with PCR.
Cell culture and transfection
Immortalized targeted replacement astrocyte (TRAE3-H and TRAE4-H) cell lines were a kind gift from Dr. Patrick Sullivan of Duke University (described in Morikawa et al. [2005]). HMC3 cells were obtained from the UNC Tissue Culture facility. Targeted replacement astrocytes were maintained at 37°C and 5% CO2 in complete medium (CM) consisting of Dulbecco’s modified Eagle medium (DMEM) high glucose (Cat# 15-013-CV; Corning), supplemented with 10% fetal bovine serum (FBS, Cat# 97068-085; VWR), 2 mM glutamine (Cat# 25005-CI; Corning), and 1X penicillin/streptomycin (Cat# 30-002-CI; Corning). CM used for targeted replacement astrocytes was additionally supplemented with 200 µg/ml Geneticin (Cat# 10131027; Gibco). HMC3 cells were maintained at 37°C and 5% CO2 in Eagle’s Minimum Essential Medium supplemented with 10% FBS and 1× penicillin/streptomycin. Cells were confirmed free of mycoplasma and monitored at regular intervals using a Mycoplasma PCR Detection Kit (G238; ABM).
Primary rat cortical astrocyte cultures were prepared from neonatal (1–2-day old) Sprague–Dawley rats (Charles River) as previously described (McCarthy and De Vellis 1980; Stogsdill et al., 2017). Briefly, cortices from both sexes were microdissected and digested with papain (10 U/brain, Cat# LK003176l; Worthington Biochemical) for 25 min at 37°C. The tissue was washed three times with astrocyte growth media (AGM): DMEM high glucose (Cat# 15-013-CV0; Corning), 10% FBS (Cat# 97068-085; VWR), 2 mM L-glutamine (Cat# 25005-CI; Corning), 1X penicillin/streptomycin (Cat# 30-002-CI; Corning), 4 µg/ml hydrocortisone (Cat# 20739; Cayman Chemical), 5 µg/ml bovine insulin (Cat# I6634; Sigma-Aldrich), and 5 µg/ml N-acetyl-L-cysteine (Cat# 20261; Cayman Chemical) and gently aspirated to remove residual papain. The tissue was triturated in AGM with three fire-polished Pasteur pipettes with progressively narrower bores and filtered through a 70-μm cell strainer (Falcon). Equal volumes of the cell suspension were plated on 75-cm2 flasks (Flacon) coated with 10 µg/ml of poly-D-lysine. Cells were plated such that one set of cortices was plated per flask. Flasks were maintained at 37°C, 5% CO2 with complete media exchanges on DIV1 and DIV2. On DIV3, confluent flasks were smacked by hand three times in prewarmed PBS until the adherent astrocyte monolayer remained, and the flasks were replenished with AGM containing 10 µM AraC to limit the proliferation of contaminating cells. On DIV6, flasks were replenished with fresh medium and subcultured on DIV7.
For light microscopy, cells were seeded in eight-well chamber slides with #1.5 high-performance cover glass bottom (Cellvis C8-1.5H-N). 200 µl of filter-sterilized 10 or 50 µg/ml poly-D-lysine was added to each well of an eight-well chamber slide for seeding primary astrocytes or targeted replacement astrocytes respectively. Slides were incubated at 37°C for 30–45 min and rinsed three times with MilliQ water before seeding cells. For seeding HMC3 cells, 200 µl of 10 µg/ml fibronectin was added to each well and the slides were incubated at 37°C for 5 min and rinsed three times with MilliQ water before seeding. The following cell numbers were used for seeding in eight-well slides: TRAE3-H/TRAE4-H, 12,000 cells per well; HMC3, 15,000 cells per well; and primary astrocytes, 25,000 cells per well.
