


Mitochondria critically rely on protein import and its tight regulation. Here, we found that the complex I assembly factor NDUFAF8 follows a two-step import pathway linking IMS and matrix import systems. A weak targeting sequence drives TIM23-dependent NDUFAF8 matrix import, and en route, allows exposure to the IMS disulfide relay, which oxidizes NDUFAF8. Import is closely surveyed by proteases: YME1L prevents accumulation of excess NDUFAF8 in the IMS, while CLPP degrades reduced NDUFAF8 in the matrix. Therefore, NDUFAF8 can only fulfil its function in complex I biogenesis if both oxidation in the IMS and subsequent matrix import work efficiently. We propose that the two-step import pathway for NDUFAF8 allows integration of the activity of matrix complex I biogenesis pathways with the activity of the mitochondrial disulfide relay system in the IMS. Such coordination might not be limited to NDUFAF8 as we identified further proteins that can follow such a two-step import pathway.

Introduction
Mitochondria are essential organelles that harbor more than 1,200 proteins to fulfill their diverse functions in energy metabolism, synthesis of biomolecules, and cellular signaling (Bock and Tait, 2020; Martinez-Reyes and Chandel, 2020; Montague et al., 2014; Morgenstern et al., 2021; Pfanner et al., 2019; Spinelli and Haigis, 2018). Mitochondrial protein amounts are controlled by carefully balancing translation rates, mitochondrial protein import, and degradation processes (Bragoszewski et al., 2017; Deshwal et al., 2020; Kummer and Ban, 2021; Ott et al., 2016; Pfanner et al., 2019; Richter-Dennerlein et al., 2015; Song et al., 2021).
The majority of mitochondrial proteins are synthesized on cytosolic ribosomes and become imported via dedicated machineries into the different mitochondrial subcompartments (Edwards et al., 2021; Endo et al., 2011; Finger and Riemer, 2020; Hansen and Herrmann, 2019; Pfanner et al., 2019). For import into the mitochondrial matrix, the outer (OMM) and inner (IMM) mitochondrial membranes and the intermembrane space (IMS) have to be traversed. Most matrix proteins are synthesized as precursor proteins containing an N-terminal mitochondrial targeting sequence (MTS) that can vary in strength (Calvo et al., 2017; Tasaki et al., 2012; Vaca Jacome et al., 2015; Vogtle et al., 2009). These MTS are poorly conserved at the primary sequence level; however, presequence properties such as a length between 20 and 60 amino acids and a positive net charge between +3 and +6 are well conserved (Calvo et al., 2017). After import, most MTS are processed by matrix proteases (MPP, Icp55, and Oct1) to reveal (often stabilizing) amino acids at the neo-N-terminus that follow the N-end rule from bacteria (Tasaki et al., 2012; Vaca Jacome et al., 2015; Vogtle et al., 2009). During import, the MTS mediates the interaction of precursor proteins with OMM receptors that are part of the translocase of the OMM (TOM; Abe et al., 2000; Callegari et al., 2020; Dekker et al., 1998; Endo and Kohda, 2002; Esaki et al., 2004; Meisinger et al., 1999; Shiota et al., 2011). After the passage of the unfolded precursor through the TOM pore, the MTS interacts with subunits of the translocase of the IMM (TIM23 complex). The TIM23 and TOM complexes are closely tethered during import, presumably to prevent exposure of the precursor protein to the IMS (Callegari et al., 2020; Chacinska et al., 2003; Demishtein-Zohary and Azem, 2017). The TIM23 complex facilitates precursor translocation across the IMM in conjunction with the membrane potential and an ATP-dependent import motor at the matrix site (Demishtein-Zohary and Azem, 2017; Mokranjac, 2020; Schulz et al., 2015). In most cases, the precursor is unfolded during transit to the matrix; however, in some rare cases, translocation of partially folded precursors through the TIM23 channel has been reported (Huang et al., 2002; Longen et al., 2014; Sato et al., 2019).
Most soluble proteins of the IMS do not contain classical N-terminal MTS. Instead, they harbor conserved cysteine residues often in so-called twin-CX9C motifs that become oxidized to disulfide bonds during the import process, thereby becoming significantly more stably folded (Chacinska et al., 2009; Edwards et al., 2021; Finger and Riemer, 2020; Habich et al., 2019b; Longen et al., 2009; Reinhardt et al., 2020). The mitochondrial disulfide relay machinery facilitates this oxidation. Its key component is the oxidoreductase MIA40/CHCHD4, which directly interacts with and oxidizes incoming substrate proteins (Banci et al., 2009; Hofmann et al., 2005; Mesecke et al., 2005; Rissler et al., 2005). In human cells, the soluble MIA40/CHCHD4 forms a permanent trimeric complex with the IMM-anchored protein AIFM1 (Elguindy and Nakamaru-Ogiso, 2015; Hangen et al., 2015; Herrmann and Riemer, 2020; Meyer et al., 2015; Romero-Tamayo et al., 2021; Salscheider et al., 2022; Susin et al., 1999). The binding of MIA40/CHCHD4 by AIFM1 is crucial for its activity and only takes place if AIFM1 dimerizes, which it does in an NADH-dependent fashion (Romero-Tamayo et al., 2021; Salscheider et al., 2022). After MIA40/CHCHD4 oxidized its substrates, it is regenerated by the sulfhydryl oxidase ALR, which eventually shuffles electrons via cytochrome c to complex IV and molecular oxygen (Allen et al., 2005; Banci et al., 2011; Bihlmaier et al., 2007; Erdogan et al., 2018; Fischer et al., 2013; Rissler et al., 2005).
The IMM contains the protein complexes of the respiratory chain. These large multisubunit complexes are assembled from proteins of dual genetic origin (i.e., they are synthesized at cytosolic or mitochondrial ribosomes) from both sides of the IMM (Priesnitz and Becker, 2018; Vercellino and Sazanov, 2022). For example, complex I of the respiratory chain consists of >40 subunits. Seven of its subunits are translated at mitochondrial ribosomes (ND1-ND6 and ND4L; Guerrero-Castillo et al., 2017; McKenzie and Ryan, 2010). Four further subunits (NDUFA8, NDUFB7, NDUFB10, and NDUFS5) that face the IMS become imported by the disulfide relay (Friederich et al., 2017; Salscheider et al., 2022). The remaining proteins are imported via the TIM23 pathway. Complex I is assembled in a modular fashion; early assembly modules form around the mitochondria-encoded subunits, while the disulfide relay-dependent subunits serve as molecular clamps that join submodules at a later stage together (Guerrero-Castillo et al., 2017; Salscheider et al., 2022).
The dual genetic origin (cytosolic and nuclear genomes) and the use of different mitochondrial import pathways by subunits of the respiratory chain necessitate close coordination (Couvillion et al., 2016; Suhm et al., 2018). This includes aligning mitochondrial translation with respiratory chain assembly by, e.g., translational activators in yeast or the MITRAC complex in human cells (Herrmann et al., 2013; Mick et al., 2012). Additionally, posttranslational modifications on the import machinery (e.g., phosphorylation on TOM complex components; Schmidt et al., 2011; Walter et al., 2021) or on precursors (e.g., proteolytic processing to stimulate cytosolic degradation; Finger et al., 2020) integrate mitochondrial functional state with biogenesis. Furthermore, different pathways have been described to coordinate impaired protein import, respiratory chain assembly, and cytosolic protein homeostasis (Boos et al., 2019; Couvillion et al., 2016; Martensson et al., 2019; Wrobel et al., 2015). To remove excess proteins including non-assembled intermediates of the respiratory chain, dedicated proteases in both IMS and matrix exist. YME1L in the IMS and Lon, CLPP, and the m-AAA protease in the matrix are the major ATP-dependent proteases involved in general quality control degrading damaged or abnormal proteins (Deshwal et al., 2020; Szczepanowska and Trifunovic, 2021). Functions of these proteases include the degradation of complex I assembly intermediates that fail to fully assemble (YME1L) and the maintenance of active complex I by contributing to the selective exchange of damaged N-modules of complex I (CLPP; Stiburek et al., 2012; Szczepanowska et al., 2020).
