


STK19 was originally identified as a manganese-dependent serine/threonine-specific protein kinase, but its function has been highly debated. Here, the crystal structure of STK19 revealed that it does not contain a kinase domain, but three intimately packed winged helix (WH) domains. The third WH domain mediated homodimerization and double-stranded DNA binding, both being important for its nuclear localization. STK19 participated in the nucleotide excision repair (NER) and mismatch repair (MMR) pathways by recruiting damage repair factors such as RPA2 and PCNA. STK19 also bound double-stranded RNA through the DNA-binding interface and regulated the expression levels of many mRNAs. Furthermore, STK19 knockdown cells exhibited very slow cell proliferation, which cannot be rescued by dimerization or DNA-binding mutants. Therefore, this work concludes that STK19 is highly unlikely to be a kinase but a DNA/RNA-binding protein critical for DNA damage repair (DDR) and cell proliferation. To prevent further confusions, we renamed this protein as TWH19 (Tandem Winged Helix protein formerly known as STK19).
Introduction
Dating back to 1994, an MHC class III region gene (RP1 or G11) was reported to encode a 364-a.a. protein in several cell lines (the 41-kD isoform; Shen et al., 1994), but another group suggested that this gene encodes two proteins lacking the N-terminal 110 amino acids and with or without a 4-a.a. insertion (coding 254 or 258 a.a., named 29-kD STK19; Sargent et al., 1994). The encoded polypeptides showed limited sequence similarity to several proteins, one of which is the tyrosine kinase-transforming protein of Fujinami sarcoma virus (18.7% identity over 157 amino acids; Sargent et al., 1994). Both the 29- and 41-kD forms of the protein were reported to possess Mn2+-dependent protein kinase activity (Gomez-Escobar et al., 1998); hence, the protein was named STK19 (Yang et al., 2001).
However, the identity and functions of STK19 are highly debated. From 2012 to 2017, several groups reported a recurrent melanoma mutation in STK19, which was proposed to drive the progression of melanoma (Bonilla et al., 2016; Chang et al., 2016; Hodis et al., 2012). This mutation lies in the N-terminal region of 41-kD STK19 at D89 but outside the coding region of 29-kD STK19. In 2019, Yin et al. reported that the D89 mutation promoted NRAS phosphorylation and melanoma formation (Yin et al., 2019). Shortly after, Rodríguez-Martinez et al. claimed that STK19 is not an NRAS-dependent melanoma driver since 41-kD STK19 is not expressed, rendering the “D89 mutation” out of the coding region and functionally irrelevant in melanoma (Rodríguez-Martínez et al., 2020). Although no NRas-specific kinase activity of STK19 was detected, these data were unable to rule out STK19 as a kinase (Rodríguez-Martínez et al., 2020). In fact, several online databases (such as UniProt and The Human Protein Atlas) and papers published after the debate still refer to STK19 as a kinase protein (Asquith and Temme, 2020; Carrozza et al., 2021; Chelius et al., 2020; Chen et al., 2021; Duan et al., 2021; Garcia-Alvarez et al., 2021; Guo et al., 2021; Marino et al., 2022; Qiu et al., 2021).
To gain insights into the identity and molecular functions of STK19, we performed a combination of biochemical, structural, and cellular studies. Results showed that STK19 was highly unlikely to be a kinase but a DNA/RNA-binding protein. STK19 can form dimers and use the surface created by dimerization to engage double-stranded (ds) DNA/RNA. STK19 contributed to DNA damage repair (DDR) by recruiting many repair factors to damaged sites. Finally, STK19-downregulated cells displayed defects in mRNA expression, metabolism, and cell proliferation.
Results
STK19 is highly unlikely to be a kinase
Compared with the 29-kD isoform, the 41-kD isoform contains an extra N-terminal 110 amino acids that are not required for kinase activity (Gomez-Escobar et al., 1998). In this work, we focused on the 29-kD STK19 isoform, which encodes a 254-a.a. protein. This protein contains two conserved domains, an N-terminal bipartite nuclear localization signal (NLS) and a C-terminal conserved putative kinase domain (Fig. S1 A). Pull-down showed that the NLS was necessary and sufficient for binding to Importin α1, an adaptor of the classical nuclear import (Fig. S1 B). Deletion of this NLS, however, only partially reduced the nuclear localization of STK19 in HeLa cells (Fig. S1 C), suggesting that additional factors are responsible for its nuclear localization.
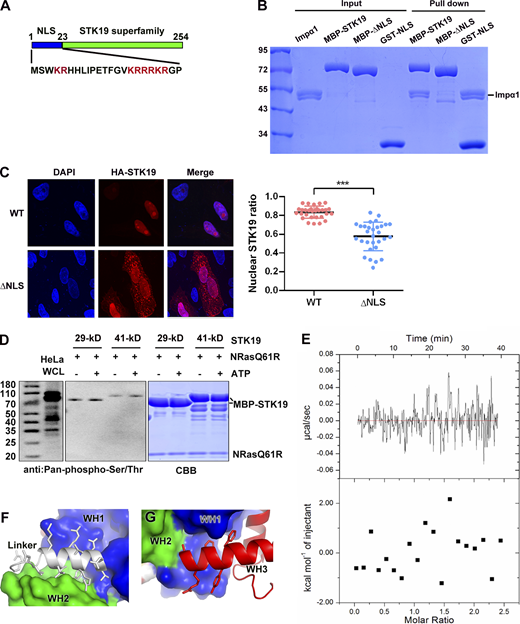
STK19 has no kinase activity. (A) Domain organization of STK19 and the sequence of the N-terminus. The STK19 superfamily is predicted to be a kinase domain. (B) Pull-down analysis of STK19 binding to Impα1 using different fragments of MBP-tag STK19. All proteins used were purified from E. coli. (C) Subcellular localization of STK19 and the mutant without the NLS in HeLa cells. The statistical analysis of the nuclear STK19 ratio is presented in the right panel. Error bars represent the SD for at least 30 cells from each group. Two-tailed unpaired Student’s t test was used. *** denotes P < 0.001. (D) An in vitro kinase assay was performed using E. coli–purified MBP-STK19 (29 or 41 kD, 8 μM) and NRAS-Q61R (4 μM) proteins in the presence or absence of 300 μM ATP for 30 min at 30°C. Protein phosphorylation was detected using a pan-pS/pT antibody. HeLa whole cell lysate (WCL) was immunoblotted as a control. The right panel shows the input proteins stained by Coomassie Brilliant Blue. The kinase assay buffer contained 20 mM MnCl2, 50 mM HEPES (pH 8.0), and 200 mM NaCl. (E) Isothermal titration calorimetry analysis of STK19 binding to ATP. The assay buffer contained 20 mM HEPES 7.5, 200 mM NaCl, 2 mM MnCl2, and 2 mM MgCl2. STK19 and ATP were used at concentrations of 30 and 350 µM, respectively. (F) The linker between WH1 and WH2. (G) The WH3 helix (α1) that is embraced by WH1. The molecular weights are in kD. Source data are available for this figure: SourceData FS1.
STK19 has no kinase activity. (A) Domain organization of STK19 and the sequence of the N-terminus. The STK19 superfamily is predicted to be a kinase domain. (B) Pull-down analysis of STK19 binding to Impα1 using different fragments of MBP-tag STK19. All proteins used were purified from E. coli. (C) Subcellular localization of STK19 and the mutant without the NLS in HeLa cells. The statistical analysis of the nuclear STK19 ratio is presented in the right panel. Error bars represent the SD for at least 30 cells from each group. Two-tailed unpaired Student’s t test was used. *** denotes P < 0.001. (D) An in vitro kinase assay was performed using E. coli–purified MBP-STK19 (29 or 41 kD, 8 μM) and NRAS-Q61R (4 μM) proteins in the presence or absence of 300 μM ATP for 30 min at 30°C. Protein phosphorylation was detected using a pan-pS/pT antibody. HeLa whole cell lysate (WCL) was immunoblotted as a control. The right panel shows the input proteins stained by Coomassie Brilliant Blue. The kinase assay buffer contained 20 mM MnCl2, 50 mM HEPES (pH 8.0), and 200 mM NaCl. (E) Isothermal titration calorimetry analysis of STK19 binding to ATP. The assay buffer contained 20 mM HEPES 7.5, 200 mM NaCl, 2 mM MnCl2, and 2 mM MgCl2. STK19 and ATP were used at concentrations of 30 and 350 µM, respectively. (F) The linker between WH1 and WH2. (G) The WH3 helix (α1) that is embraced by WH1. The molecular weights are in kD. Source data are available for this figure: SourceData FS1.
Using an assay performed according to the reported conditions (Yin et al., 2019), the kinase activity of STK19 was not detected, either by western blot or by mass spectrometry (MS; Fig. S1 D and Data S1). In addition, no binding between ATP and STK19 was observed by isothermal titration analysis (Fig. S1 E). To understand the functions of STK19, we crystalized the putative kinase domain encompassing a.a. 25–254 (25-C) and determined its crystal structure at 1.65 Å resolution using selenomethionine derivative crystals and single wavelength anomalous diffraction (Table 1). Each asymmetric unit contains two copies of STK19 containing residues 32–254. The protein is folded with 10 α helices and nine small β strands that form three antiparallel β sheets (Fig. 1 A). At a glance, this is highly different from known catalytic domain structures of the kinase, which always contains two subdomains, an ATP-binding N-lobe and a substrate-binding C-lobe (Eswaran and Knapp, 2010; Huse and Kuriyan, 2002; Fig. 1 B). Three-dimensional structure blast using DALI revealed that the protein can be divided into three “winged” helix domains (WHDs; Fig. 1 C), but WHD has never been reported to possess kinase activity (Harami et al., 2013; Soler et al., 2007). Surface representation shows that STK19 does not contain a possible ATP-binding pocket (Fig. 1 D). The presumed “ATP-binding site” residue K203 (corresponding to K317 in 41-kD STK19) is located at a protruding loop but not in a concave pocket (Fig. 1 D). These biochemical and structural data suggest that STK19 is highly unlikely to be a kinase.