Plasmid DNA was transfected into all cell types using Lipofectamine 2000 (Cat# 11668019; Invitrogen) according to the manufacturer’s instructions. A 2:1 ratio of Lipofectamine to plasmid DNA was used for all transfections. siRNAs were transfected using DharmaFECT 1 (Cat# T-2001; Horizon Discovery) according to the manufacturer’s instructions. 5 µM siRNA was used for each transfection. Non-targeting (NT) siRNA (Cat# D-001810-04), APOE siRNA #1 (Cat# D-006470-02), and APOE siRNA #2 (Cat# D-006470-04) were purchased from Horizon Discovery. After transfection, cells were incubated in their corresponding “imaging media” with the same composition but lacking phenol red and antibiotics.
iAstro preparation and transfection
Cells and culture conditions
Human KOLF2.1J wildtype (wt) induced pluripotent stem cells (hiPSCs) were obtained from Bill Skarnes (The Jackson Laboratory) and were used to generate a hiPSCs-PB-TO-hSOX9/NFIA.1 cell line. Cells were incubated at 37°C, 5% CO2, and cultured under feeder-free conditions in StemFlex medium (A3349401; Gibco) on Vitronectin (VTN-N)-coated plates (VTN-N Recombinant Human Protein, Truncated, A14700; Gibco). The cells were passaged using ReLeSR (#100-0484; STEMCELL Technologies) in the presence of 0.5× ROCK inhibitor (A2644501; Gibco), except for thawing and cryopreservation, in which 1X ROCK inhibitor was employed.
Generation of PB-TO-hSOX9/NFIA.1-hiPSCs
An hiPSCs-PB-TO-hSOX9/NFIA.1 stable cell line was generated as previously described (Fernandopulle et al., 2018). Briefly, KOLF2.1J wt cells were transfected with 500 ng piggyBac plasmid carrying a bipartite hSOX9/NFIA.1 cassette (plasmids gifted from Michael Ward; NINDS). Transfected cells were selected for stable integration using Puromycin treatment (0.7 µg/ml) and propagated as a non-clonal pool.
Differentiation into iAstrocytes
hiPSCs-PB-TO-hNGN2 were rapidly differentiated into astrocytes using a method adapted from previous publications (Krencik and Zhang, 2011; Li et al., 2018). In brief, hiPSCs were dissociated into single cells using Accutase (A6964; Sigma-Aldrich) at high confluency (50–60%) on VTN-N coated plates in the presence of Induction Media (KO DMEM [10829018; Gibco], 1X GlutaMAX Supplement [35050061; Gibco], 0.5X B-27 [A1486701; Thermo Fisher Scientific], 0.5× N-2 [17502048; Thermo Fisher Scientific], 0.2% heparin [2 mg/ml heparin; #07980; STEMCELL Technologies], 2 μM SB431542 (16-141-0; Tocris Biosciences], 2 μM DMH-1 [41-261-0; Tocris Biosciences], 2 μM XAV939 [37-481-0; Tocris Biosciences], and 1 µM Doxycycline [D9891; Sigma-Aldrich]) in the presence of 50 nM Chroman-1 (71-631-0; Tocris Biosciences). Between 18 and 24 h, the media was replaced by induction media without the addition of Chroman-1. After 48 h, cells were passaged in Accutase and seeded onto VTN-N-coated plates in the presence of Chroman-1 in induction media. The following day the media was replaced with iAstrocyte Media without Chroman-1 (DMEM/F12 GlutaMAX [10565018; Gibco], 0.5× B-27 [A1486701; Thermo Fisher Scientific], 0.5X N-2 [17502048; Thermo Fisher Scientific], 0.2% heparin [2 mg/ml heparin; #07980; STEMCELL Technologies], and 1 µM Doxycycline [D9891; Sigma-Aldrich]), and it was fully replaced every second day. After 7 days of differentiation, cells were passaged using TrypLE Express Enzyme (1×) (12605010; Thermo Fisher Scientific) in iAstrocyte Media in the presence of Chroman-1 and seeded onto poly-L-ornithine (P3655; Sigma-Aldrich; 10× stock: 50 mg in 50 ml Borate Buffer) and laminin (5 µg/ml) (23017015; Gibco)-coated plates. The following day the media was changed to iAstrocyte maturation media (iAstrocyte Media with the addition of 10 ng/ml human CNTF [450-13; Peprotech], 10 ng/ml human LIF [300-05; Peprotech], and 1 µM Doxycycline [D9891; Sigma-Aldrich]) and fully replaced every second day. At day 12, ∼35,000 cells were seeded into eight-well chambers (C8-1.5H-N; Cellvis) previously coated with poly-L-ornithine and laminin as described above. Expression of astrocyte markers was tested at day 14 after induction, ALDH1L1 (1:500, ab190298; Abcam) and GFAP (1:500, MA5-12023; Thermo Fisher Scientific).