Complex I assembly is supported by a plethora of assembly factors whose precise contributions to assembly are often poorly understood. For example, in the matrix, the assembly factor NDUFAF5 contributes to Q module assembly. NDUFAF5 is an unusual assembly factor as it structurally belongs to the family of S-adenosylmethionine (SAM)-dependent methyltransferases (Gerards et al., 2010; Rhein et al., 2016; Sugiana et al., 2008). However, instead of being a methyltransferase, it catalyzes the hydroxylation of an arginine residue in the Q module subunit NDUFS7, and this posttranslational modification is crucial in early complex I assembly (Gerards et al., 2010; Rhein et al., 2016; Sugiana et al., 2008). To fulfill its function, NDUFAF5 has been proposed to cooperate with the proteins NDUFAF8 (previously C17orf89; Alston et al., 2020; Floyd et al., 2016) and PYURF (Pei et al., 2022; Rensvold et al., 2022) in the mitochondrial matrix. PYURF stabilizes NDUFAF5. In line, loss of PYURF leads to loss of NDUFAF5 and complex I deficiency (Arroyo et al., 2016; Rensvold et al., 2022). NDUFAF8 has been proposed to stabilize NDUFAF5 as well. Similar to PYURF, loss of NDUFAF8 results in NDUFAF5 degradation and causes a stalled Q module assembly, leading to the accumulation of an assembly intermediate containing NDUFS2, NDUFS3, and NDUFA5 and consequently an isolated complex I deficiency (Alston et al., 2020; Floyd et al., 2016). While PYURF contains an MTS for import into the matrix, NDUFAF8, confusingly, has all the hallmarks of a disulfide relay substrate and thus should be localized to the IMS. It remains unclear how this discrepancy can be aligned with the function of NDUFAF8 in NDUFAF5 stabilization in the matrix.
Here, we characterize the complex maturation pathway of NDUFAF8. We find that this pathway provides a mechanism to coordinate the efficiency of IMS and matrix import pathways with early steps of complex I assembly in the matrix. NDUFAF8 contains four conserved cysteines and a very weak MTS. This MTS only facilitates slow matrix import and thereby allows access of import intermediates of NDUFAF8 to the disulfide relay. NDUFAF8 thereby acquires disulfide bonds before its transfer to the matrix. The import and maturation of NDUFAF8 are closely surveyed as the accumulation of reduced NDUFAF8 in the matrix is counteracted by the protease CLPP and of excess NDUFAF8 in the IMS by YME1L. In the matrix, using its non-cleavable MTS, NDUFAF8 interacts with its partner NDUFAF5 to stabilize and activate it. Matrix NDUFAF8 levels closely correlate with its capability to support complex I assembly. They are determined by the efficiency of NDUFAF8 oxidation in the IMS. This, in turn, depends on a functional disulfide relay and the NADH/NAD+ ratio in the IMS linking the metabolic state in the IMS to matrix protein stabilities and activities. Besides NDUFAF8, other proteins appear to follow a similar road into the matrix including the ribosomal protein CHCHD1 (in yeast, Mrp10), COA6 isoform 2, CHCHD2, CHCHD9, and CHCHD10 suggesting that this mode of IMS-matrix coordination is important for different matrix processes. Collectively, we characterized a two-step mitochondrial protein import pathway that allows sensing and integrating the functionality of IMS and matrix import machineries.
Results
NDUFAF8 localizes to the mitochondrial IMS and matrix
Complex I assembly relies on the action of different assembly factors. Among them are the assembly factors NDUFAF8 and NDUFAF5. Both proteins are poorly characterized but have been shown to interact with each other and to mediate the maturation of the Q-module, specifically its subunit NDUFS7 (Alston et al., 2020; Floyd et al., 2016; Rhein et al., 2016). NDUFAF5 contains a classical MTS and is a matrix protein, in line with its function in the biogenesis of the matrix-exposed Q module (Rhein et al., 2016). Conversely, NDUFAF8 localization is less clear. On the one hand, it harbors four conserved cysteines in a twin-CX9C pattern indicating the protein as a typical substrate of the mitochondrial disulfide relay and suggesting localization in the IMS (Fig. 1 A). On the other hand, NDUFAF8 interacts with NDUFAF5 (although it remained unclear where in mitochondria they interacted; Alston et al., 2020; Floyd et al., 2016), and careful sequence analysis indicated a presumably very weak MTS for mitochondrial matrix targeting in NDUFAF8. Although the TargetP algorithm predicted for NDUFAF8 only a probability of 0.0524 for matrix targeting, a sliding prediction of the TargetP score along the N-terminal region of NDUFAF8 using the iMTS algorithm (Boos et al., 2018) indicated a certain likelihood for the presence of matrix targeting information within the first 19 amino acids with a net charge of +4 (Fig. 1 A).
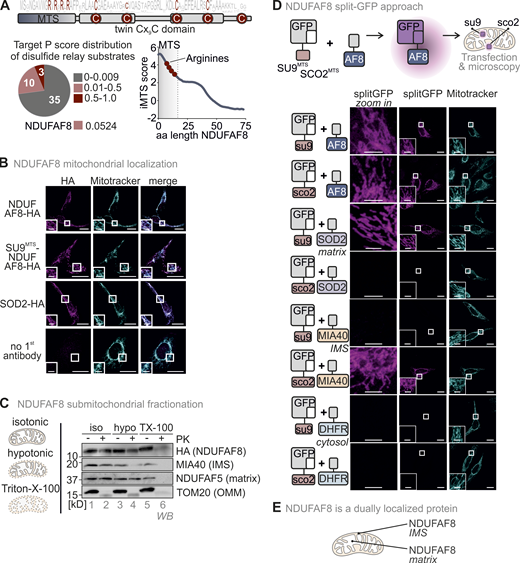
NDUFAF8 is dually localized to IMS and matrix. (A) Domain layout of NDUFAF8. NDUFAF8 contains four highly conserved cysteines in a twin CX9C motif. These cysteines reside in two alpha-helices. Four arginine residues and no negatively charged amino acid are found in the N-terminal 19 amino acids of NDUFAF8. According to TargetP, this region does not serve as MTS; however a sliding TargetP score prediction indicates a certain propensity of this region to serve as MTS. MTS, mitochondrial targeting signal; aa, amino acid. (B) Immunofluorescence analysis to localize NDUFAF8. NDUFAF8-HA localizes to mitochondria. HEK293 cells stably and inducibly expressing the indicated NDUFAF8-HA variants were fixated, permeabilized, and stained using a primary antibody against the HA epitope (HA) and mitotracker. Cells were analyzed by fluorescence microscopy. Bar corresponds to 20 µm. (C) Submitochondrial fractionation to detect the localization of NDUFAF8-HA. Mitochondria isolated from HEK293 cells expressing NDUFAF8-HA were exposed to three different buffer conditions, an isotonic buffer which leaves the OMM intact, a hypotonic buffer (hypo), which leads to swelling of the matrix and thereby disruption of the OMM, and a TX-100 containing buffer that solubilizes mitochondria completely. Afterward, mitochondria were treated with proteinase K (PK). TOM20, MIA40/CHCHD4, and NDUFAF5 served as control proteins localizing to OMM, IMS, and matrix, respectively. The signal of NDUFAF8 becomes weaker in the IMS fraction (lane 4) and disappears in the matrix fraction (lane 6) indicating NDUFAF8 resides in IMS and matrix as a dually localized protein. (D) Split-GFP assay to detect the localization of NDUFAF8. GFP can be split into two non-fluorescent parts, a larger part (GFP1-10) and a smaller part (GFP11). If these parts come together in the same compartment, they can self-reassemble to reconstitute a fluorescent GFP. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS respectively (see “split GFP” signal). DHFR-GFP11 co-expression did not result in the reconstitution of GFP with any of the mitochondria localized GFP1-10s. In the case of NDUFAF8, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10, indicating NDUFAF8 to be a protein localized to IMS and matrix. Bar corresponds to 20 µm. (E) Model. NDUFAF8 is dually localized to IMS and matrix. Source data are available for this figure: SourceData F1.