Crystal data collection and refinement statistics
| STK19 SeMet dataset | |
|---|---|
| Cell axial lengths (Å) | a = 55.3, b = 96.6, c = 98.8, α = β = γ = 90 |
| Spacegroup | P212121 |
| Data collection | |
| Resolution range (Å) | 50.00–1.65 (1.69–1.65) |
| Number of observed reflections | 800,904 (45,192) |
| Number of unique reflections | 64,082 (4,673) |
| Completeness (%) | 99.3 (99.2) |
| Multiplicity | 12.5 (9.7) |
| Anomalous completeness | 99.1 (98.9) |
| Anomalous multiplicity | 6.5 (5.0) |
| Rpim | 0.019 (0.310) |
| CC1/2 | 0.999 (0.821) |
| Mean I/Isigma | 26.1 (2.4) |
| Refinement | |
| Resolution range (Å) | 50.0–1.65 (1.69–1.65) |
| Number of working reflections | 60,759 (4,453) |
| Number of test reflections | 3,247 (216) |
| Rwork | 0.149 (0.233) |
| Rfree | 0.194 (0.333) |
| R.m.s. deviation bond lengths (Å) | 0.007 |
| R.m.s. deviation bond angles (°) | 1.487 |
| Average B-factors (Å2) (# of atoms) | 36.5 (3,918) |
| Ramachandran plot | |
| Favored regions (%) | 97.0 |
| General allowed regions (%) | 3.0 |
| Disallowed regions (%) | 0.0 |

STK19 is folded with three intimately packed WHDs and is highly unlikely to be a kinase. (A) The crystal structure of STK19 (32–254). It contains 10 α helices and nine small β strands that form three antiparallel β sheets. (B) Representative structure of a kinase (1CMK). Kinase proteins contain at least two subdomains, an ATP-binding N-lobe and a substrate-binding C-lobe. (C) DALI search match correlation matrix. Red indicates a positive correlation and blue indicates a negative correlation. The results showed that the three WHDs can be separately but not simultaneously searched/superimposed to known structures. (D) Stereo view of the electrostatic surface potential map of STK19 viewed from different angles. There is no concave pocket suitable for negatively charged ATP. (E) The crystal structure of STK19 (32–254) consists of three WHDs, which are colored blue, green and red, respectively. The linker between WH1 and WH2 is shown in white. (F) The architecture of the individual WHD of STK19. (G) Structural superimposition of the three WHDs.
STK19 is folded with three intimately packed WHDs and is highly unlikely to be a kinase. (A) The crystal structure of STK19 (32–254). It contains 10 α helices and nine small β strands that form three antiparallel β sheets. (B) Representative structure of a kinase (1CMK). Kinase proteins contain at least two subdomains, an ATP-binding N-lobe and a substrate-binding C-lobe. (C) DALI search match correlation matrix. Red indicates a positive correlation and blue indicates a negative correlation. The results showed that the three WHDs can be separately but not simultaneously searched/superimposed to known structures. (D) Stereo view of the electrostatic surface potential map of STK19 viewed from different angles. There is no concave pocket suitable for negatively charged ATP. (E) The crystal structure of STK19 (32–254) consists of three WHDs, which are colored blue, green and red, respectively. The linker between WH1 and WH2 is shown in white. (F) The architecture of the individual WHD of STK19. (G) Structural superimposition of the three WHDs.
STK19 is folded with three intimately packed WHDs and binds dsDNA in a sequence-nonspecific manner
We named the three WHDs WH1 (32–104), WH2 (120–179), and WH3 (180–254; Fig. 1 E). Each WHD comprises three α helices and three β strands in the canonical order α1-β1-α2-α3-β2-β3 (Fig. 1 F). The three WHDs are intimately packed and approximately arranged in a line, with WH1 in the middle (Fig. 1 E). WH1 and WH2 are connected by a linker helix (105–119), which forms substantial hydrophobic contacts with both WH1 and WH2 (Fig. S1 F). WH2 is followed by a long WH3 helix (α1) that sequentially lines up WH2, WH1, and WH3 (Fig. S1 G). These three domains are considerably different from each other, especially the α1 helix and the W1 wing (a loop between β2 and β3; Fig. 1 G). WHDs often play a role in DNA binding (Harami et al., 2013). Electrophoretic mobility shift assays (EMSA) were used to test whether STK19 is a DNA binder. The results showed that both full-length (FL) STK19 and its C-terminal domain (25-C) displayed substantial DNA-binding ability (Fig. 2 A). Using EMSA, the apparent binding affinity (Kd) between 25-C and DNA1 was determined to be 3.8 μM (Fig. 2 B). DNA longer than 25 bp bound to a similar extent, but binding of 10-bp dsDNA was hardly detectable (Fig. 2 C). STK19 barely bound to single-stranded DNA (Fig. 2 D) and was not dependent on a specific sequence (Fig. 2 E). Overall, these results showed that STK19 bound to dsDNA in a sequence-nonspecific manner.
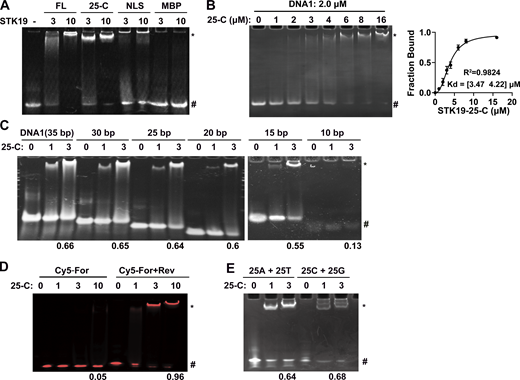
STK19 binds dsDNA in a sequence-nonspecific manner. Refer to Table S1 for DNA sequences. All EMSA experiments were repeated at least twice. (A) EMSA analysis of dsDNA (DNA1) in the presence of different proteins. The numbers (3 and 10) represent the molar ratio of STK19 to DNA. “*” and “#” denote protein-bound and free DNA, respectively. (B) EMSA analysis of the binding affinity between STK19 25-C and DNA1. Different concentrations of STK19 were incubated with 2.0 μM DNA1 at 4°C for 2 h prior to gel separation. Error bars represent the SD for three biological replicates. The data were fitted using GraphPad and yielded an apparent 3.8 μM binding affinity with a 95% confidence interval of 3.5–4.2 μM. (C) EMSA analysis of dsDNA of different lengths in the presence of STK19 25-C. The numbers at the bottom represent the ratio of bound DNA (bound/total). The numbers (1 and 3) represent the molar ratio of STK19 to DNA. (D) STK19 (25-C) binding to 35-bp single-stranded DNA (5′ Cy5-labeled) in the presence or absence of the reverse strand (unlabeled). The numbers at the bottom represent the ratio of bound DNA (bound/total). The numbers (1, 3, and 10) represent the molar ratio of STK19 to DNA. (E) EMSA analysis of different 25-bp dsDNA sequences in the presence of STK19 25-C. Source data are available for this figure: SourceData F2.
STK19 binds dsDNA in a sequence-nonspecific manner. Refer to Table S1 for DNA sequences. All EMSA experiments were repeated at least twice. (A) EMSA analysis of dsDNA (DNA1) in the presence of different proteins. The numbers (3 and 10) represent the molar ratio of STK19 to DNA. “*” and “#” denote protein-bound and free DNA, respectively. (B) EMSA analysis of the binding affinity between STK19 25-C and DNA1. Different concentrations of STK19 were incubated with 2.0 μM DNA1 at 4°C for 2 h prior to gel separation. Error bars represent the SD for three biological replicates. The data were fitted using GraphPad and yielded an apparent 3.8 μM binding affinity with a 95% confidence interval of 3.5–4.2 μM. (C) EMSA analysis of dsDNA of different lengths in the presence of STK19 25-C. The numbers at the bottom represent the ratio of bound DNA (bound/total). The numbers (1 and 3) represent the molar ratio of STK19 to DNA. (D) STK19 (25-C) binding to 35-bp single-stranded DNA (5′ Cy5-labeled) in the presence or absence of the reverse strand (unlabeled). The numbers at the bottom represent the ratio of bound DNA (bound/total). The numbers (1, 3, and 10) represent the molar ratio of STK19 to DNA. (E) EMSA analysis of different 25-bp dsDNA sequences in the presence of STK19 25-C. Source data are available for this figure: SourceData F2.
STK19 binds dsDNA using two basic patches in the protein homodimer
The two chains in the asymmetric unit interact through WH3 and form an overall “V”-shaped architecture (Fig. 3 A). The dimer interface residues have low B factors as the inner core residues (Fig. 3 B) and have well-defined electron densities (Fig. 3 C), suggesting that the dimer may exist in solution. Dimerization is mediated by the third WHD (Fig. S2 A), and the dimer interface buries an area of ∼550 Å2 in each monomer. At the heart of the interface, E206 and R250 formed two salt bridges with R250 and E206 in the other monomer, respectively (Fig. 3 D and Fig. S2 B). Additionally, L248 formed hydrophobic contact with Y204 in the other monomer (Fig. 3 D). The two monomers in the dimer are similar to each other and to the AlphaFold-predicted structure, except for two loop regions (Fig. S2, C and D). The purification yields of dimer interface mutants R250A, E206K, L248A, and Y204E decreased to varying degrees, which is unlikely if these were protein surface mutations (Fig. S2 E). The more severe the mutation interfered with dimer formation (E206K and R250A), the less the purification yield. The melting temperatures (Tm) were similar between WT and the mutants (except for R250A, no yield) when measured at a low concentration (no dimerization) by differential scanning fluorimetry (DSF, Fig. S2 F). At a high concentration, E206K and L248A were less stable but Y204E was similarly stable (Fig. S2 F). These results indicated that dimerization may be important for protein stability at high concentration and the purification yield.
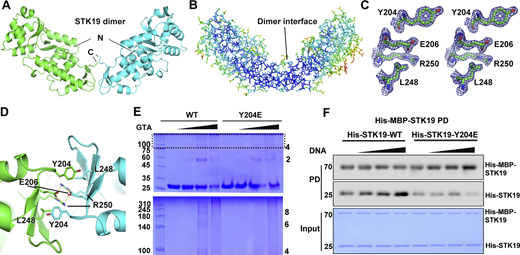
STK19 can form dimers that are enhanced by DNA binding. (A) The STK19 dimer in the asymmetric unit. The N and C termini are labeled. (B) B-factor representation of the STK19 dimer. Blue to red represent B-factors from low to high. (C) Stereo-view of the 2Fo-Fc simulated annealing omit map around Y204, E206, L248, and R250, contoured at 2σ. (D) The dimer interface with core interacting residues shown as sticks. Salt bridges are shown as yellow dotted lines. (E) STK19 dimer and oligomers in the presence of the crosslinking agent glutaraldehyde (0%, 0.05%, 0.1%, and 0.2%). STK19 formed other oligomeric species corresponding to even but not odd numbers of oligomers. (F) His-MBP-STK19 (0.2 µM) pull-down of his-tagged STK19 WT (2 µM) and Y204E (2 µM) in the presence of increasing concentrations of 15-bp dsDNA (0, 0.5, 2, 8 µM). The bound proteins (PD) were stained by their His-tag. The molecular weights are in kD. Source data are available for this figure: SourceData F3.