iAstrocyte transfection
iAstrocyte were transfected with 100 ng of APOE3-mEm using Lipofectamine 3000 (L3000001; Thermo Fisher Scientific) according to the manufacturer’s instructions. iAstrocyte Maturation Media was replaced with unsupplemented DMEM/F12 GlutaMAX before adding the transfection mix and incubating for 4 h. Media containing transfection mix was then replaced with fresh iAstrocyte Maturation Media. Cells were imaged 16 h after transfection.
Immunofluorescence sample preparation
Cells were washed twice in 1× PHEM buffer (60 mM PIPES, 27.3 mM HEPES, 8.22 mM MgSO4, 10 mM EGTA, and pH 7.0) and fixed for 10 min at room temperature in room temperature–equilibrated 4% PFA in 1× PHEM. After fixation, cells were washed three times in 1× PHEM and then permeabilized in 0.01% saponin in PHEM for 10 min at room temperature. Permeabilization buffer was then replaced with a blocking buffer (10% Normal Donkey Serum, 3% bovine serum albumin [BSA], 300 mM glycine, 0.01% saponin in PHEM) in which cells were incubated 30–45 min at room temperature. The blocking solution was removed and cells were washed twice with 0.01% saponin in PHEM before applying the primary antibody solution. Primary antibodies were diluted in antibody dilution solution (3% BSA, 0.01% saponin in PHEM). Cells were incubated in primary antibody solution for 24–48 h at 4°C. After primary antibody incubation, cells were washed with 0.01% saponin three times for 10 min each at room temperature. Secondary antibodies were diluted in antibody dilution solution together with 1 µg/ml BODIPY 493/503 to stain for LDs. Cells were incubated in secondary antibody solution for 1 h at room temperature and then washed three times in PHEM for 10 min each at room temperature. Cells were imaged immediately after preparing or stored at 4°C and imaged within a week of preparation.
Immunogold electron microscopy
Targeted replacement cells were grown on Nunc Lab-Tek Permanox chamber slides. Cells were treated with 400 µM OA for 5 h and fixed for 1 h at room temperature in 4% paraformaldehyde in 0.15 M sodium phosphate buffer (PB). After three washes in 0.15 M PB, the cells were immunostained using a modified protocol for pre-embedding immunogold-silver staining developed by Yi et al. (2001). Cell permeabilization and free-aldehyde inactivation were achieved using 0.1% saponin in 0.2 M glycine in 0.15 M PB for 30 min followed by a 1-h incubation in an Aurion Goat Blocking Solution (Aurion; Electron Microscopy Sciences). The cells were incubated in anti-APOE antibody overnight at 4°C (Rabbit anti-APOE, ab52607; Abcam) diluted 1:50 with IEM buffer (PB with 0.2% BSA-Ac/0.01% saponin). After washes in IEM buffer, the cells were incubated overnight at 4°C in a 1:100 dilution of Goat anti-rabbit IgG (H+L) Ultrasmall EM grade 0.8 nm colloidal gold (cat #25121-0.6 ml; Aurion; Electron Microscopy Sciences). Following washes in IEM buffer, the cells were then washed with PB, postfixed in 2% glutaraldehyde in 0.15 M PB, and held at 4° for several days. Cells were rinsed in 0.15 M PB and silver-enhanced for 90 min using an Aurion R-Gent SE-EM Silver Enhancement Kit (cat #25521, Aurion; Electron Microscopy Sciences), followed by postfixation in 0.1% osmium tetroxide for 15 min, gradient dehydration in ethanol and embedment in Polybed 812 epoxy resin (Polysciences, Inc.). Grid-mounted 80-nm ultrathin sections were stained with 4% uranyl acetate and examined by transmission electron microscopy using a JEOL JEM-1230 transmission electron microscope operating at 80 kV (JEOL USA) equipped with a Gatan Orius SC1000 CCD camera and Gatan Microscopy Suite 3.0 software (Gatan, Inc.).