NDUFAF8 is dually localized to IMS and matrix. (A) Domain layout of NDUFAF8. NDUFAF8 contains four highly conserved cysteines in a twin CX9C motif. These cysteines reside in two alpha-helices. Four arginine residues and no negatively charged amino acid are found in the N-terminal 19 amino acids of NDUFAF8. According to TargetP, this region does not serve as MTS; however a sliding TargetP score prediction indicates a certain propensity of this region to serve as MTS. MTS, mitochondrial targeting signal; aa, amino acid. (B) Immunofluorescence analysis to localize NDUFAF8. NDUFAF8-HA localizes to mitochondria. HEK293 cells stably and inducibly expressing the indicated NDUFAF8-HA variants were fixated, permeabilized, and stained using a primary antibody against the HA epitope (HA) and mitotracker. Cells were analyzed by fluorescence microscopy. Bar corresponds to 20 µm. (C) Submitochondrial fractionation to detect the localization of NDUFAF8-HA. Mitochondria isolated from HEK293 cells expressing NDUFAF8-HA were exposed to three different buffer conditions, an isotonic buffer which leaves the OMM intact, a hypotonic buffer (hypo), which leads to swelling of the matrix and thereby disruption of the OMM, and a TX-100 containing buffer that solubilizes mitochondria completely. Afterward, mitochondria were treated with proteinase K (PK). TOM20, MIA40/CHCHD4, and NDUFAF5 served as control proteins localizing to OMM, IMS, and matrix, respectively. The signal of NDUFAF8 becomes weaker in the IMS fraction (lane 4) and disappears in the matrix fraction (lane 6) indicating NDUFAF8 resides in IMS and matrix as a dually localized protein. (D) Split-GFP assay to detect the localization of NDUFAF8. GFP can be split into two non-fluorescent parts, a larger part (GFP1-10) and a smaller part (GFP11). If these parts come together in the same compartment, they can self-reassemble to reconstitute a fluorescent GFP. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS respectively (see “split GFP” signal). DHFR-GFP11 co-expression did not result in the reconstitution of GFP with any of the mitochondria localized GFP1-10s. In the case of NDUFAF8, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10, indicating NDUFAF8 to be a protein localized to IMS and matrix. Bar corresponds to 20 µm. (E) Model. NDUFAF8 is dually localized to IMS and matrix. Source data are available for this figure: SourceData F1.
NDUFAF8 localization has not been assessed so far, and it was also not detected in any submitochondrial-compartment-targeted large-scale proteomic approach. Thus, we first investigated its precise submitochondrial localization using immunofluorescence, mitochondrial fractionation, and split-GFP approaches. A challenge we faced in this study was the lack of a suitable antibody against endogenous NDUFAF8. We thus had to rely on stably and inducibly expressed C-terminally HA-tagged NDUFAF8 for analysis of protein levels in cells or on the analysis of in vitro translated NDUFAF8 in in vitro import experiments into isolated mitochondria. In a set of control experiments, we therefore first verified that the HA-tag did not influence import kinetics and submitochondrial localization of NDUFAF8 (Fig. S1, A–D), which we had also previously confirmed for other disulfide relay substrates in human cell culture (Fischer et al., 2013). In the immunofluorescence approach, NDUFAF8-HA colocalized with mitotracker as a mitochondrial marker (Fig. 1 B). In the fractionation approach, we analyzed mitochondria isolated from HEK293 cells expressing NDUFAF8-HA. We found NDUFAF8 to be a mitochondrial protein that was not accessible to protease digestion of intact mitochondria and only in part accessible to protease digestion upon hypoosmotic opening of the OMM (Fig. 1 C, lanes 2 and 4). It was protease-sensitive as soon as the IMM was opened (Triton X-100 [TX-100]-treated mitochondria), indicating a potential dual localization of NDUFAF8 between IMS and matrix (Fig. 1 C, lane 6). As expected NDUFAF5 behaved like a matrix protein. In an orthogonal split-GFP approach, we equipped full-length NDUFAF8 with the small portion of the split-GFP system (NDUFAF8-GFP11) and coexpressed it together with the large portion (GFP1-10), which we targeted either to the IMS (SCO2MTS-GFP1-10) or the matrix (SU9MTS-GFP1-10, Fig. 1 D and Fig. S1 E). We thereby found self-reassembly of split-GFP in the matrix and the IMS. The controls for matrix (SOD2) and IMS (MIA40) self-reassembled exclusively in matrix and IMS, respectively, underlining the validity of the approach. We, therefore, conclude that NDUFAF8 is a protein dually localized in IMS and mitochondrial matrix (Fig. 1 E).
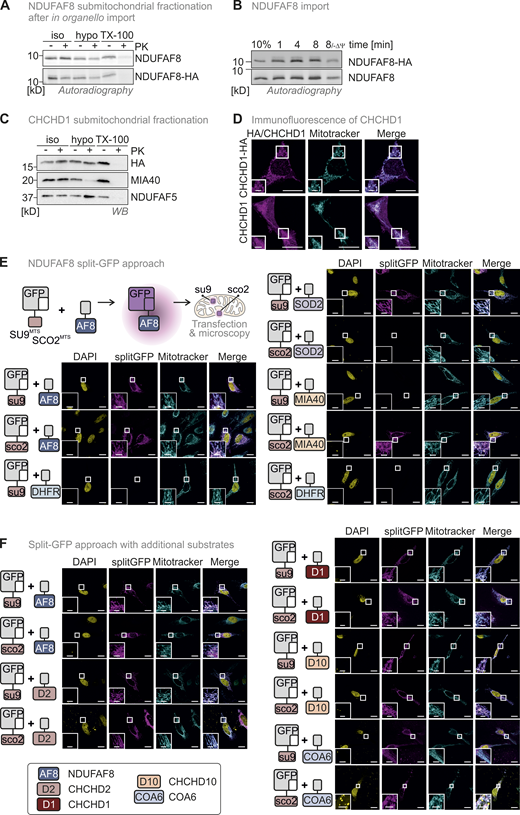
(Related toFigs. 1 and 8,). NDUFAF8 is representative of a class of dually localized proteins. Lack of an antibody against endogenous NDUFAF8 necessitated the use of HA-tagged NDUFAF8 throughout this study. A–D assess whether the tag influences import kinetics and (sub)-mitochondrial localization of NDUFAF8 and the similar behaving protein CHCHD1. E–F show the complete data sets for the split-GFP assays performed in this study (Figs. 1 and 8). (A) Submitochondrial fractionation to detect the localization of NDUFAF8 and NDUFAF8-HA after in organello import into mitochondria isolated from HEK293 cells. Experiment was performed as described in Fig. 2 G. NDUFAF8 and NDUFAF8-HA behave similarly in this assay, indicating that the HA tag does not influence the distribution of NDUFAF8 within mitochondrial subcompartments. (B) In organello import assay with NDUFAF8 and NDUFAF8-HA. Experiment was performed as described in Fig. 2 A. NDUFAF8 and NDUFAF8-HA behave similarly in this assay indicating that the HA tag does not influence the import kinetics of NDUFAF8. (C) Submitochondrial fractionation to detect the localization of CHCHD1 and CHCHD1-HA in mitochondria isolated from HEK293 cells. Experiment was performed as described in Fig. 1 C. CHCHD1 and CHCHD1-HA behave similarly in this assay, indicating that the HA tag does not influence the distribution of CHCHD1 within mitochondrial subcompartments. (D) Immunofluorescence analysis to localize CHCHD1 and CHCHD1-HA. CHCHD1 and CHCHD1-HA localize to mitochondria. Experiment was performed as described in Fig. 1 B. The HA tag did not influence the mitochondrial localization of CHCHD1. (E) Split-GFP assay to detect the localization of NDUFAF8. GFP can be split into two non-fluorescent parts, a larger part (GFP1-10) and a smaller part (GFP11). If these parts come together in the same compartment they can self-reassemble to reconstitute a fluorescent GFP. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS, respectively (see “split GFP” signal). DHFR-GFP11 coexpression did not result in the reconstitution of GFP with any of the mitochondria-localized GFP1-10s. In the case of NDUFAF8, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10, indicating NDUFAF8 to be a protein localized to IMS and matrix. DAPI stain and merge serve as orientation. Bar corresponds to 20 µm. (F) Split-GFP assay to detect the localization of different disulfide relay substrates. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 (AF8), CHCHD2 (D2), CHCHD1 (D1), CHCHD10 (D10), or COA6 isoform 2. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For all selected disulfide relay substrates, GFP reassembled indicating them to be proteins localized to IMS and matrix. DAPI stain and merge serve as orientation. Bar corresponds to 20 µm. Source data are available for this figure: SourceData FS1.