STK19 can form dimers that are enhanced by DNA binding. (A) The STK19 dimer in the asymmetric unit. The N and C termini are labeled. (B) B-factor representation of the STK19 dimer. Blue to red represent B-factors from low to high. (C) Stereo-view of the 2Fo-Fc simulated annealing omit map around Y204, E206, L248, and R250, contoured at 2σ. (D) The dimer interface with core interacting residues shown as sticks. Salt bridges are shown as yellow dotted lines. (E) STK19 dimer and oligomers in the presence of the crosslinking agent glutaraldehyde (0%, 0.05%, 0.1%, and 0.2%). STK19 formed other oligomeric species corresponding to even but not odd numbers of oligomers. (F) His-MBP-STK19 (0.2 µM) pull-down of his-tagged STK19 WT (2 µM) and Y204E (2 µM) in the presence of increasing concentrations of 15-bp dsDNA (0, 0.5, 2, 8 µM). The bound proteins (PD) were stained by their His-tag. The molecular weights are in kD. Source data are available for this figure: SourceData F3.
STK19 can form homodimer to bind dsDNA. (A) Architecture of the STK19 dimer. The α3 and W1 of three WHDs in the dimer. Y204 in W1WH3 is shown as yellow sticks. (B) Interaction details between two chains of STK19. This panel is generated using PDBsum. (C) Alignment of two chains in the asymmetric unit. The two most prominent changes are all in WH1, namely, the loop (L1) between α1 and β1 and W1. These changes are likely due to the different crystal packing environments faced by the two monomers. (D) Alignment of the STK19 crystal structure (cyan, shown is a dimer) with AlphaFold-predicted 41-kD STK19. AlphaFold-predicted STK19 is shown in green except for the N-terminal 110 amino acids (magenta). (E) Expression and Ni-NTA purification profile of STK19 mutants. S: soluble fraction after lysis and spin. B: Proteins that were bound to beads after supernatant incubation and washing. (F) DSF analysis of STK19 WT or its mutants (Y204E, E206K, L248A, 3KA, R200A) at different concentrations. The two panels on the right show the Tm change (∆Tm) with respect to the WT Tm. R200A was used as an NC. Shown are the mean of three biological replicates. Two-tailed unpaired Student’s t test was used. The yield of R250A was not sufficient to perform this assay. (G) MBP pull-down analysis of the STK19 dimer. The bound proteins (PD) were stained by their 6X-His expression tag (or His-MBP tag). (H) EMSA analysis of DNA1 in the presence of STK19 WT and Y204E. The numbers below represent the molar ratio of bound DNA. (I) The linear dsDNA (17 bp, extracted from PDB 6FBQ) was modeled to bind one basic patch in the dimer. Binding to two basic patches will likely bend the dsDNA. The two long helixes were colored magenta. The molecular weights are in kD. Source data are available for this figure: SourceData FS2.
STK19 can form homodimer to bind dsDNA. (A) Architecture of the STK19 dimer. The α3 and W1 of three WHDs in the dimer. Y204 in W1WH3 is shown as yellow sticks. (B) Interaction details between two chains of STK19. This panel is generated using PDBsum. (C) Alignment of two chains in the asymmetric unit. The two most prominent changes are all in WH1, namely, the loop (L1) between α1 and β1 and W1. These changes are likely due to the different crystal packing environments faced by the two monomers. (D) Alignment of the STK19 crystal structure (cyan, shown is a dimer) with AlphaFold-predicted 41-kD STK19. AlphaFold-predicted STK19 is shown in green except for the N-terminal 110 amino acids (magenta). (E) Expression and Ni-NTA purification profile of STK19 mutants. S: soluble fraction after lysis and spin. B: Proteins that were bound to beads after supernatant incubation and washing. (F) DSF analysis of STK19 WT or its mutants (Y204E, E206K, L248A, 3KA, R200A) at different concentrations. The two panels on the right show the Tm change (∆Tm) with respect to the WT Tm. R200A was used as an NC. Shown are the mean of three biological replicates. Two-tailed unpaired Student’s t test was used. The yield of R250A was not sufficient to perform this assay. (G) MBP pull-down analysis of the STK19 dimer. The bound proteins (PD) were stained by their 6X-His expression tag (or His-MBP tag). (H) EMSA analysis of DNA1 in the presence of STK19 WT and Y204E. The numbers below represent the molar ratio of bound DNA. (I) The linear dsDNA (17 bp, extracted from PDB 6FBQ) was modeled to bind one basic patch in the dimer. Binding to two basic patches will likely bend the dsDNA. The two long helixes were colored magenta. The molecular weights are in kD. Source data are available for this figure: SourceData FS2.
In crosslinking experiments, odd oligomers were not observed in crosslinked products, and Y204E was less crosslinked (Fig. 3 E). Binding between MBP-tagged STK19 and untagged STK19 could be detected in pull-down assays, and the binding was reduced by the Y204E mutation (Fig. S2 G). Furthermore, increasing the concentration of 15-bp dsDNA in pull-down increased the binding of untagged WT STK19 but not the Y204E mutant (Fig. 3 F), arguing that dimerization is likely involved in DNA binding. Taken together, STK19 can form dimers in solution.
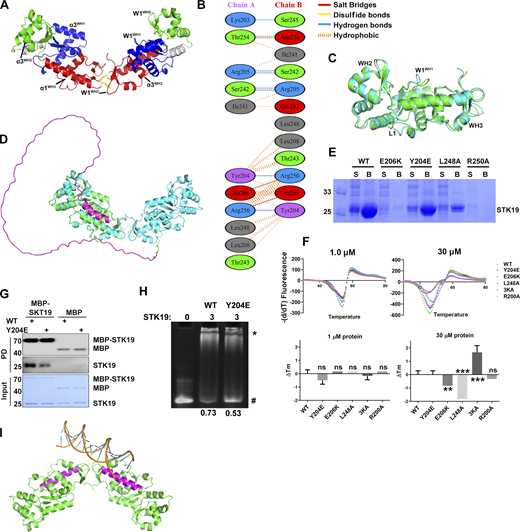
The observation that STK19 bound to dsDNA irrespective of its sequence (Fig. 2 E) implied that it may interact with the acidic phosphate backbone of DNA. The electrostatic surface potential map shows a basic patch in STK19, mostly centered on the α1 helix of WH3 (α1WH3; Fig. 4 A). However, this region only spans ∼25 Å, which is quite small compared with the ∼67 Å span of a 20-bp dsDNA (Mandelkern et al., 1981). In the protein homodimer, two basic patches are adjacent in space and the distance between Cα atoms of K186 is 58.9 Å (Fig. 4 B). In view that the dimerization interface mutant Y204E reduced DNA binding (Fig. S2 H), it was speculated that the dsDNA bound to the STK19 dimer rather than a monomer. Computer modeling showed that bending of dsDNA is likely required to bind STK19 (Fig. S2 I). Sequence alignment of STK19 from different species showed that K186, R192, and K201 in α1WH3 are well conserved (Fig. 4 C). R192 is mostly buried and thus unlikely to be involved in DNA binding (Fig. 4 A). To prevent possible mutational compensation, the adjacent K190 was simultaneously mutated when performing the K186 mutation. Either the K201A or K186A/K190A mutation dramatically reduced STK19’s DNA-binding ability, and simultaneously mutating three lysine residues (3KA, K186A/K190A/K201A) totally abolished the DNA-binding activity (Fig. 4 D). Chromatin precipitation results showed that WT STK19 bound to isolated chromatin, but 3KA did not (Fig. 4 E). The above results indicate that STK19 may use two basic patches flanking the homodimer to bind dsDNA.
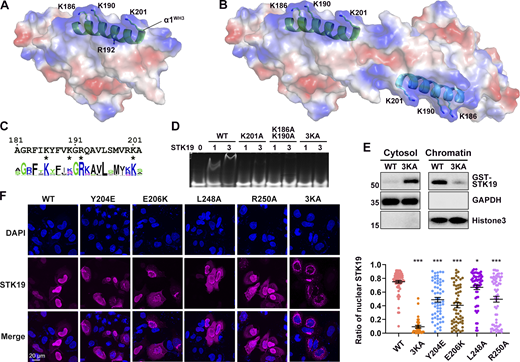
DNA is likely bound to two basic patches in the protein homodimer. (A) Electrostatic surface potential map of the STK19 monomer. Red and blue represent negatively and positively charged surfaces, respectively. The α1 of WH3 (α1WH3), three surface-exposed basic residues, and one buried basic residue R192 in this helix are shown. (B) Electrostatic surface potential map of the dimer. (C) Sequence conservation of α1WH3. The discussed basic residues are indicated with an “*” below the sequence. (D) EMSA analysis of DNA1 in the presence of STK19 WT or different lysine mutants. (E) Western blot analysis of STK19 localization. Chromatins were precipitated by spinning lysed cell nuclei at 2,000 g. The molecular weights are in kD. (F) Representative confocal images of the subcellular localization of STK19 mutants in HeLa cells and statistical analysis of the nuclear STK19 ratio. Error bars represent the SEM for at least 30 cells from each group. All groups were compared to WT, and two-tailed unpaired Student’s t test was used. * denotes P < 0.05; *** denotes P < 0.001. Source data are available for this figure: SourceData F4.
DNA is likely bound to two basic patches in the protein homodimer. (A) Electrostatic surface potential map of the STK19 monomer. Red and blue represent negatively and positively charged surfaces, respectively. The α1 of WH3 (α1WH3), three surface-exposed basic residues, and one buried basic residue R192 in this helix are shown. (B) Electrostatic surface potential map of the dimer. (C) Sequence conservation of α1WH3. The discussed basic residues are indicated with an “*” below the sequence. (D) EMSA analysis of DNA1 in the presence of STK19 WT or different lysine mutants. (E) Western blot analysis of STK19 localization. Chromatins were precipitated by spinning lysed cell nuclei at 2,000 g. The molecular weights are in kD. (F) Representative confocal images of the subcellular localization of STK19 mutants in HeLa cells and statistical analysis of the nuclear STK19 ratio. Error bars represent the SEM for at least 30 cells from each group. All groups were compared to WT, and two-tailed unpaired Student’s t test was used. * denotes P < 0.05; *** denotes P < 0.001. Source data are available for this figure: SourceData F4.