Western blot
Cells were lysed on ice in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich) for 10 min. Adherent samples were scraped, collected, and centrifuged at 17,000 × g for 10 min at 4°C to isolate the postnuclear supernatant. Protein concentrations in samples were measured using a Detergent Resistant Bradford Assay (Thermo Fisher Scientific). Samples were prepared for SDS-PAGE in 6× Laemmli buffer and denatured for 5 min at 95°C. Equal protein amounts were loaded into each well of a NuPAGE 10%, Bis-Tris, 1.0 mm, Mini Protein Gel (Thermo Fisher Scientific). After SDS-PAGE, proteins were wet-transferred to nitrocellulose for 1 h at 100 V. Membranes were blocked in 5% milk in TBS for 30 min at room temperature and then incubated with primary antibodies diluted in 3% BSA in TBST overnight at 4°C. Membranes were washed three times with TBST and then incubated with secondary antibody solution for 1 h at room temperature, washed 3× with TBST, and imaged using an Odyssey CLx (LI-COR Biosciences).
ELISA
Intracellular and secreted APOE concentrations were determined using a human APOE ELISA kit (Cat# ab108813; Abcam) per the manufacturer’s instructions. Three independent biological replicates were conducted. For secreted APOE, 50 µl taken from 1 ml neat culture media was assayed. For intracellular APOE, cells were lysed in 100 µl of ELISA lysis buffer (10 mM Tris/Cl pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 0.5% tween; protease inhibitor cocktail), and 50 µl of lysate diluted 1:10 was assayed. Absorbance values were collected at 450 nm on a Synergy HT plate reader (BioTek).
Fluorescence protease protection assay
A fluorescence protease protection assay was performed using a modified version of the protocol outlined in Lorenz et al. (2006). Primary rat astrocytes were seeded on DIV7 and transfected with TagBFP2-KDEL and either mEm N1, APOE3-mEm, GFP-PLIN2, or LiveDrop-mEm. After transfection, media was replaced with imaging media supplemented with 50 ng/ml BODIPY 665/676. 24 h after transfection, cells were washed twice with KHMN (110 mM potassium acetate, 20 mM HEPES, 2 mM MgCl2, and 10 mM NaCl) buffer and placed in 150 µl KHMN. For each well, a field of view containing two to three cells transfected with TagBFP2-KDEL and mEm or the LD protein marker of interest was selected, and a baseline image was taken. Optimization experiments determined 30 µM digitonin was the lowest concentration that showed full loss of cytoplasmic mEmerald signal in most cells after 1 min, and this concentration was used in subsequent experiments. For each experiment, 150 µl of 60 µM digitonin in KHMN buffer was added to 150 µl of KHMN in the well for a final concentration of 30 µM digitonin and cells were allowed to permeabilize for 1 min. Cells were refocused and the “post-permeabilization” image was taken. 300 µl of 100 µg/ml proteinase K in KHMN was then added to the well for a final concentration of 50 µg/ml proteinase K. After 1 min, cells were refocused and the “post-PK” timepoint image was taken. Three to five wells per condition were performed per independent experiment, and data were collected and pooled from three independent experiments.