(Related toFigs. 1 and 8,). NDUFAF8 is representative of a class of dually localized proteins. Lack of an antibody against endogenous NDUFAF8 necessitated the use of HA-tagged NDUFAF8 throughout this study. A–D assess whether the tag influences import kinetics and (sub)-mitochondrial localization of NDUFAF8 and the similar behaving protein CHCHD1. E–F show the complete data sets for the split-GFP assays performed in this study (Figs. 1 and 8). (A) Submitochondrial fractionation to detect the localization of NDUFAF8 and NDUFAF8-HA after in organello import into mitochondria isolated from HEK293 cells. Experiment was performed as described in Fig. 2 G. NDUFAF8 and NDUFAF8-HA behave similarly in this assay, indicating that the HA tag does not influence the distribution of NDUFAF8 within mitochondrial subcompartments. (B) In organello import assay with NDUFAF8 and NDUFAF8-HA. Experiment was performed as described in Fig. 2 A. NDUFAF8 and NDUFAF8-HA behave similarly in this assay indicating that the HA tag does not influence the import kinetics of NDUFAF8. (C) Submitochondrial fractionation to detect the localization of CHCHD1 and CHCHD1-HA in mitochondria isolated from HEK293 cells. Experiment was performed as described in Fig. 1 C. CHCHD1 and CHCHD1-HA behave similarly in this assay, indicating that the HA tag does not influence the distribution of CHCHD1 within mitochondrial subcompartments. (D) Immunofluorescence analysis to localize CHCHD1 and CHCHD1-HA. CHCHD1 and CHCHD1-HA localize to mitochondria. Experiment was performed as described in Fig. 1 B. The HA tag did not influence the mitochondrial localization of CHCHD1. (E) Split-GFP assay to detect the localization of NDUFAF8. GFP can be split into two non-fluorescent parts, a larger part (GFP1-10) and a smaller part (GFP11). If these parts come together in the same compartment they can self-reassemble to reconstitute a fluorescent GFP. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS, respectively (see “split GFP” signal). DHFR-GFP11 coexpression did not result in the reconstitution of GFP with any of the mitochondria-localized GFP1-10s. In the case of NDUFAF8, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10, indicating NDUFAF8 to be a protein localized to IMS and matrix. DAPI stain and merge serve as orientation. Bar corresponds to 20 µm. (F) Split-GFP assay to detect the localization of different disulfide relay substrates. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8 (AF8), CHCHD2 (D2), CHCHD1 (D1), CHCHD10 (D10), or COA6 isoform 2. Combinations of GFP11 and GFP1-10-containing constructs were transfected and analyzed by fluorescence microscopy. For all selected disulfide relay substrates, GFP reassembled indicating them to be proteins localized to IMS and matrix. DAPI stain and merge serve as orientation. Bar corresponds to 20 µm. Source data are available for this figure: SourceData FS1.
NDUFAF8 employs its weak MTS but not its conserved cysteines for import into the mitochondrial matrix
The presence of two conflicting import signals in NDUFAF8 and its dual localization prompted us to investigate its mitochondrial import pathway. In vitro translated–full-length NDUFAF8 could be efficiently imported into mitochondria isolated from HEK293 cells (Fig. 2 A), notably in a membrane potential-dependent fashion and without any processing (Fig. 2 A, lane 5). “Shaving” mitochondria using proteinase K incubation prior to import did not affect the amounts of NDUFAF8 imported into mitochondria indicating that there is no strong dependency on OMM receptors that are normally required for MTS-dependent import (Fig. S2 A). We then tested for the relevance of the potential MTS in the N-terminal region of NDUFAF8 for its mitochondrial import. We generated fusion proteins of the N-terminal 19 residues of NDUFAF8 (NDUFAF8MTS) with mouse dihydrofolate reductase (DHFR) or a non-folded mutant variant of DHFR (DHFRmut; Vestweber and Schatz, 1988). Both versions of DHFR, when fused to NDUFAF8MTS, were imported into isolated mitochondria albeit with different efficiencies (Fig. 2 B and Fig. S2 B). DHFR alone was not imported, while the import of the SU9MTS-DHFR control progressed much more efficiently (SU9MTS being a strong MTS). The import of NDUFAF8MTS-DHFR and NDUFAF8MTS-DHFRmut was dependent on the membrane potential (Fig. 2 B).
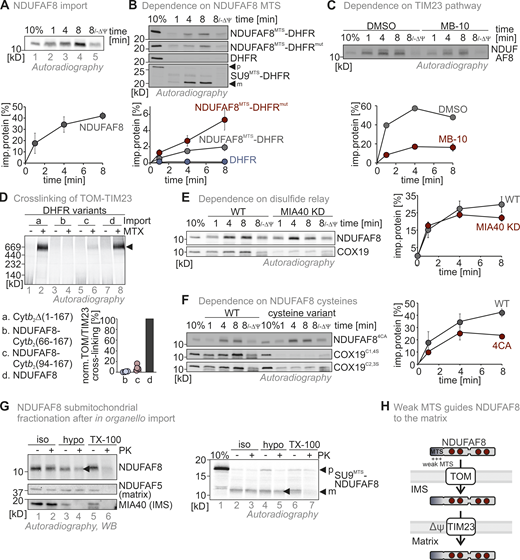
NDUFAF8 relies on its weak MTS for import into the matrix. (A) In organello import assay with NDUFAF8. In vitro translated radioactive NDUFAF8 was incubated with mitochondria isolated from HEK293 cells. Non-imported proteins were removed by treatment with Proteinase K. An import reaction was performed on mitochondria treated with CCCP to dissipate the mitochondrial membrane potential (–ΔΨ). Imported proteins were analyzed by reducing SDS-PAGE and autoradiography. Signals were quantified using ImageQuantTL, and the amount of imported protein was plotted. NDUFAF8 can be imported into mitochondria and relies on the mitochondrial membrane potential for import. N = 3 replicates; error bars indicate SD. (B) In organello import assay with NDUFAF8-DHFR fusion constructs. Experiment was performed as described in A. To test the capacity of the N-terminal 19 amino acids of NDUFAF8 (AF8MTS) to serve as MTS, they were fused to the cytosolic proteins DHFR or DHFRmut. The latter protein carries mutations that prevent it from stable folding and thereby enable mitochondrial import also by weaker MTS. AF8MTS facilitated the import of both forms of DHFR albeit with different efficiencies. DHFR alone was not imported into mitochondria. N = 3 replicates; error bars indicate SD. (C) In organello import assay with NDUFAF8 to test for its dependence on the TIM23 import pathway. The experiment was performed as described in A. To test for the dependence of NDUFAF8 on the TIM23 channel, mitochondria were incubated with 100 µM Mitoblock-10 (MB-10) to inhibit this pathway. NDUFAF8 import into mitochondria was strongly impaired. N = 1–3 replicates; error bars indicate SD. (D) In organello import-BN-PAGE assay with NDUFAF8-variants to analyze the binding of the DHFR-fused NDUFAF8 variants in the TOM-TIM23 supercomplex during import. The indicated radiolabeled NDUFAF8-DHFR variants and as control the cytochrome b2 (1-167)-DHFR fusion construct were imported into wild-type yeast mitochondria. The import was performed in the absence of the presence of methotrexate (MTX) as indicated. Protein complexes were analyzed by BN-PAGE and autoradiography. NDUFAF8-DHFR but not the extended precursor fusion constructs established the TOM-TIM23 supercomplex during import. N = 4 replicates. (E) In organello import assay with NDUFAF8 to test for its dependence on the disulfide relay import pathway. Experiment was performed as described in D. To test for the dependence of NDUFAF8 on the mitochondrial disulfide relay, import into mitochondria isolated from wild-type or MIA40 knockdown (MIA40 KD) was analyzed. COX19 served as a control protein that is highly dependent on MIA40/CHCHD4. NDUFAF8 import into mitochondria was not affected by lack of MIA40/CHCHD4. N = 3 replicates; error bars indicate SD. (F) In organello import assay with NDUFAF8-variants to test for its dependence on its cysteines. Experiment was performed as described in A. COX19 served as a control protein that is highly dependent on its cysteine residues for import. To test for the dependence of NDUFAF8 on its cysteines, a wild-type and a cysteine-to-alanine mutant of NDUFAF8 (NDUFAF8-4CA, mutation of all four conserved cysteines) were compared. Initial NDUFAF8 import appears to be independent of its cysteines, but amounts are lower after 8 min of import. Wild-type quantification are the data shown in A. N = 3 replicates; error bars indicate SD. (G) Submitochondrial fractionation to detect the localization of NDUFAF8 after in organello import into mitochondria isolated from HEK293 cells. Experiment was performed as described in A and Fig. 1 C. The signal of NDUFAF8 becomes weaker in the IMS fraction (lane 4) and disappears in the matrix fraction (lane 6) indicating NDUFAF8 resides in IMS and matrix as a dually localized protein. Conversely, NDUFAF8 equipped with a strong SUMTS does only disappear in the matrix fraction indicating its sole localization to the matrix. (H) Model. NDUFAF8 is imported into the matrix independently of the mitochondrial disulfide relay but “detours” through the IMS because of its weak MTS. Source data are available for this figure: SourceData F2.