Immunofluorescence results showed that the overexpressed STK19 was mainly localized in the nucleus of HeLa cells (Fig. 4 F). The 3KA mutant, albeit containing an intact NLS, dramatically relocalized from the nucleus to the cytoplasm (Fig. 4 F). In fact, nuclear localization of STK19 was more dependent on its DNA-binding residues than its NLS (compare Fig. 4 F and Fig. S1 C). DSF showed that the thermal stability of 3KA was similar to that of WT at a low concentration but slightly increased at a high concentration (Fig. S2 F). The cytoplasmic localization of dimerization mutants, including Y204E, E206K, R250A, and L248A, was also notably increased (Fig. 4 F), further supporting the notion that STK19 dimerizes to bind DNA. Overall, both dimerization and DNA-binding mutants can affect STK19 localization and possibly its function.
DNA binding is critical for STK19-mediated DDR
To investigate the molecular functions of STK19, we sought to identify STK19 WT-interacting proteins by GST pull-down and MS analysis (Data S2). Proteins involved in nucleotide excision repair (NER) and mismatch repair (MMR) were significantly enriched by STK19 WT (Fig. 5 A), suggesting that STK19 may participate in MMR and NER. Tandem mass tag (TMT)–based quantitative proteomic showed that STK19 knockdown (KD) cells downregulated several proteins involved in MMR and NER (Fig. S3, A and B). Upon UVB treatment, which induces NER, both the mRNA and protein expression levels of STK19 increased in B16-F10, HeLa, and 293T cells (Fig. 5 B and Fig. S3 C). Depletion of STK19 (Cas9-mediated polyclonal KD, which depletes both 29-kD and barely detectable 41-kD) led to more pronounced γ∙H2AX expression, a marker of DNA damage, either by immunofluorescence or western blotting (Fig. 5, C and D). Re-expression of STK19 WT in the STK19 KD cells reduced the level of γ∙H2AX to a greater extent than re-expression of 3KA (Fig. 5 E), suggesting that the DNA-binding activity of STK19 is required for its NER function. In the presence of methylnitronitrosoguanidine (MNNG), a compound that triggers MMR, more H2AX foci were also observed in STK19 KD cells (Fig. S3 D). In contrast, after cells were treated with double-strand-break-inducing x-ray, neutral comet assay revealed hardly any tail moment difference between control siRNA and siSTK19 (Fig. S3 E). These results indicated that STK19 was required for single-stranded repair including NER and MMR but not double-stranded repair.
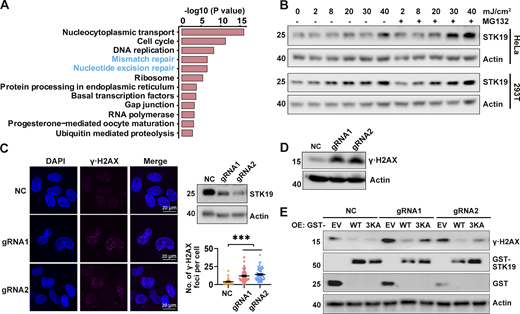
DNA binding is critical for STK19-mediated DDR. (A) Pathways enriched by proteins that WT bound at least fivefold more than EV (empty vector). Samples were derived from 293T cells overexpressing EV or GST-STK19-WT, respectively. Pull-down was performed in the presence of DNase and RNase for 12 h to suppress indirect interactions via DNA/RNA. (B) STK19 protein levels under different doses of UVB exposure, with or without proteasome inhibitor MG132 treatment. Cells were lysed 2 h after UVB treatment. (C) The immunofluorescence of γ∙H2AX in HeLa cells and the statistical analysis of γ∙H2AX foci numbers. NC indicates that gRNA NC was used. gRNA1 and gRNA2 targeted the seventh and fourth exons of 41-kD STK19 gene, respectively, and therefore can KD both the 29- and 41-kD isoforms. KD efficiency of 29-kD STK19 is shown in the top right panel. Error bars represent the SEM of at least 30 measurements. All groups were compared with NC, and two-tailed unpaired Student’s t test was used. *** denotes P < 0.001. (D) The expression level of γ∙H2AX in HeLa cells treated with 8 mJ/cm2 UVB. The cells were harvested 5 h after treatment. (E) The level of γ∙H2AX in NC or KD cells transfected with gRNA-resistant expression of GST-tagged WT or 3KA. Cells were lysed 34 h after transfection with different plasmids. The molecular weights are in kD. Source data are available for this figure: SourceData F5.
DNA binding is critical for STK19-mediated DDR. (A) Pathways enriched by proteins that WT bound at least fivefold more than EV (empty vector). Samples were derived from 293T cells overexpressing EV or GST-STK19-WT, respectively. Pull-down was performed in the presence of DNase and RNase for 12 h to suppress indirect interactions via DNA/RNA. (B) STK19 protein levels under different doses of UVB exposure, with or without proteasome inhibitor MG132 treatment. Cells were lysed 2 h after UVB treatment. (C) The immunofluorescence of γ∙H2AX in HeLa cells and the statistical analysis of γ∙H2AX foci numbers. NC indicates that gRNA NC was used. gRNA1 and gRNA2 targeted the seventh and fourth exons of 41-kD STK19 gene, respectively, and therefore can KD both the 29- and 41-kD isoforms. KD efficiency of 29-kD STK19 is shown in the top right panel. Error bars represent the SEM of at least 30 measurements. All groups were compared with NC, and two-tailed unpaired Student’s t test was used. *** denotes P < 0.001. (D) The expression level of γ∙H2AX in HeLa cells treated with 8 mJ/cm2 UVB. The cells were harvested 5 h after treatment. (E) The level of γ∙H2AX in NC or KD cells transfected with gRNA-resistant expression of GST-tagged WT or 3KA. Cells were lysed 34 h after transfection with different plasmids. The molecular weights are in kD. Source data are available for this figure: SourceData F5.
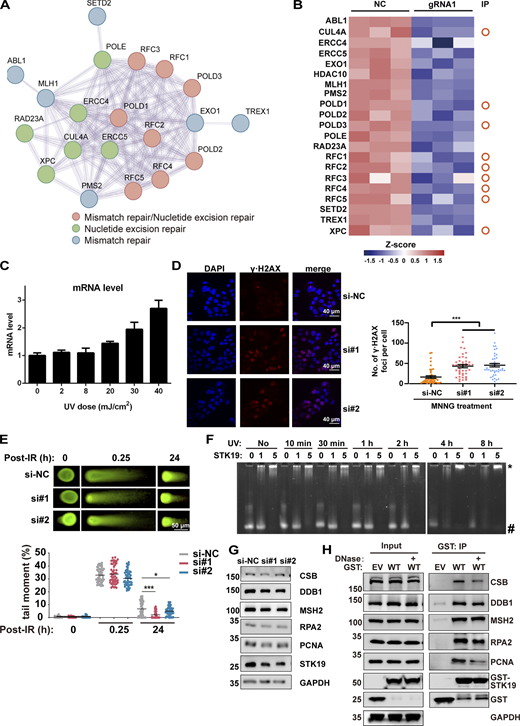
STK19 is involved in DDR. (A) Protein–protein interaction analysis of DDR proteins that were significantly downregulated by STK19 KD. The color of the bubbles illustrates the different types of DDR. (B) Heatmap showing the DDR proteins downregulated by STK19 KD. The circles on the right denote the proteins that were previously immunoprecipitated (IP) by WT STK19. (C) UVB treatment induced a dose-dependent increase of STK19 mRNA levels in B16-F10 cells. Cells were analyzed 2 h after UVB treatment. Error bars represent SEM. (D) The immunofluorescence of γ∙H2AX in HeLa cells after MNNG treatment and the statistical analysis of γ∙H2AX foci numbers. HeLa cells were transfected with two siRNAs targeting STK19 coding region (si#1 and si#2) or nontargeting control (si-NC). After 72 h, 25 μM MNNG was added and incubated for 6 h before cells were imaged. Error bars represent the SEM of at least 30 measurements. Two-tailed unpaired Student’s t test was used. (E) The quantification of neutral comet assays in HCT116 cells treated with siRNAs targeting STK19 (si#1 and si#2) and nontargeting control (si-NC) at the indicated time after IR treatment. DNA damage was quantified via the tail moment using the CometScore software. Data represent mean ± SEM (unpaired two-tailed Student’s t test). KD of STK19 slightly enhanced (but not impaired) double-strand break repair at 24 h. (F) STK19 binding to UVB-irradiated DNA1 at different times. DNA was notably degraded after UVB treatment for 4 h. (G) Western blot analysis of DDR factor expression levels before and after siRNA-mediated STK19 KD in HeLa cells. (H) GST-STK19 WT pull-down of CSB, DDB1, MSH2, RPA2, and PCNA with or without DNase treatment. 293T cells overexpressing GST-STK19 WT were lysed. Samples were added with or without DNase during overnight pull-down. EV: empty vector. Compared with Fig. 6 C, PCNA degradation was weaker in this experiment (repeated three times), possibly due to less protease expression or the usage of new protease inhibitors. The molecular weights are in kD. Source data are available for this figure: SourceData FS3.
STK19 is involved in DDR. (A) Protein–protein interaction analysis of DDR proteins that were significantly downregulated by STK19 KD. The color of the bubbles illustrates the different types of DDR. (B) Heatmap showing the DDR proteins downregulated by STK19 KD. The circles on the right denote the proteins that were previously immunoprecipitated (IP) by WT STK19. (C) UVB treatment induced a dose-dependent increase of STK19 mRNA levels in B16-F10 cells. Cells were analyzed 2 h after UVB treatment. Error bars represent SEM. (D) The immunofluorescence of γ∙H2AX in HeLa cells after MNNG treatment and the statistical analysis of γ∙H2AX foci numbers. HeLa cells were transfected with two siRNAs targeting STK19 coding region (si#1 and si#2) or nontargeting control (si-NC). After 72 h, 25 μM MNNG was added and incubated for 6 h before cells were imaged. Error bars represent the SEM of at least 30 measurements. Two-tailed unpaired Student’s t test was used. (E) The quantification of neutral comet assays in HCT116 cells treated with siRNAs targeting STK19 (si#1 and si#2) and nontargeting control (si-NC) at the indicated time after IR treatment. DNA damage was quantified via the tail moment using the CometScore software. Data represent mean ± SEM (unpaired two-tailed Student’s t test). KD of STK19 slightly enhanced (but not impaired) double-strand break repair at 24 h. (F) STK19 binding to UVB-irradiated DNA1 at different times. DNA was notably degraded after UVB treatment for 4 h. (G) Western blot analysis of DDR factor expression levels before and after siRNA-mediated STK19 KD in HeLa cells. (H) GST-STK19 WT pull-down of CSB, DDB1, MSH2, RPA2, and PCNA with or without DNase treatment. 293T cells overexpressing GST-STK19 WT were lysed. Samples were added with or without DNase during overnight pull-down. EV: empty vector. Compared with Fig. 6 C, PCNA degradation was weaker in this experiment (repeated three times), possibly due to less protease expression or the usage of new protease inhibitors. The molecular weights are in kD. Source data are available for this figure: SourceData FS3.