FRAP
FRAP experiments were conducted in primary rat cortical astrocytes transfected with APOE3-mEm and stained for LDs with BODIPY 665/676 as described above. Experiments were performed after treating cells for 4 h in antibiotic and phenol red-free AGM supplemented with 200 µM OA for 4 h (OA pulse) or cells treated with OA for 4 h followed by a 2-h chase in OA-free AGM. In each experiment, bleaching was conducted in 8–10 cells per condition, and only one APOE ring was bleached and analyzed per cell. Images were acquired once per second. Imaging started 5 s before bleaching and continued until 5 min after bleach.
OA pulse-chase assays
For APOE knockdown OA pulse-chase assays, 6,000 TRAE3-H cells were seeded in each well. 24 h after seeding, cells were transfected with 5 µM of the indicated siRNA with Dharmafect 1 using the manufacturer’s protocol in imaging media. 48 h after transfection, media was replaced with imaging media supplemented with 200 ng/ml BODIPY 493/503 (for baseline wells) or imaging media supplemented with 400 µM sodium oleate (for +OA and washout wells) and 200 ng/ml BODIPY 493/503. After 5 h of OA treatment, pulse-chase wells were replaced with imaging media, and “baseline” and “+OA” wells were imaged immediately.
For experiments comparing TRAE3-H and TRAE4-H cells, 12,000 cells were seeded in each well. 48 h after seeding, the OA pulse-chase assay was conducted as described above.
For rescue experiments, 6,000 TRAE3-H cells were seeded in each well. 24 h after seeding, cells were transfected with siRNA as indicated above. 24 h after siRNA transfection, cells were treated with the indicated lentivirus (UNC Lenti-shRNA Core Facility) diluted 1:10 in imaging media supplemented with 10 µg/ml polybrene. 48 h after transduction, the OA pulse-chase was performed, with cells prepared for immunofluorescence at each timepoint.
Lipid peroxidation assays
Cells first underwent the OA pulse-chase treatment as described above. 18 h after the CM chase, cells were washed once in HBSS (Cat# 14025092; Thermo Fisher Scientific) and then loaded with 2 µM BODIPY C11 for 30 min. After loading, cells were washed once with HBSS and incubated in HBSS for 2 h. Cells were then treated with 200 µM cumene hydroperoxide or 0.2% EtOH vehicle control for 2 h and immediately imaged live.
Light microscopy image acquisition
Confocal and Airyscan images were acquired using an inverted Zeiss 800/Airyscan single-point scanning confocal microscope equipped with 405, 488, 561, and 647 nm diode lasers, two gallium arsenide phosphide (GaAsp) detectors, and one Airyscan detector. Images were acquired using a Plan-Apo 63×/1.4 NA oil objective lens using ZEN Blue software. All live cell imaging was conducted at 37°C and 5% CO2.
Light microscopy image analysis
LD number and size analysis was performed using a semiautomated pipeline in Fiji (Schindelin et al., 2012). Individual cells were first manually segmented and the area outside of each cell was removed using the “Clear Outside” function. To segment LDs, the BODIPY channel was first passed through a Gaussian filter followed by a Laplacian of Gaussian (LoG) filter. The radius of the LoG filter and the Gaussian blur were heuristically optimized for each image to maximize segmentation accuracy. Autothresholding using the Otsu algorithm was then applied, followed by the binary operations “Open,” “Fill Holes,” and “Watershed.” Then, “Analyze Particles” was used to measure the number and size of segmented LDs, as well as the sum area of all LDs in the cell.
For colocalization analysis, the APOE was segmented by first applying a median filter with a radius of three pixels to the APOE channel followed by autothresholding using the “Moments” algorithm. The Golgi was masked by applying a median filter with a radius of two pixels to the GM130 channel followed by autothresholding using the “Huang2” algorithm. The Image Calculator function was used to create a mask of pixels where APOE and GM130 masks overlap. The Mander’s coefficient was calculated by dividing the area of the overlap mask by the area of the APOE mask.