NDUFAF8 relies on its weak MTS for import into the matrix. (A) In organello import assay with NDUFAF8. In vitro translated radioactive NDUFAF8 was incubated with mitochondria isolated from HEK293 cells. Non-imported proteins were removed by treatment with Proteinase K. An import reaction was performed on mitochondria treated with CCCP to dissipate the mitochondrial membrane potential (–ΔΨ). Imported proteins were analyzed by reducing SDS-PAGE and autoradiography. Signals were quantified using ImageQuantTL, and the amount of imported protein was plotted. NDUFAF8 can be imported into mitochondria and relies on the mitochondrial membrane potential for import. N = 3 replicates; error bars indicate SD. (B) In organello import assay with NDUFAF8-DHFR fusion constructs. Experiment was performed as described in A. To test the capacity of the N-terminal 19 amino acids of NDUFAF8 (AF8MTS) to serve as MTS, they were fused to the cytosolic proteins DHFR or DHFRmut. The latter protein carries mutations that prevent it from stable folding and thereby enable mitochondrial import also by weaker MTS. AF8MTS facilitated the import of both forms of DHFR albeit with different efficiencies. DHFR alone was not imported into mitochondria. N = 3 replicates; error bars indicate SD. (C) In organello import assay with NDUFAF8 to test for its dependence on the TIM23 import pathway. The experiment was performed as described in A. To test for the dependence of NDUFAF8 on the TIM23 channel, mitochondria were incubated with 100 µM Mitoblock-10 (MB-10) to inhibit this pathway. NDUFAF8 import into mitochondria was strongly impaired. N = 1–3 replicates; error bars indicate SD. (D) In organello import-BN-PAGE assay with NDUFAF8-variants to analyze the binding of the DHFR-fused NDUFAF8 variants in the TOM-TIM23 supercomplex during import. The indicated radiolabeled NDUFAF8-DHFR variants and as control the cytochrome b2 (1-167)-DHFR fusion construct were imported into wild-type yeast mitochondria. The import was performed in the absence of the presence of methotrexate (MTX) as indicated. Protein complexes were analyzed by BN-PAGE and autoradiography. NDUFAF8-DHFR but not the extended precursor fusion constructs established the TOM-TIM23 supercomplex during import. N = 4 replicates. (E) In organello import assay with NDUFAF8 to test for its dependence on the disulfide relay import pathway. Experiment was performed as described in D. To test for the dependence of NDUFAF8 on the mitochondrial disulfide relay, import into mitochondria isolated from wild-type or MIA40 knockdown (MIA40 KD) was analyzed. COX19 served as a control protein that is highly dependent on MIA40/CHCHD4. NDUFAF8 import into mitochondria was not affected by lack of MIA40/CHCHD4. N = 3 replicates; error bars indicate SD. (F) In organello import assay with NDUFAF8-variants to test for its dependence on its cysteines. Experiment was performed as described in A. COX19 served as a control protein that is highly dependent on its cysteine residues for import. To test for the dependence of NDUFAF8 on its cysteines, a wild-type and a cysteine-to-alanine mutant of NDUFAF8 (NDUFAF8-4CA, mutation of all four conserved cysteines) were compared. Initial NDUFAF8 import appears to be independent of its cysteines, but amounts are lower after 8 min of import. Wild-type quantification are the data shown in A. N = 3 replicates; error bars indicate SD. (G) Submitochondrial fractionation to detect the localization of NDUFAF8 after in organello import into mitochondria isolated from HEK293 cells. Experiment was performed as described in A and Fig. 1 C. The signal of NDUFAF8 becomes weaker in the IMS fraction (lane 4) and disappears in the matrix fraction (lane 6) indicating NDUFAF8 resides in IMS and matrix as a dually localized protein. Conversely, NDUFAF8 equipped with a strong SUMTS does only disappear in the matrix fraction indicating its sole localization to the matrix. (H) Model. NDUFAF8 is imported into the matrix independently of the mitochondrial disulfide relay but “detours” through the IMS because of its weak MTS. Source data are available for this figure: SourceData F2.
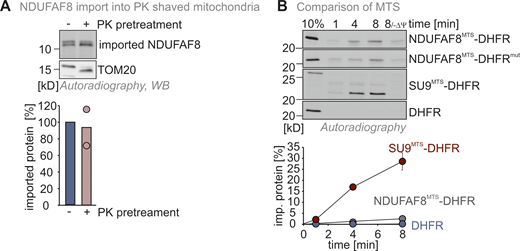
(Related toFig. 2,). NDUFAF8 import does not rely on OMM receptors. (A) In organello import assay with NDUFAF8 into mitochondria devoid of OMM proteins facing the cytosol. Experiment was performed as described in Fig. 2 A except that mitochondria were pretreated with proteinase K (PK) to remove surface receptors at the OMM. Import of NDUFAF8 into mitochondria was thereby not affected. N = 2 biological replicates. (B) In organello import assay with NDUFAF8-DHFR fusion constructs. Data are from Fig. 2 B. Quantification also includes the data for the SU9MTS-DHFR construct. N = 3 replicates; error bars indicate SD. Source data are available for this figure: SourceData FS2.
(Related toFig. 2,). NDUFAF8 import does not rely on OMM receptors. (A) In organello import assay with NDUFAF8 into mitochondria devoid of OMM proteins facing the cytosol. Experiment was performed as described in Fig. 2 A except that mitochondria were pretreated with proteinase K (PK) to remove surface receptors at the OMM. Import of NDUFAF8 into mitochondria was thereby not affected. N = 2 biological replicates. (B) In organello import assay with NDUFAF8-DHFR fusion constructs. Data are from Fig. 2 B. Quantification also includes the data for the SU9MTS-DHFR construct. N = 3 replicates; error bars indicate SD. Source data are available for this figure: SourceData FS2.
Next, we assessed the dependence of the import of full-length NDUFAF8 on the TIM23 pathway. We first employed the inhibitor MitoBloCK-10 (Miyata et al., 2017). MitoBloCK-10 attenuates protein-associated motor (PAM) complex activity by inhibiting TIMM44 binding to the mitochondrial precursor protein and to mitochondrial HSP70. NDUFAF8 import was delayed when isolated mitochondria were treated with MitoBloCK-10 (Fig. 2 C). We then assessed whether NDUFAF8 could provoke the formation of the TOM-TIM23 tethering complex, which is commonly observed during matrix precursor import, indicating that translocation processes across OMM and IMM occur in a synchronized manner (Fig. 2 D; Chacinska et al., 2003; Gomkale et al., 2021; Martensson et al., 2019). We used yeast mitochondria, in which the arrest of N-terminally DHFR-fused precursor proteins in the TOM-TIM23 supercomplex is well established (Chacinska et al., 2003; Martensson et al., 2019). During import into isolated mitochondria, methotrexate (MTX) is added, which leads to tight folding of DHFR. This prevents further import of the precursor–DHFR fusion protein as DHFR is stuck at the cytosolic face of the TOM channel. Subsequent analysis of the import complex on BN-PAGE revealed at a height of ∼600 kD a complex composed of TOM, TIM23, and precursor-DHFR. Using an artificial cytochrome b2 (1-167)-DHFR construct as control, we indeed detected this complex in the presence of MTX in our setup (Fig. 2 D, lane 2). When we used a full-length NDUFAF8–DHFR fusion, we also detected such a complex (Fig. 2 D, lane 8), indicating that the NDUFAF8-DHFR precursors span both the TOM and TIM23 complex and that both complexes are in proximity during NDUFAF8 import. We speculate that the arrest of the precursor in a TOM-TIM23 supercomplex is influenced by the distance between MTS and DHFR (as this indicates that initial translocation steps are concluded). To test whether the NDUFAF8-DHFR fusion also adhered to this rule, we elongated the NDUFAF8-DHFR construct by adding amino acids from the cytochrome b2 precursor between NDUFAF8 and DHFR (either 73 or 101 amino acids). As expected, this decreased the amounts of the TOM-TIM23 tethering complex to the fusion construct length in both cases (Fig. 2 D, lanes 4 and 6). Thus, despite its weak MTS, NDUFAF8 import shares the hallmarks of classical TIM23 import pathway substrates.
Besides the MTS, NDUFAF8 contains four conserved cysteines in a twin-CX9C motif pointing to a dependence of its import on the mitochondrial disulfide relay in the IMS. Import of full-length NDUFAF8 into mitochondria isolated from cells with low MIA40/CHCHD4 amounts exhibited similar kinetics compared to wild-type mitochondria (Fig. 2 E). By comparison, the import of the MIA40/CHCHD4 substrate COX19 (Fischer et al., 2013; Habich et al., 2019a) into these mitochondria was hampered. Mutating the four conserved cysteines of NDUFAF8 to alanine (NDUFAF8-4CA) did only have a minor effect on the import of NDUFAF8 into mitochondria except for later import time points (Fig. 2 F). This was again different for COX19, where two different cysteine mutants were strongly impaired in their import.