STK19 facilitates the recruitment of DNA repair factors to chromatin in NER
Using EMSA, no binding difference in STK19 binding was observed when DNA was treated with different doses of UVB (Fig. S3 F), suggesting that STK19 is probably not directly involved in the recognition of damaged DNA in NER. In the presence of UVB treatment, STK19 and DNA repair factors such as CSB, MSH2, RPA2, and PCNA were notably enriched in the chromatin faction to mediate DDR (Fig. 6 A). STK19 KD inhibited the chromatin recruitment of all these repair factors, although the expression of these proteins was not affected by STK19 KD (Fig. 6 A and Fig. S3 G). Chromatin recruitment of DDB1, a global genome NER (GG-NER) factor that participates in DNA lesion recognition, was not affected by UVB or STK19 KD. In UVB-treated HeLa cells expressing endogenous STK19, overexpression of STK19 WT weakly promoted the chromatin recruitment of upstream repair factors DDB1, MSH2, and RPA2, and notably enhanced chromatin recruitment of the downstream repair factor PCNA (recruiting polymerase for DNA synthesis; Fig. 6 B). In contrast, overexpression of 3KA did not enhance chromatin recruitment of these repair factors. These results suggested that STK19 likely bound to the periphery of damaged DNA to stabilize upstream DDR factors and recruit downstream DDR factors to facilitate NER.
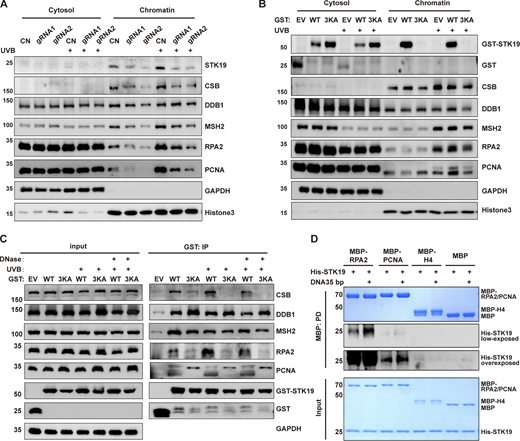
STK19 promotes chromatin recruitment of DNA repair factors in NER. (A) Western blot analysis of chromatin localization of different DNA damage repair key factors after STK19 knockdown, with or without UVB irradiation. HeLa cells were treated with UVB irradiation (30 mJ/cm2). After 2 h, cells were lysed and chromatins were precipitated. (B) Western blot analysis of chromatin localization of different damage repair factors after overexpression of GST-STK19 WT or 3KA, with or without UVB irradiation. HeLa cells overexpressing GST-STK19 WT or 3KA were irradiated with 30 mJ/cm2 UVB and then chromatin fractionation was performed 2 h later. Cleavage of chromatin-bound PCNA was likely due to plasmid transfection. (C) GST-STK19 WT or 3KA pull-down of CSB, DDB1, MSH2, RPA2, and PCNA with or without UVB irradiation. 293T cells overexpressing GST-STK19 WT or 3KA were lysed 2 h after UVB treatment (30 mJ/cm2). GST-tag helps to reduce the extent of mutant degradation and ensures a similar expression level for WT and mutant STK19. Samples were added with or without DNase during overnight pull-down. EV: empty vector; IP: immunoprecipitation. (D) MBP pull-down analysis of STK19 interactions with E. coli–purified RPA2 and PCNA. MBP-tagged full-length RPA2/PCNA was expressed in E. coli. Bound his-STK19 was visualized with his-tag antibody. The molecular weights are in kD. Source data are available for this figure: SourceData F6.
STK19 promotes chromatin recruitment of DNA repair factors in NER. (A) Western blot analysis of chromatin localization of different DNA damage repair key factors after STK19 knockdown, with or without UVB irradiation. HeLa cells were treated with UVB irradiation (30 mJ/cm2). After 2 h, cells were lysed and chromatins were precipitated. (B) Western blot analysis of chromatin localization of different damage repair factors after overexpression of GST-STK19 WT or 3KA, with or without UVB irradiation. HeLa cells overexpressing GST-STK19 WT or 3KA were irradiated with 30 mJ/cm2 UVB and then chromatin fractionation was performed 2 h later. Cleavage of chromatin-bound PCNA was likely due to plasmid transfection. (C) GST-STK19 WT or 3KA pull-down of CSB, DDB1, MSH2, RPA2, and PCNA with or without UVB irradiation. 293T cells overexpressing GST-STK19 WT or 3KA were lysed 2 h after UVB treatment (30 mJ/cm2). GST-tag helps to reduce the extent of mutant degradation and ensures a similar expression level for WT and mutant STK19. Samples were added with or without DNase during overnight pull-down. EV: empty vector; IP: immunoprecipitation. (D) MBP pull-down analysis of STK19 interactions with E. coli–purified RPA2 and PCNA. MBP-tagged full-length RPA2/PCNA was expressed in E. coli. Bound his-STK19 was visualized with his-tag antibody. The molecular weights are in kD. Source data are available for this figure: SourceData F6.
In agreement with the stabilizing/recruiting function, STK19 pull-down experiments showed that STK19 interacted with all these DDR factors with or without UVB treatment (Fig. 6 C). Even without UVB treatment to prevent possible UV-induced crosslinking, DNase did not abolish their interaction (Fig. S3 H), possibly due to DNA protection by the complexes formed by STK19 and other DDR factors, or that once formed, the STK19 complexes were stable even without DNA. The 3KA mutant bound to DDB1, MSH2, and PCNA, suggesting that STK19 can form DNA-independent complexes with these proteins. On the other hand, the interaction with CSB (lesion recognition) and RPA2 (binds the undamaged strand) was reduced by 3KA mutation (or DNase) since 3KA cannot be recruited (by dsDNA adjacent to the lesion) to form a complex with lesion-localized CSB and RPA2. The recruitment of WT STK19 may corecruit the bound DDR factors DDB1, MSH2, and PCNA to form a larger repair complex. STK19 further induced PCNA degradation even in the absence of UVB, but such degradation was not observed in chromatin precipitation experiments with shorter assay time. In addition, STK19 directly bound Escherichia coli–purified PCNA and RPA2 in pull-down experiments, and the binding was potentiated by dsDNA (Fig. 6 D). These results suggest that STK19 may interact with DNA and different DDR factors directly or indirectly, thereby stabilizing/recruiting these factors to damaged DNA.
STK19 is also an RNA-binding protein involved in other cellular processes
Except for DDR proteins, STK19 interacted with many proteins involved in RNA transcription, splicing, translation, and degradation (Fig. S4 A), and interaction with these RNA metabolism proteins was reduced by the 3KA mutation (Fig. S4 B). EMSA results showed that STK19 bound similarly to dsDNA and dsRNA (Fig. 7 A). In cells, HeLa-expressed GST-STK19 bound much more RNA than DNA (Fig. 7 B). Furthermore, RNA binding also relied on the DNA-binding residues (Fig. 7 C), suggesting a shared binding interface. TMT proteomics by Gene Set Enrichment Analysis (GSEA) revealed that many proteins, especially those involved in metabolic pathways, were downregulated after STK19 siRNA treatment (Fig. 7, D and E). Six downregulated proteins were tested by quantitative PCR (qPCR) analysis, and the results showed that their mRNA levels were all downregulated to varying degrees (Fig. 7 F). Among these proteins, PTBP2 and RBM47 regulate multiple aspects of RNA metabolism, such as splicing, stability, and translation (Shivalingappa et al., 2021; Stern et al., 2009). These results suggest that STK19 binds dsRNA and regulates the expression levels of some mRNAs, which may (partially) explain the observed proteomic changes in GSEA analysis.
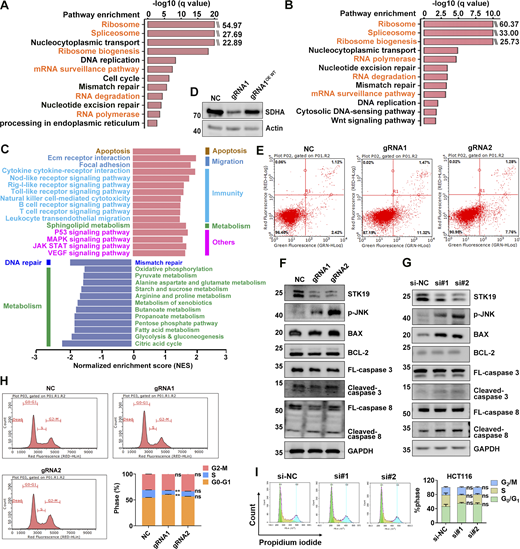
STK19 is implicated in multiple cellular pathways. (A) Pathways enriched by proteins that WT bound at least fivefold more than EV (empty vector). Samples were derived from 293T cells overexpressing EV or GST-STK19-WT, respectively. Pull-down was performed in the presence of RNase for 4 h. (B) Pathways enriched by proteins that WT bound at least twofold more than 3KA with RNase for 4 h 293T cells overexpressing GST-STK19-WT or GST-STK19-3KA were subjected to GST pull-down and MS analysis. (C) Selected pathways from GSEA based on the KEGG database that represent the most significant changes between Cas9/gRNA-mediated control and STK19 KD in HeLa. Compared with control cells, the upregulated and downregulated pathways in STK19 KD cells are represented by red (right) and blue (left) bars. All cells were collected for TMT experiments. (D) Western blot analysis of SDHA expression before and after Cas9/gRNA-mediated STK19 KD in HeLa. Overexpression of STK19 WT (OE WT) rescued the expression level of SDHA. (E) Flow cytometry analysis of apoptosis in Cas9/gRNA-mediated control (NC) and STK19 KD HeLa cells by annexin V-FITC apoptosis detection kit (C1062S; Beyotime). Red and green fluorescence represent propidium iodide and Annexin V, respectively. Cells were first treated with puromycin then cultured without puromycin for 24 h and analyzed. (F) Western blot analysis of apoptotic factors before and after Cas9/gRNA-mediated STK19 KD in HeLa. Cells were first treated with puromycin then washed and cultured without puromycin for 24 h and analyzed. (G) Western blot analysis of apoptotic factors before and after siRNA-mediated STK19 KD in HCT116 cells. (H) Flow cytometry analysis of cell cycle changes by Cas9/gRNA-mediated STK19 KD in HeLa cells. (I) Flow cytometry analysis of cell cycle changes by siRNA-mediated STK19 KD in HCT116 cells. All groups were compared to NC (H) or si-NC (I), and two-tailed unpaired Student’s t test was used. The molecular weights are in kD. Source data are available for this figure: SourceData FS4.