To measure LD protein enrichment, the above algorithm was first applied to create an accurate mask of LDs in the cell. Then, the LD mask was expanded twice using the “Dilate Function” to include protein surrounding the surface of the LD. A second mask was created by subtracting the expanded LD mask from a mask of the entire cell. The mean intensity of the LD protein of interest was measured in the expanded LD mask as the subtracted whole-cell mask. LD ratios were calculated by dividing the mean intensity of the LD mask by the mean intensity of the Cell-LD mask.
and the coefficients A (the mobile fraction) and k (the rate constant) were extracted from each.
Lipidomics
Targeted replacement astrocytes were seeded in 6-cm dishes; three plates were collected per condition for each biological replicate. Two independent biological replicates were performed. Cells were washed three times with ice-cold PBS, scraped, and collected into 1.5-ml Eppendorf tubes. Cells were pipetted up and down 80–100 times on ice and counted using a Countess II automated cell counter (Thermo Fisher Scientific) for normalization; four counts per plate were collected and averaged to ensure accuracy. Cells were then pelleted at 1,000 × g for 10 min and flash-frozen in liquid nitrogen. Cell pellets were extracted via a liquid–liquid partition with water (200 µl), methanol (300 µl), and methyl-tertbutyl ether (1 ml). An internal standard, Equisplash by Avanti Polar Lipids, was spiked into the methanol used for extraction at 1.5 µg/ml. Samples were shaken for 15 min and then centrifuged at 20,000 rcf for 10 min. The top layer was dried and reconstituted with 150 µl of isopropanol for analysis.
Lipidomics analysis was performed using a Q Exactive HF-X (Thermo Fisher Scientific) coupled to a Waters Acquity H-Class UPLC. A 100 mm × 2.1 mm, 2.1 µm Waters BEH C18 column was used with the following mobile phases: A-60/40 ACN/H20 B-90/10 IPA/ACN; both mobile phases had 10 mM ammonium formate and 0.1% formic acid. A flow rate of 0.2 ml/min with a gradient starting at 32% B, which increased to 40% B at 1 min (held until 1.5 min) and then 45% B at 4 min, was used. This was increased to 50% B at 5 min, 60% B at 8 min, 70% B at 11 min, and 80% B at 14 min (held for 2 min). At 16 min, the composition switched back to starting conditions and was held for 4 min. Samples were analyzed in positive/negative switching ionization mode with top five data-dependent fragmentation utilizing a stepped collision energy of 25, 35, and 45 V. A resolution of 60,000 was utilized with a scan range of 200–1,200 m/z. Data-dependent acquisition was acquired at a resolution of 15,000 and an isolation window of 1.5 m/z.
Lipidomics data processing
LC-MS data were analyzed by LipidSearch 4.2 by Thermo Fisher Scientific and the peak area was normalized to the area of deuterated internal standards from the Avanti Equisplash mix as well as the number of cells. Lipids were identified by MS2 fragmentation with the following parameters: precursor tolerance 5 ppm, product tolerance 8 ppm, m-score threshold 2.0, and product relative intensity threshold 0.1%. The higher-energy C-trap dissociation and labeled databases were used for identification. The identifications were generated individually for each sample and then aligned by grouping the samples. Data were normalized to both internal standards and cell counts for each sample.
Statistical analysis
RStudio was used to perform all statistical analyses (RStudio Team, 2022). All plots were created using ggplot2 (Wickham 2009). The Rosner–Glynn–Lee variation of the Wilcoxon rank-sum test for clustered data was applied to assess statistical significance between groups from data collected from multiple independent experiments performed at different times. This test treats data from each independent experiment as a “cluster” and accounts for variability both within and between independent experimental trials (Rosner et al., 2006). The test was applied using the “clusrank” package in R and was used for LD enrichment in Figs. 1 and S4, LD parameter analysis in Figs. 5, 6, and 7, and colocalization analysis in Fig. 1 (Jiang et al., 2020).