We then assessed where the newly imported NDUFAF8 was localized. Using a fractionation approach after import into isolated mitochondria, we found NDUFAF8 to localize to both IMS and matrix (Fig. 2 G, lanes 4 and 6). Conversely, a version of NDUFAF8 that was equipped with the strong SU9MTS led to the localization of mature NDUFAF8 only to the matrix (Fig. 2 G, right panel, lanes 4 and 6). Collectively, we conclude that the N-terminal region of NDUFAF8 serves as a rather weak MTS that guides NDUFAF8 via the TIM23 pathway to the matrix although a part of the protein also appears to accumulate in the IMS directly after import. The conserved cysteines of NDUFAF8 and the mitochondrial disulfide relay do not appear to influence mitochondrial uptake kinetics (Fig. 2 H).
The weak NDUFAF8 MTS allows the mitochondrial disulfide relay to introduce disulfide bonds into NDUFAF8 en route to the matrix
Although NDUFAF8 import was not dependent on its cysteines, the predicted NDUFAF8 structure indicated the presence of two disulfide bonds between cysteines 25 and 61, and 35 and 51, respectively (Fig. 3 A). Moreover, NDUFAF8 steady-state levels strongly correlated with the presence of these cysteines (Fig. 3 B). This might indicate that the oxidative folding of NDUFAF8 is important for its stability. Since the disulfide relay is the only machinery for oxidative protein folding in mitochondria, we further investigated its impact on NDUFAF8.
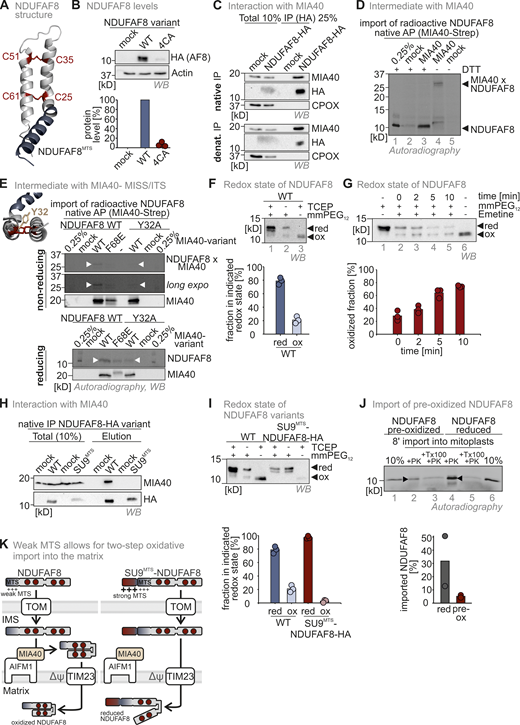
The weak NDUFAF8 MTS allows the mitochondrial disulfide relay to introduce disulfide bonds into NDUFAF8 en route to the matrix. (A) Modeled structure of NDUFAF8 highlighting two disulfide bonds between the four conserved cysteines in NDUFAF8. (B) Protein levels in HEK293 cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using ImageLab, and the amount of protein was plotted. NDUFAF8 lacking its four conserved cysteines is present at very low levels. N = 3 replicates; error bars indicate SD. (C) Assessment of MIA40/CHCHD4–NDUFAF8 interaction. NDUFAF8-HA was immunoprecipitated (IP) under native and denaturing conditions after stopping thiol-disulfide exchange reactions by NEM incubation. Precipitates were tested for MIA40/CHCHD4, HA, and as negative control the IMS protein CPOX by reducing SDS-PAGE and immunoblotting. 10% of the total lysate was loaded as input control for the HA blot while 2.5% input was loaded for the MIA40/CHCHD4 and CPOX blots. Under both precipitation conditions, NDUFAF8-HA coprecipitates MIA40/CHCHD4 indicating both proteins to interact via a covalent interaction. (D) Assessment of MIA40/CHCHD4–NDUFAF8 interaction during mitochondrial import. In vitro translated radioactive NDUFAF8 was incubated with mitochondria isolated from HEK293 cells expressing Strep-MIA40/CHCHD4. After import, MIA40/CHCHD4 was affinity-purified (AP) using streptactin beads under native conditions. Precipitates were analyzed by reducing (+DTT) and non-reducing SDS-PAGE and autoradiography. 0.25% of the total lysate was loaded as input control. During import NDUFAF8-HA and MIA40/CHCHD4-Strep form a disulfide linked complex that is reduced by the addition of reductant. (E) Assessment of MIA40/CHCHD4–NDUFAF8 interaction during mitochondrial import. In vitro translated radioactive NDUFAF8-WT and NDUFAF8-Y32A (mutant of the potential MISS/ITS site) were incubated with mitochondria isolated from HEK293 cells expressing MIA40/CHCHD4-Strep or MIA40/CHCHD4-F68E-Strep. After import, MIA40/CHCHD4 was affinity purified using streptactin beads (AP) under native conditions. Precipitates were analyzed by reducing (+DTT) and non-reducing SDS-PAGE and autoradiography. 0.25% of the total lysate was loaded as input control. During import, NDUFAF8-HA and MIA40/CHCHD4-Strep form a disulfide-linked complex only when the MISS/ITS site in NDUFAF8 is intact. (F) Redox state analysis of NDUFAF8. To test for the redox state of NDUFAF8-HA, cells were lysed and either treated with the strong reductant TCEP (lanes 1 and 3) or left untreated (lane 2). Then lysates were incubated with the maleimide mmPEG12 that modifies free thiols but not thiols in disulfide bonds as indicated. Lysates were analyzed by SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of reduced and oxidized protein was plotted. NDUFAF8-HA is mainly present in the reduced state. Approximately 20% of the protein is oxidized at steady state. N = 3 replicates; error bars indicate SD. (G) Redox state analysis of NDUFAF8 over time. Experiment was performed as in E except that cells were pretreated with the ribosome inhibitor emetine before the redox state determination. Over time, the fraction of oxidized NDUFAF8-HA increases under these conditions indicating either further oxidation of NDUFAF8 or specific degradation of the reduced form of the protein. N = 3 replicates; error bars indicate SD. (H) Assessment of the MIA40/CHCHD4–SU9MTS-NDUFAF8 interaction. NDUFAF8-HA and SU9MTS-NDUFAF8-HA were immunoprecipitated (IP) under native conditions after stopping thiol-disulfide exchange reactions by NEM incubation. Precipitates were tested for MIA40/CHCHD4 and HA. 10% of the total lysate was loaded as input control for the HA blot while 2.5% input was loaded for the MIA40/CHCHD4 blot. While NDUFAF8-HA coprecipitates with MIA40/CHCHD4, SU9MTS-NDUFAF8-HA cannot interact with MIA40/CHCHD4. (I) Redox state analysis of NDUFAF8-variants. Experiment was performed as in E except that cells expressing either NDUFAF8-HA or SU9MTS-NDUAF8 were analyzed. Equipping NDUFAF8 with a strong MTS (SU9MTS) results in a completely reduced protein at steady state, indicating that the weak MTS of NDUFAF8 is required to allow at least partial disulfide bond formation to occur. N = 3 replicates; error bars indicate SD. (J) In organello import assay with preoxidized or reduced NDUFAF8 into mitoplasts (mitochondria without the OMM). Experiment was performed as described in Fig. 2 A for 8 min import time. Preoxidized NDUFAF8 can be imported into mitoplasts. N = 2 replicates; error bars indicate SD. PK, proteinase K. (K) Model for the two-step import of NDUFAF8. The weak MTS of NDUFAF8 allows for TIM23-dependent import of NDUFAF8 into the mitochondrial matrix. It also allows for interaction with the mitochondrial disulfide relay component MIA40/CHCHD4 in the IMS that introduces two disulfide bonds into a fraction of NDUFAF8 molecules. A stronger MTS would not allow this interaction and would result in the accumulation of completely reduced NDUAF8 in the matrix. Source data are available for this figure: SourceData F3.