STK19 is implicated in multiple cellular pathways. (A) Pathways enriched by proteins that WT bound at least fivefold more than EV (empty vector). Samples were derived from 293T cells overexpressing EV or GST-STK19-WT, respectively. Pull-down was performed in the presence of RNase for 4 h. (B) Pathways enriched by proteins that WT bound at least twofold more than 3KA with RNase for 4 h 293T cells overexpressing GST-STK19-WT or GST-STK19-3KA were subjected to GST pull-down and MS analysis. (C) Selected pathways from GSEA based on the KEGG database that represent the most significant changes between Cas9/gRNA-mediated control and STK19 KD in HeLa. Compared with control cells, the upregulated and downregulated pathways in STK19 KD cells are represented by red (right) and blue (left) bars. All cells were collected for TMT experiments. (D) Western blot analysis of SDHA expression before and after Cas9/gRNA-mediated STK19 KD in HeLa. Overexpression of STK19 WT (OE WT) rescued the expression level of SDHA. (E) Flow cytometry analysis of apoptosis in Cas9/gRNA-mediated control (NC) and STK19 KD HeLa cells by annexin V-FITC apoptosis detection kit (C1062S; Beyotime). Red and green fluorescence represent propidium iodide and Annexin V, respectively. Cells were first treated with puromycin then cultured without puromycin for 24 h and analyzed. (F) Western blot analysis of apoptotic factors before and after Cas9/gRNA-mediated STK19 KD in HeLa. Cells were first treated with puromycin then washed and cultured without puromycin for 24 h and analyzed. (G) Western blot analysis of apoptotic factors before and after siRNA-mediated STK19 KD in HCT116 cells. (H) Flow cytometry analysis of cell cycle changes by Cas9/gRNA-mediated STK19 KD in HeLa cells. (I) Flow cytometry analysis of cell cycle changes by siRNA-mediated STK19 KD in HCT116 cells. All groups were compared to NC (H) or si-NC (I), and two-tailed unpaired Student’s t test was used. The molecular weights are in kD. Source data are available for this figure: SourceData FS4.
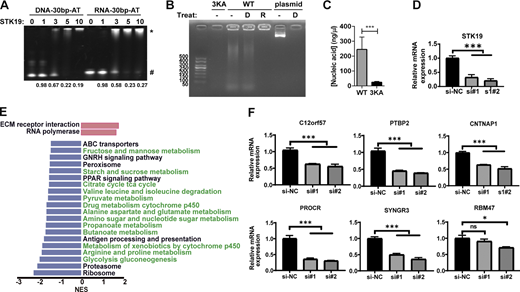
STK19 also binds dsRNA and influences mRNA expression levels. (A) Comparative analysis of STK19 binding to 30-bp dsDNA and 30-bp dsRNA by EMSA. The numbers at the bottom represent the ratio of unbound DNA (unbound/total). (B) Gel electrophoresis of STK19-bound nucleic acids treated with DNase (D) or RNase (R). A 6 k bps dsDNA plasmid was used as a control to illustrate the activity of DNase. (C) The level of nucleic acid bound by GST-STK19 or the 3KA mutant. Error bars represent the SD of three biological repeats, two-tailed unpaired Student’s t test was used. (D) qPCR analysis of STK19 mRNA expression levels in STK19 siRNA KD HeLa cells. Error bars represent the SEM of three biological repeats. All groups were compared with si-NC, and two-tailed unpaired Student’s t test was used. (E) Selected pathways from GSEA based on the KEGG database that represented the most significant changes between NC (si-NC) and STK19 KD (si#1) in HeLa cells. Compared with NC, the upregulated and downregulated pathways in STK19 KD cells are represented by red (right) and blue (left) bars. Downregulated metabolic pathways are colored green. (F) qPCR analysis of mRNA levels for six downregulated proteins in TMT proteomic analysis. Error bars represent the SEM of three biological repeats. Two-tailed unpaired Student’s t test was used. * denotes P < 0.05; *** denotes P < 0.001. Source data are available for this figure: SourceData F7.
STK19 also binds dsRNA and influences mRNA expression levels. (A) Comparative analysis of STK19 binding to 30-bp dsDNA and 30-bp dsRNA by EMSA. The numbers at the bottom represent the ratio of unbound DNA (unbound/total). (B) Gel electrophoresis of STK19-bound nucleic acids treated with DNase (D) or RNase (R). A 6 k bps dsDNA plasmid was used as a control to illustrate the activity of DNase. (C) The level of nucleic acid bound by GST-STK19 or the 3KA mutant. Error bars represent the SD of three biological repeats, two-tailed unpaired Student’s t test was used. (D) qPCR analysis of STK19 mRNA expression levels in STK19 siRNA KD HeLa cells. Error bars represent the SEM of three biological repeats. All groups were compared with si-NC, and two-tailed unpaired Student’s t test was used. (E) Selected pathways from GSEA based on the KEGG database that represented the most significant changes between NC (si-NC) and STK19 KD (si#1) in HeLa cells. Compared with NC, the upregulated and downregulated pathways in STK19 KD cells are represented by red (right) and blue (left) bars. Downregulated metabolic pathways are colored green. (F) qPCR analysis of mRNA levels for six downregulated proteins in TMT proteomic analysis. Error bars represent the SEM of three biological repeats. Two-tailed unpaired Student’s t test was used. * denotes P < 0.05; *** denotes P < 0.001. Source data are available for this figure: SourceData F7.
GSEA analysis by Cas9-gRNA-mediated STK19 KD also revealed the downregulation of many metabolic pathways (Fig. S4 C). Succinate dehydrogenase complex subunit A (SDHA), a component in the TCA cycle and oxidative phosphorylation, was significantly downregulated in STK19 KD cells (Fig. S4 D). Further transfection of STK19 WT rescued the expression level of SDHA. Consistent with the upregulation of apoptotic pathway in GSEA (Fig. S4 C), flow cytometry results showed an increase in apoptosis after STK19 KD (Fig. S4 E). Western blotting revealed obvious p-JNK activation but no changes in the levels of apoptotic mediators, including BCL-2; FL and cleaved caspase-3; and FL and cleaved caspase-8 (Fig. S4, F and G). Increased BAX levels were observed in STK19-KD HCT116 cells but not HeLa cells. Significant reduction of S phase was observed only in one of the gRNA-treated HeLa cells, but not in siRNA-treated HCT116 cells (Fig. S4, H and I). These results suggest that STK19 may regulate other cellular processes such as metabolism and apoptosis, possibly via a mRNA regulation mechanism.
STK19 is critical for cell proliferation
We were unable to obtain Cas9-gRNA-mediated STK19 single-colony knockout cells (HeLa, 293T, and B16-F10) because these knockout cells barely proliferated. Polyclonal STK19 KD cells were viable and always re-expressed STK19, typically 3–4 days after KD (Fig. S5 A). The proliferation of polyclonal STK19 KD cells was indeed much lower than that of control KD cells, especially considering the re-expression of STK19 in this assay (Fig. 8 A). Transiently transfected WT notably enhanced the growth of STK19 KD cells (Fig. 8 B). 3KA and the dimerization mutants, which were completely and partially cytoplasm mislocalized, were ineffective and barely effective in rescuing cell proliferation, respectively (Fig. 8 B). Analysis of The Cancer Genome Atlas (TCGA) database showed that STK19 was overexpressed in colorectal cancer (CRC) compared with normal colon tissues (Fig. 8 C). Moreover, the Kaplan–Meier survival analysis indicated that STK19 expression was negatively correlated with overall survival and progression-free survival in CRC patients (Fig. 8, D and E). The colony formation and proliferation of the human CRC cell line HCT116 were dramatically inhibited by STK19 silencing mediated by two siRNAs (Fig. 8, F and G). Proliferation by STK19 siRNA KD cells can be enhanced by transfection of STK19 WT, but not 3KA (Fig. S5 B). Taken together, these results suggested that STK19 was critical for cell proliferation by binding DNA/RNA and may play a role in CRC.
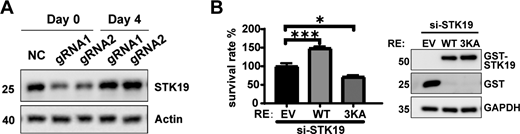
STK19 is involved in cell proliferation. (A) The expression of STK19 in HeLa polyclonal KD cells over time. STK19 was re-expressed after 4 days of culture. Days 0 represent cells that were incubated with 6 µg/ml puromycin (fresh KD cells). (B) Cell viability of STK19 KD HCT116 cells rescued with siRNA-resistant WT or 3KA. Error bars represent the SEM of three biological replicates. *P < 0.05, ***P < 0.001. Two-tailed unpaired Student’s t test was used. The right shows the expression level of GST-STK19-WT or 3KA. RE: rescue. EV: empty vector. The molecular weights are in kD. Source data are available for this figure: SourceData FS5.
STK19 is involved in cell proliferation. (A) The expression of STK19 in HeLa polyclonal KD cells over time. STK19 was re-expressed after 4 days of culture. Days 0 represent cells that were incubated with 6 µg/ml puromycin (fresh KD cells). (B) Cell viability of STK19 KD HCT116 cells rescued with siRNA-resistant WT or 3KA. Error bars represent the SEM of three biological replicates. *P < 0.05, ***P < 0.001. Two-tailed unpaired Student’s t test was used. The right shows the expression level of GST-STK19-WT or 3KA. RE: rescue. EV: empty vector. The molecular weights are in kD. Source data are available for this figure: SourceData FS5.