For LD enrichment analysis in Fig. 2, data we pooled from three independent experiments and P values were calculated using standard Wilcox rank sum test and Bonferonni-corrected. Comparisons of FRAP data in Fig. 3 were conducted using Wilcox rank sum tests. For LD enrichment analysis in Fig. 4, the Dunn test was used to calculate pairwise P values, and significance groups were assigned based on these pairwise P values. Lipid peroxidation data in Fig. 9 was compared using Wilcox rank sum tests and Bonferonni-corrected for multiple comparisons.
For lipidomics experiments, two independent biological replicates were performed. Within each biological replicate, three separate plates were collected for each condition and considered as technical replicates. Normalized lipidomics data was log2 transformed, grouped by lipid class, and Z-score normalized in R. For saturation profile comparisons, species within individual lipid classes (e.g., TG, PE) were grouped by their degree of unsaturation and then Z-score normalized. The means of the three technical replicates for each condition and the biological replicates were used in heatmap visualizations. For boxplots, each technical replicate was treated as an individual data point, and conditions were statistically compared using Wilcox rank sum tests and Bonferonni-corrected for multiple comparisons.
Online supplemental material
Figs. S1 and S2 show supporting data for Fig. 1. Fig. S3 shows supporting data for Fig. 3. Fig. S4 shows supporting data for Fig. 7. Fig. S5 shows supporting data for Figs. 5 and 7. Video 1 and Video 2 show partial and full rings of APOE on LDs at ER-LD contact sites and correspond to Fig. 3. Video 3 and Video 4 show FRAP movies corresponding to Fig. 3. Table S1 contains raw lipidomics data from a biological replicate of one of the non-targeting siRNA versus APOE siRNA lipidomics experiments presented in Fig. 5. Table S2 contains raw lipidomics data from a biological replicate of two of the non-targeting siRNA versus APOE siRNA lipidomics experiments presented in Fig. 5. Table S3 contains raw lipidomics data from a biological replicate of one of the APOE3 versus APOE4 lipidomics experiments presented in Fig. 7. Table S4 contains raw lipidomics data from a biological replicate of two of the APOE3 versus APOE4 lipidomics experiments presented in Fig. 7.
Acknowledgments
We thank Patrick Sullivan (Duke University, Durham, NC, USA) and David Holtzman (Washington University in St. Louis, St. Louis, MO, USA) for providing the targeted replacement astrocyte cell lines; Victoria Madden of the Microscopy Services Laboratory for assistance with immunogold EM experiments; the University of North Carolina Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with mass spectrometry analysis; Wendy Salmon, director of the Hooker Imaging Core, for assistance with fast Airyscan imaging; Sunghyun Lee and Tal Kafri for lentivirus production; and Gregory Miner and current Cohen Lab members for helpful discussions and feedback on the manuscript.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R35GM133460 (S. Cohen), T32GM119999 (I.A. Windham), and T32GM133364 (A.E. Powers), by the National Institute on Aging under award number F31AG069419 (I.A. Windham), and by the Alzheimer’s Association under award number 2018-AARG-590347. Mass spectrometry work conducted by the University of North Carolina Mass Spectrometry Core Facility was supported by the National Science Foundation under Grant No. CHE-1726291.
Author contributions: I.A. Windham and S. Cohen conceptualized the project, designed the research, acquired funding, and wrote the manuscript. A.E. Powers performed ELISA experiments. J.V. Ragusa contributed to HMC3 experiments and prepared primary astrocyte cultures. E.D. Wallace performed lipid extraction, data processing, and mass spectrometry analysis. M.C. Zanellati and V.H. Williams generated the iAstrocyte lines and prepared iAstrocytes for the experiments in this study. C.H. Wagner contributed to APOE truncation experiments. K.K. White performed immunogold electron microscopy. I.A. Windham performed all other experiments in the manuscript. I.A. Windham performed image analysis and wrote scripts for image and statistical analysis. S. Cohen supervised the research.