The weak NDUFAF8 MTS allows the mitochondrial disulfide relay to introduce disulfide bonds into NDUFAF8 en route to the matrix. (A) Modeled structure of NDUFAF8 highlighting two disulfide bonds between the four conserved cysteines in NDUFAF8. (B) Protein levels in HEK293 cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using ImageLab, and the amount of protein was plotted. NDUFAF8 lacking its four conserved cysteines is present at very low levels. N = 3 replicates; error bars indicate SD. (C) Assessment of MIA40/CHCHD4–NDUFAF8 interaction. NDUFAF8-HA was immunoprecipitated (IP) under native and denaturing conditions after stopping thiol-disulfide exchange reactions by NEM incubation. Precipitates were tested for MIA40/CHCHD4, HA, and as negative control the IMS protein CPOX by reducing SDS-PAGE and immunoblotting. 10% of the total lysate was loaded as input control for the HA blot while 2.5% input was loaded for the MIA40/CHCHD4 and CPOX blots. Under both precipitation conditions, NDUFAF8-HA coprecipitates MIA40/CHCHD4 indicating both proteins to interact via a covalent interaction. (D) Assessment of MIA40/CHCHD4–NDUFAF8 interaction during mitochondrial import. In vitro translated radioactive NDUFAF8 was incubated with mitochondria isolated from HEK293 cells expressing Strep-MIA40/CHCHD4. After import, MIA40/CHCHD4 was affinity-purified (AP) using streptactin beads under native conditions. Precipitates were analyzed by reducing (+DTT) and non-reducing SDS-PAGE and autoradiography. 0.25% of the total lysate was loaded as input control. During import NDUFAF8-HA and MIA40/CHCHD4-Strep form a disulfide linked complex that is reduced by the addition of reductant. (E) Assessment of MIA40/CHCHD4–NDUFAF8 interaction during mitochondrial import. In vitro translated radioactive NDUFAF8-WT and NDUFAF8-Y32A (mutant of the potential MISS/ITS site) were incubated with mitochondria isolated from HEK293 cells expressing MIA40/CHCHD4-Strep or MIA40/CHCHD4-F68E-Strep. After import, MIA40/CHCHD4 was affinity purified using streptactin beads (AP) under native conditions. Precipitates were analyzed by reducing (+DTT) and non-reducing SDS-PAGE and autoradiography. 0.25% of the total lysate was loaded as input control. During import, NDUFAF8-HA and MIA40/CHCHD4-Strep form a disulfide-linked complex only when the MISS/ITS site in NDUFAF8 is intact. (F) Redox state analysis of NDUFAF8. To test for the redox state of NDUFAF8-HA, cells were lysed and either treated with the strong reductant TCEP (lanes 1 and 3) or left untreated (lane 2). Then lysates were incubated with the maleimide mmPEG12 that modifies free thiols but not thiols in disulfide bonds as indicated. Lysates were analyzed by SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of reduced and oxidized protein was plotted. NDUFAF8-HA is mainly present in the reduced state. Approximately 20% of the protein is oxidized at steady state. N = 3 replicates; error bars indicate SD. (G) Redox state analysis of NDUFAF8 over time. Experiment was performed as in E except that cells were pretreated with the ribosome inhibitor emetine before the redox state determination. Over time, the fraction of oxidized NDUFAF8-HA increases under these conditions indicating either further oxidation of NDUFAF8 or specific degradation of the reduced form of the protein. N = 3 replicates; error bars indicate SD. (H) Assessment of the MIA40/CHCHD4–SU9MTS-NDUFAF8 interaction. NDUFAF8-HA and SU9MTS-NDUFAF8-HA were immunoprecipitated (IP) under native conditions after stopping thiol-disulfide exchange reactions by NEM incubation. Precipitates were tested for MIA40/CHCHD4 and HA. 10% of the total lysate was loaded as input control for the HA blot while 2.5% input was loaded for the MIA40/CHCHD4 blot. While NDUFAF8-HA coprecipitates with MIA40/CHCHD4, SU9MTS-NDUFAF8-HA cannot interact with MIA40/CHCHD4. (I) Redox state analysis of NDUFAF8-variants. Experiment was performed as in E except that cells expressing either NDUFAF8-HA or SU9MTS-NDUAF8 were analyzed. Equipping NDUFAF8 with a strong MTS (SU9MTS) results in a completely reduced protein at steady state, indicating that the weak MTS of NDUFAF8 is required to allow at least partial disulfide bond formation to occur. N = 3 replicates; error bars indicate SD. (J) In organello import assay with preoxidized or reduced NDUFAF8 into mitoplasts (mitochondria without the OMM). Experiment was performed as described in Fig. 2 A for 8 min import time. Preoxidized NDUFAF8 can be imported into mitoplasts. N = 2 replicates; error bars indicate SD. PK, proteinase K. (K) Model for the two-step import of NDUFAF8. The weak MTS of NDUFAF8 allows for TIM23-dependent import of NDUFAF8 into the mitochondrial matrix. It also allows for interaction with the mitochondrial disulfide relay component MIA40/CHCHD4 in the IMS that introduces two disulfide bonds into a fraction of NDUFAF8 molecules. A stronger MTS would not allow this interaction and would result in the accumulation of completely reduced NDUAF8 in the matrix. Source data are available for this figure: SourceData F3.
Immunoprecipitation of NDUFAF8-HA from HEK293 cells coprecipitated MIA40/CHCHD4 (Fig. 3 C). This interaction resisted a denaturing immunoprecipitation, indicating the presence of a mixed disulfide bond linkage between NDUFAF8 and MIA40/CHCHD4 (Fig. 3 C). We tested the presence of a mixed disulfide bond in an orthogonal approach by importing NDUFAF8 into isolated mitochondria containing MIA40/CHCHD4-Strep. After import, these mitochondria were lysed and MIA40/CHCHD4-Strep was precipitated. On non-reducing SDS-PAGE, we observed a disulfide-linked conjugate of MIA40/CHCHD4 and NDUFAF8 (Fig. 3 D, lane 4). NDUFAF8 was released from this mixed disulfide when the precipitate was treated with the reductant dithiothreitol (DTT, Fig. 3 D, lane 3). The interaction of MIA40/CHCHD4 depends on the recognition of a so-called MISS/ITS in its substrates (Milenkovic et al., 2009; Sideris et al., 2009). The MISS/ITS is defined by a hydrophobic amino acid at position ±3/4 from a cysteine residue. Tyrosin 32 (Y32) is close to C35 and might constitute such a signal in NDUFAF8. We thus tested the interaction of MIA40/CHCHD4 with NDUFAF8-Y32A. While NDUFAF8-WT formed a mixed disulfide intermediate during import, the Y32A variant failed to do so, indicating it to be part of the MISS/ITS of NDUFAF8 (Fig. 3 E, white arrowheads). Thus, MIA40/CHCHD4 and NDUFAF8 covalently interact with each other during NDUFAF8 import.
Next, we tested for NDUFAF8 oxidation by MIA40/CHCHD4 by alkylation assays. These assays rely on the accessibility of reduced, but not oxidized, thiol residues to the alkylating reagent methyl-polyethylenglycol-maleimide (mmPEG24) that causes a gel shift (Fig. 3 F, lanes 1 [reduced] and 3 [oxidized]). Using this assay, we were surprised to find at steady state only a small fraction of NDUFAF8 (∼20%) in the oxidized state (Fig. 3 F, lane 2). We next addressed whether this share increased over time. In an emetine chase assay, in which cytosolic translation is inhibited and consequently only the fraction of already synthesized proteins is observed, we found that indeed the share of oxidized NDUFAF8 increased with time (Fig. 3 G). This indicates that NDUFAF8 is oxidized by the disulfide relay but only very slowly.
NDUFAF8 carries a comparably weak MTS (Fig. 1 A). To test whether the strength of the MTS affected disulfide formation in NDUFAF8, we fused the protein to the strong SU9MTS. This NDUFAF8 variant did not interact with MIA40/CHCHD4 anymore (Fig. 3 H). Moreover, unlike wild-type NDUFAF8, which was at least partly oxidized at steady state, SU9MTS-NDUFAF8 was completely reduced (Fig. 3 I), indicating the need for a weak MTS on NDUFAF8 to allow its oxidation during import into the matrix.
Oxidation of NDUFAF8 in the IMS would require the transport of oxidized (and thus folded) NDUFAF8 across the IMM. To test this directly, we imported NDUFAF8 either as a reduced precursor or preoxidized protein into mitochondria lacking their OMM (mitoplasts). Although mitoplast import of oxidized NDUFAF8 was clearly weaker than that of reduced precursor, it took place in appreciable amounts supporting that oxidized (but also reduced) NDUFAF8 can translocate into the matrix (Fig. 3 J).