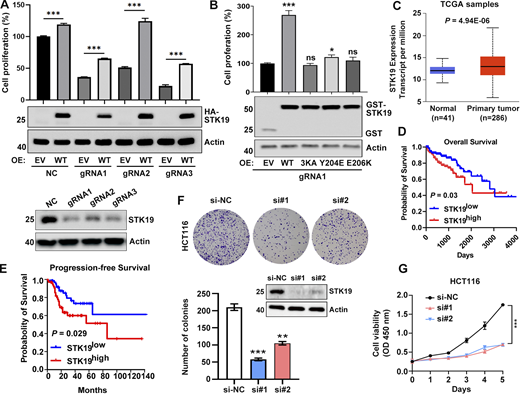
STK19 is critical for the proliferation of HeLa and HCT116 cells. (A) Cell viability of NC and STK19 KD HeLa cells. NC (control gRNA/CRISPR/Cas9 mediated NC) and KD (gRNA/CRISPR/Cas9 mediated STK19 KD). After seeding in 96-well plates, cells were transfected with EV (empty vector) or gRNA-resistant HA-STK19 and cultured for 3 days prior to sulforhodamine B staining. Error bars represent SEM. Two-tailed unpaired Student’s t test was used. ***P < 0.001. STK19 KD efficiency before seeding was analyzed by western blotting and shown in the bottom panel. (B) Cell viability of STK19 KD cells (gRNA1) transfected with gRNA-resistant WT, 3KA, or dimerization mutants. Error bars represent SEM. All groups were compared with EV, and two-tailed unpaired Student’s t test was used. OE: overexpression. (C) Boxplot showing relative mRNA expression of STK19 between normal and colorectal cancer samples in TCGA samples using UALCAN web portal (https://ualcan.path.uab.edu). (D) Kaplan–Meier plot depicting overall survival of CRC patients with tumors expressing high (red) or low (blue) levels of STK19 in TCGA samples using the online database OncoLnc (Gehan-Breslow-Wilcoxon test). (E) Kaplan–Meier plot depicting overall survival of CRC patients with tumors expressing high (red) or low (blue) levels of STK19 using the online database GEPIA2 (log-rank test). (F) Western blot analysis of STK19 protein levels in HCT116 cells transduced with two different siRNAs targeting STK19 coding region (si#1 and si#2) or nontargeting control (si-NC). Representative images and quantification of colony formation assay for HCT116 cells with STK19 KD. Error bars represent the SEM of three biological replicates. Two-tailed unpaired Student’s t test was used. **P < 0.01; ***P < 0.001. (G) Cell growth of HCT116 cells with STK19 KD detected by CCK8 assay compared with their control (si-NC). Error bars represent the SEM of three biological replicates. Two-tailed unpaired Student’s t test was used. ***, P < 0.001. The molecular weights are in kD. Source data are available for this figure: SourceData F8.
STK19 is critical for the proliferation of HeLa and HCT116 cells. (A) Cell viability of NC and STK19 KD HeLa cells. NC (control gRNA/CRISPR/Cas9 mediated NC) and KD (gRNA/CRISPR/Cas9 mediated STK19 KD). After seeding in 96-well plates, cells were transfected with EV (empty vector) or gRNA-resistant HA-STK19 and cultured for 3 days prior to sulforhodamine B staining. Error bars represent SEM. Two-tailed unpaired Student’s t test was used. ***P < 0.001. STK19 KD efficiency before seeding was analyzed by western blotting and shown in the bottom panel. (B) Cell viability of STK19 KD cells (gRNA1) transfected with gRNA-resistant WT, 3KA, or dimerization mutants. Error bars represent SEM. All groups were compared with EV, and two-tailed unpaired Student’s t test was used. OE: overexpression. (C) Boxplot showing relative mRNA expression of STK19 between normal and colorectal cancer samples in TCGA samples using UALCAN web portal (https://ualcan.path.uab.edu). (D) Kaplan–Meier plot depicting overall survival of CRC patients with tumors expressing high (red) or low (blue) levels of STK19 in TCGA samples using the online database OncoLnc (Gehan-Breslow-Wilcoxon test). (E) Kaplan–Meier plot depicting overall survival of CRC patients with tumors expressing high (red) or low (blue) levels of STK19 using the online database GEPIA2 (log-rank test). (F) Western blot analysis of STK19 protein levels in HCT116 cells transduced with two different siRNAs targeting STK19 coding region (si#1 and si#2) or nontargeting control (si-NC). Representative images and quantification of colony formation assay for HCT116 cells with STK19 KD. Error bars represent the SEM of three biological replicates. Two-tailed unpaired Student’s t test was used. **P < 0.01; ***P < 0.001. (G) Cell growth of HCT116 cells with STK19 KD detected by CCK8 assay compared with their control (si-NC). Error bars represent the SEM of three biological replicates. Two-tailed unpaired Student’s t test was used. ***, P < 0.001. The molecular weights are in kD. Source data are available for this figure: SourceData F8.
Discussion
In the first report that suggested STK19 is a kinase, the authors mutated K186, K201, and K203 of STK19 and found that the kinase activity was inhibited (Gomez-Escobar et al., 1998). Our results showed that these mutations are not active site mutations but protein surface mutations that should affect DNA binding and result in cytoplasmic mislocalization. Furthermore, HA-tag immunoprecipitated STK19 was used to demonstrate the kinase activity of STK19 (Gimple and Wang, 2019; Gomez-Escobar et al., 1998; Yin et al., 2020), but we found that many kinases bound to STK19 in cells (Data S2). The experiments of Rodríguez-Martinez et al. at best can rule out the possibility of STK19 being an NRas-specific kinase, but it remained possible that STK19 is a kinase. Our structural and biochemical analysis firmly established that STK19 is highly unlikely to be a kinase but a DNA/RNA-binding protein. To avoid further mistakes and to prevent the development of more kinase inhibitors for STK19 (Qian et al., 2020; Yin et al., 2019), we renamed this protein as TWH19 (Tandem Winged Helix protein previously known as STK19) hereafter and urge timely correction to its annotations.
WHD usually uses α3 and W1 to interact with DNA or protein (Harami et al., 2013). The α3 of WH3 is mostly buried by WH1, and the W1 of WH3 is involved in protein dimerization (Fig. S2 A). Instead, WH3 uses its α1 to interact with DNA. In WH1 and WH2, the α3 and W1 elements are generally exposed, rendering them possible for protein–protein interactions. Some of the TWH19-interacting proteins such as RPA2 and PCNA may bind to WH1 or WH2 or both domains. Therefore, the architecture of TWH19 may allow it to serve as a multivalent scaffold for DNA and DDR factors. Nonetheless, it is unlikely that TWH19 directly interacted with every DDR factor, but its interaction with some DDR factors may be mediated through DNA or direct-interacting DDR factors. The multivalence of TWH19 can be further increased by its dimerization, which is supported by multiple lines of evidence including crystal structure b-factors, purification yield, crosslinking, and pull-down. Dimer interface mutants partially mislocalized TWH19 and barely rescued the proliferation of TWH19 KD cells, suggesting that TWH19 functions as dimers in cells.
Two previous genomic screening studies indicated that TWH19 is involved in transcription-coupled NER (TCNER) (Boeing et al., 2016), a subtype of NER that is activated during transcription blockade, or NER (Olivieri et al., 2020), but whether TWH19 participates in other repair pathways is not tested. Here, we showed that TWH19 played a role in both NER and MMR but not double-stranded repair. This should not be surprising since many DDR proteins such as RPA2 and PCNA are shared in multiple DNA repair pathways (Pearl et al., 2015). TWH19 is possibly involved in both GG-NER and TCNER since it interacted with both the GG-NER player DDB1 and the TCNER player CSB (Iovine et al., 2011; Tiwari et al., 2021). In TCNER, TWH19 may bind to dsDNA adjacent to the damage site after upstream factors CSB and RPA2 bind to DNA lesions, preventing their dissociation and recruiting downstream factors such as PCNA (Fig. 6). PCNA promotes the processivity of DNA polymerase during DNA repair and is degraded after damage repair to prevent genomic instability (Cazzalini et al., 2014). Recruitment of MSH2, an MMR factor, can activate E3 ubiquitin ligases CRL4 to degrade repair factors after repair in NER (Tanaka et al., 2017), but whether PCNA is degraded by CRL4 is unclear. TWH19 may additionally regulate NER and MMR by regulating the expression level of several repair factors in these pathways (Fig. S3 B), but further validation is needed.
Tumor cells are particularly sensitive to DDR inhibitors (O’Connor, 2015). Radiotherapy and chemotherapy induce DNA damage to drive cancer cells into apoptosis and senescence (Ou and Schumacher, 2018). Several PARP inhibitors have been licensed to treat cancers (Gourley et al., 2019). Here, the DDR protein TWH19 was also shown to be critical for the proliferation and, to a lesser extent, the apoptosis of human cells. As TWH19 is amplified or overexpressed in several cancer types (AACR Project GENIE Consortium, 2017; Uhlen et al., 2017), it may play an oncogenic role in affected cancer and serve as a new anticancer target. Another striking observation in cells depleted of TWH19 (via either siRNA or gRNA) was the downregulation of metabolic pathways, which may explain the decrease in cell proliferation. These KD observations are consistent with the DDR function of TWH19, whereby cells respond to unrepaired DNA damage (possibly via JNK activation; Li and Chen, 2018; Milanese et al., 2019; Surova and Zhivotovsky, 2013; Williams and Schumacher, 2016). However, the cell cycle, which is often arrested in the presence of unrepaired DNA damage (Fousteri and Mullenders, 2008), was barely altered by TWH19 KD (Fig. S4, H and I), suggesting that the KD phenotypes may not be sorely due to DNA damage responses. Alternatively, TWH19 may regulate these cellular processes and cell fate via a DDR-independent route, i.e., binding to RNA and regulating mRNA levels. Further studies are needed to elucidate how TWH19 exerts its molecular functions to mediate these diverse cellular processes.
Materials and methods
Antibodies and plasmids
The DNA constructs and antibodies used in this paper are listed in Table S2.
Cloning, expression, and purification
Human STK19 (accession # NM_004197.2), STK19-C (25–254), 3KA (STK19K186A, K190A, K201A) RPA2, PCNA, or H4 were cloned into a psyno-1 vector expressing an N-terminal cleavable 6His-MBP fusion to facilitate its protein expression. His-STK19-WT and its Y204E, E206K, or L248A mutations were cloned into a psyno-1 vector expressing an N-terminal 6His. All proteins were expressed in the E. coli BL21 (DE3) strain and grown in LB broth medium. Protein expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside, and the culture was grown overnight at 18°C. The protein was purified by Ni-NTA beads and eluted with a buffer containing 30 mM Tris, pH 8.0, 300 mM NaCl, 300 mM imidazole, and 10% glycerol. The eluate was collected, added with 2 mM DTT, and further purified by size exclusion chromatography using a Superdex 200 Increase gel filtration column (GE Healthcare) in 20 mM Tris, pH 8.0, 200 mM NaCl, 2 mM DTT, and 10% glycerol.
Crystallization and data collection
Crystals of selenomethionine (SeMet)-labeled STK19 (25-C) were grown using the hanging-drop vapor-diffusion method at 16°C by mixing an equal volume of STK19-C (10 mg/ml) and crystallization buffer. The crystallization conditions contained 0.1 M 2-(N-morpholino)ethanesulfonic acid, pH 5.5, 0.2 M NaCl, and 2.0 M (NH4)2SO4. All crystals were cryo-protected using 20% (vol/vol) glycerol-supplemented crystallization buffer and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at the Shanghai Synchrotron Radiation Facility beamline BL17U1. The data collection statistics are given in Table 1.
Structure solution and refinement
The structure was solved by selenium single-wavelength anomalous dispersion using the program Autobuild and manually built using the program COOT. Refinement was performed using the program Refmac. Data at intervals of 50.00–1.65 Å resolution were used, and at the end of the refinement, the R-value was 0.149 (Rfree = 0.194) for all reflections. Coordinates were deposited PDB with accession codes 7XRB.
Pull-down assay
To assess different interactions, maltose binding protein (MBP)-tagged proteins were immobilized on dextrin beads (Smart Lifesciences). Soluble proteins at the indicated concentrations were incubated with the immobilized proteins in a total volume of 1 ml for 2 h at 4°C. After three washing steps, bound proteins were separated by 12% SDS/PAGE and visualized by Coomassie Blue staining. Each experiment was repeated at least twice and checked for consistency. The pull-down buffer contained 20 mM Tris, pH 8.0, 200 mM NaCl, 10% glycerol, 0.005% Triton X-100, and 5 mM DTT.
EMSA
DNA (25 μM, 2 μl) was incubated with increasing amounts of proteins in a buffer (50 mM NaCl, 20 mM Tris8.0, 10% glycerol, 1 mM DTT, and 2 mM MgCl2) for 30 min on ice (total 20 μl). After incubation, all samples were loaded onto an 8% nondenaturing PAGE gel. Gel electrophoresis was performed with cold TBE buffer (180 mM Tris/boric acid buffer, pH 7.6, and 2 mM EDTA) at 80 V for 60 min. Gels were stained with ethanol bromide and photographed with a gel documentation system (GenoSens 1880).
Cell culture and transfection
STK19 or its mutants were cloned into pcDNA3.1(+) or pEBG. HEK293T, HeLa, and B16-F10 cell lines were cultured in DMEM supplemented with 10% fetal bovine serum standard (Newzerum) at 37°C. HEK293T cells were transfected with polyethylenimine (PEI) transfection reagent (Invitrogen). HeLa cells were transfected with liposome transfection reagent (Yeasen). The cells were cultured at different times after transfection and treated differently. For example, cells were harvested and lysed directly for western blot and exposure (Clinx Science Instruments).
Immunofluorescent staining and confocal microscopy
Samples were fixed with 4% formaldehyde in PBS for 15 min and permeabilized with 0.2% Triton X-100 (Sangon Biotech) in PBS for 15 min. After blocking with 5% BSA in PBS for 1 h, samples were incubated with primary antibodies overnight at 4°C. After washing, the samples were incubated with fluorescently tagged secondary antibodies for 1 h at room temperature. Confocal images were acquired by ZEN blue software under a confocal microscope (LSM 880 with airyscan; Zeiss) with a 63×/1.40 oil-immersion objective lens (Zeiss) at room temperature. The fluorochromes used were Alexa Fluor 546 and DAPI. Images were prepared in Fiji and Adobe Photoshop. γ∙H2AX foci numbers were quantified using the Analyze Particles plugin in ImageJ.
Pull-down detection of STK19-interacting proteins in cells
Cells were harvested and lysed in lysis buffer (P0013; Beyotime Biotechnology) containing 1% protease inhibitors (B14002; Bimake) with or without DNase/RNase. Glutathione beads (Smart Lifesciences) and the supernatant from cell lysates were mixed and incubated at 4 °C for 6 h. The beads were then washed with lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40) four times, and the bound proteins were analyzed with the respective antibodies.
UVB irradiation
HeLa, 293T, or B16-F10 cells were irradiated with UVB at the indicated doses. To avoid UVB absorption by the medium, the cells were washed with PBS prior to irradiation, and fresh medium was replaced thereafter. The cells were then returned to the incubator and allowed to recover for 2 h before harvesting or fixing.
Chromatin fractionation
Cells were harvested and resuspended in lysis buffer A containing 10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100, and protease inhibitor cocktail. After centrifuging at 4°C for 4 min at 1,500 g, the supernatant was collected as the cytoplasm. The pellet was gently resuspended in buffer B (3 mM EDTA, 0.2 mM EGTA, and 1 mM DTT) with protease inhibitor and placed on ice for 10 min. Cells were then centrifuged at 4°C for 4 min at a speed of 2,000 g and the pellet was collected as the chromatin fraction.
Lentiviral-mediated polyclonal KD
For CRISPR-Cas9-mediated gene knockout, gRNA sequences (5′-GTCCTCACCGTCCGAGATGC-3′ or 5′-CCCATCGTGCTGAGGAGCCA-3′) were introduced into the V2T construct via PCR. Constructs encoding Cas9 and gRNA were cotransfected with viral packaging plasmids (V2T: pMDLg/pRRE:pRes-Rev:pCMV-VSVG = 3:1:1:1) into HEK293T cells. After 48 h, the viral supernatants were collected, filtered, and added to 6 μg/ml polybrene (YEASEN) before infecting HeLa cells. After 48 h of infection, the target cells were cultured in selection medium containing 8 μg/ml puromycin (BBI Life Sciences). Since these cells always repressed STK19 in the absence of puromycin, the efficiency of KD was enforced by puromycin selection before being used in any experiment. A portion of the puromycin-treated cells was used to determine the level of KD by immunoblotting.
RT-qPCR
Total RNA was extracted with a SteadyPure universal RNA extraction kit (Accurate Biotechnology) as described in the manufacturer’s manual. RNA was reverse-transcribed into cDNA and analyzed by RT-qPCR using a SYBR Green qPCR kit (Accurate Biotechnology). Three independent replicates were analyzed per sample and the relative gene expression was normalized to the internal tubulin (TUBB) control using the 2−ΔΔCt method. Error bars represent the SEM of three biological replicates. Two-tailed unpaired Student’s t test was used in all groups. *P < 0.05; **P < 0.01; ***P < 0.001. The qPCR primers used are listed in Table S3.
MS analysis
After GST pull-down with or without DNase/RNase, the proteins that interact with STK19 were boiled on beads and subsequently collected for in-gel digestion. After separation by SDS-PAGE, the gel was stained using colloidal Coomassie G-250 and the gel band was cut into 1 mm³ small pieces. The gel particles were washed with 50% ethanol and water and then dehydrated in 100% acetonitrile until the gel particles became opaque white in color. After drying, the gel was reduced by 5 mM DTT at 56°C for 1 h and alkylated with 15 mM iodoacetamide in the dark at room temperature for an additional 30 min. Then, the gel was digested with trypsin at 37°C overnight and the tryptic peptides were extracted by a decreasing gradient of trifluoroacetic acid and an increasing gradient of acetonitrile. After being desalted using C18 ZipTip, the peptides were analyzed using an EASY-nLC 1000 instrument coupled to a Q Exactive plus mass spectrometer.
To delineate the proteomic profile after STK19 KD, TMT-based quantitative proteomics was carried out. Three biological replicates of STK19 negative control (NC) and KD cells were first lysed in radioimmunoprecipitation assay buffer (150 mM NaCl, 50 mM Tris, 1% NP-40 [vol/vol], 0.5% sodium deoxycholate [wt/vol], pH = 7.5) containing protease and phosphatase inhibitors, followed by 3 min of sonication (3 s on and 10 s off, amplitude 25%). The lysate was centrifuged at 20,000 g for 10 min and the supernatant was collected. The protein concentration was determined using the Bradford assay. 50 µg of protein from each sample was reduced with 10 mM tris(2-carboxyethyl)phosphine at 56°C for 1 h and alkylated with 20 mM iodoacetamide for 30 min to block the free cysteine residues. After protein precipitation by methanol/chloroform, trypsin digestion was carried out overnight at 37°C. Peptides were labeled with TMT according to the TMT 10plex reagent instructions (Thermo Fisher Scientific). After a 1-h incubation at room temperature, 5% hydroxylamine was added to quench the reaction. Peptides labeled with nine different TMT labels were mixed, dried, and desalted. Then, the desalted TMT-labeled peptides were fractionated by HPLC (Agilent 1260; Agilent), and the obtained 120 fractions were combined into 20 parts. The 20 combined fractions were desalted with C18 ZipTip and analyzed by an EASY-nLC 1200 instrument coupled to a Q Exactive HF-X mass spectrometer.
All the raw files were searched against the Swiss-Prot human protein sequence database in MaxQuant (version 1.6). TMT isobaric labeling-based relative quantification algorithms were used for protein quantification.
Statistical analysis
All results were repeated at least three times. Statistical analyses were performed using GraphPad Prism 8 software. Comparisons between groups were analyzed by unpaired two-tailed Student’s t test where indicated. The statistical parameters (including n, SEM, and SD) are shown in the figure legends. Data distribution was assumed to be normal, but this was not formally tested. *P < 0.05; **P < 0.01; ***P <0.001. P values < 0.05 are considered to be significant.
Online supplemental material
The associated supplemental files contain five figures, three tables, and two Excel files. Fig. S1 shows that STK19 has no kinase activity. Fig. S2 shows that STK19 can form a homodimer to bind dsDNA. Fig. S3 shows that STK19 is involved in DDR. Fig. S4 shows that STK19 is implicated in multiple cellular pathways. Fig. S5 shows that STK19 is involved in cell proliferation. Table S1 contains different DNA sequences used in this study. Table S2 contains a summary of antibodies and plasmids used in this work. Table S3 is the list of primer sequences used for qPCR. Data S1 contains the MS data about kinase activity detection of STK19 on NRas. Data S2 shows the list of proteins bound to STK19 and TMT MS data.
Data availability
All the MS raw data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX (Ma et al., 2019) partner repository with the dataset identifier PXD034123. The data are available from the corresponding author upon reasonable request.
Acknowledgments
We thank the staff from the BL17B1/BL18U1/BL19U1/BL19U2/BL01B beamline of the National Facility for Protein Science in Shanghai at Shanghai Synchrotron Radiation Facility for assistance during data collection (Zhang et al., 2019).
This work was supported by the National Key R&D Program of China (No. 2018YFC2000305), and the National Natural Science Foundation of China (No. 82273850, 31870826, 81570060, and 82073221).
Author contributions: Q. Sun conceived the project. Q. Sun, Y. Li, Y. Gong, and L. Dai prepared the manuscript. Y. Li, Y. Gong, Y. Zhou, Y. Xiao, and W. Huang performed all the experiments. Q. Sun, L. Dai, and Y. Zhao supervised the project. S. Zhang, Y. Tu, and Q. Zhou provided relevant resources.