Collectively, these results indicate that in the IMS, MIA40/CHCHD4 recognizes NDUFAF8 as a substrate and introduces disulfide bonds during NDUFAF8 import. Subsequently, oxidized (but also reduced) NDUFAF8 translocates across the IMM to the matrix. Import does not rely on the disulfide relay, but it is the weak unconventional MTS of NDUFAF8 that enables the oxidation step in the IMS because the addition of the strong SU9MTS suppresses IMS localization as well as oxidation (Fig. 3 K). This two-step import pathway also explains why we detected NDUFAF8 as a dually localized protein in IMS and matrix (Fig. 1, C and D; and Fig. 2 G).
Mitochondrial proteases control NDUFAF8 stability in different mitochondrial subcompartments depending on the NDUFAF8 redox state
We found NDUFAF8 variants that lack the two disulfide bonds (NDUFAF8-4CA or SU9MTS-NDUFAF8) at low levels at steady state (Fig. 4 A). These variants were also very unstable when analyzed in emetine chase experiments (Fig. 4 B). These data from intact cells were further corroborated by chase experiments after NDUFAF8 import into isolated mitochondria (Fig. 4 C). Full-length NDUFAF8 was thereby more stable than SU9MTS-NDUFAF8 or NDUFAF8-4CA. Thus, although NDUFAF8 appeared to be in general a protein with a comparatively high turnover in intact cells, its turnover kinetics was further increased when the protein was in the reduced redox state.
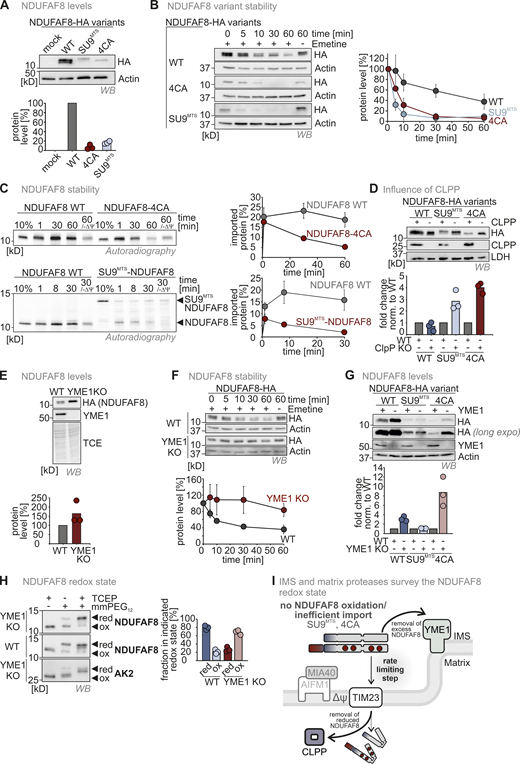
Mitochondrial proteases monitor NDUFAF8 levels in IMS and matrix depending on their redox state. (A) Protein levels in HEK293 cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS are present at very low levels. N = 3 replicates; error bars indicate SD. (B) Assessment of stability of different NDUFAF8 variants in HEK293 cells. Cells were pretreated with the ribosome inhibitor emetine for the indicated times and then lysed. Lysates were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS are very unstable compared to NDUFAF8-HA. N = 3 replicates; error bars indicate SD. (C) Assessment of stability of different NDUFAF8 variants after import into isolated mitochondria. In vitro-translated radioactive NDUFAF8-variants were incubated with mitochondria isolated from HEK293 cells. Non-imported proteins were removed by treatment with Proteinase K. An import reaction was performed into mitochondria treated with CCCP and valinomycin to dissipate the mitochondrial membrane potential (−ΔΨ). Imported proteins were analyzed by reducing SDS-PAGE and autoradiography. Signals were quantified using ImageQuantTL and the amount of imported protein was plotted. NDUFAF8-variants lacking its four conserved cysteines or equipped with a SU9MTS are very unstable compared to NDUFAF8. N = 3 replicates; error bars indicate SD. (D) Protein levels in HEK293 wild-type and CLPP knockout cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS were stabilized by the loss of CLPP. This was not the case for wild-type NDUFAF8. This indicates that CLPP degrades reduced NDUFAF8. N = 3 replicates; error bars indicate SD. (E) Protein levels in HEK293 wild-type and YME1L knockout cell lines expressing NDUFAF8-HA. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA levels are increased in YME1L knockout cells. N = 3 replicates; error bars indicate SD. (F) Assessment of stability of NDUFAF8 in HEK293 wild-type and YME1L knockout cells. Cells were pretreated with the ribosome inhibitor emetine for the indicated times and then lysed. Lysates were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA became stabilized by the loss of YME1L. N = 3 replicates; error bars indicate SD. (G) Protein levels in HEK293 wild-type and YME1L knockout cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA and NDUFAF8-4CA-HA but not SU9MTS-NDUFAF8-HA were present at increased levels in YME1L knockout cells. Thus, only NDUFAF8 variants that are exposed to the IMS become stabilized by the loss of YME1L. N = 3 replicates; error bars indicate SD. (H) Redox state analysis of NDUFAF8 in HEK293 wild-type and YME1 knockout cells. Experiment performed as described in Fig. 3 F. Adenylate kinase 2 (AK2) served as control for an IMS protein with a disulfide bond. NDUFAF8-HA is mainly present in the oxidized state in YME1L knockout cells. This indicates that YME1L targets oxidized NDUFAF8 in the IMS. N = 3 replicates; error bars indicate SD. (I) Model for protease surveillance of NDUFAF8 import. In the wild-type situation, NDUFAF8 becomes at least in part oxidized by the mitochondrial disulfide relay. A large fraction of the oxidized protein is constantly degraded by YME1L, presumably because import mediated by the weak MTS of NDUFAF8 is slow and accumulation of oxidized NDUFAF8 in the IMS has to be prevented. If no oxidation of NDUFAF8 takes place, reduced NDUFAF8 is targeted by CLPP in the matrix, strongly decreasing the half-life of reduced NDUFAF8. Source data are available for this figure: SourceData F4.
Mitochondrial proteases monitor NDUFAF8 levels in IMS and matrix depending on their redox state. (A) Protein levels in HEK293 cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS are present at very low levels. N = 3 replicates; error bars indicate SD. (B) Assessment of stability of different NDUFAF8 variants in HEK293 cells. Cells were pretreated with the ribosome inhibitor emetine for the indicated times and then lysed. Lysates were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS are very unstable compared to NDUFAF8-HA. N = 3 replicates; error bars indicate SD. (C) Assessment of stability of different NDUFAF8 variants after import into isolated mitochondria. In vitro-translated radioactive NDUFAF8-variants were incubated with mitochondria isolated from HEK293 cells. Non-imported proteins were removed by treatment with Proteinase K. An import reaction was performed into mitochondria treated with CCCP and valinomycin to dissipate the mitochondrial membrane potential (−ΔΨ). Imported proteins were analyzed by reducing SDS-PAGE and autoradiography. Signals were quantified using ImageQuantTL and the amount of imported protein was plotted. NDUFAF8-variants lacking its four conserved cysteines or equipped with a SU9MTS are very unstable compared to NDUFAF8. N = 3 replicates; error bars indicate SD. (D) Protein levels in HEK293 wild-type and CLPP knockout cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. NDUFAF8-HA-variants lacking its four conserved cysteines or equipped with a SU9MTS were stabilized by the loss of CLPP. This was not the case for wild-type NDUFAF8. This indicates that CLPP degrades reduced NDUFAF8. N = 3 replicates; error bars indicate SD. (E) Protein levels in HEK293 wild-type and YME1L knockout cell lines expressing NDUFAF8-HA. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA levels are increased in YME1L knockout cells. N = 3 replicates; error bars indicate SD. (F) Assessment of stability of NDUFAF8 in HEK293 wild-type and YME1L knockout cells. Cells were pretreated with the ribosome inhibitor emetine for the indicated times and then lysed. Lysates were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA became stabilized by the loss of YME1L. N = 3 replicates; error bars indicate SD. (G) Protein levels in HEK293 wild-type and YME1L knockout cell lines expressing different NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. NDUFAF8-HA and NDUFAF8-4CA-HA but not SU9MTS-NDUFAF8-HA were present at increased levels in YME1L knockout cells. Thus, only NDUFAF8 variants that are exposed to the IMS become stabilized by the loss of YME1L. N = 3 replicates; error bars indicate SD. (H) Redox state analysis of NDUFAF8 in HEK293 wild-type and YME1 knockout cells. Experiment performed as described in Fig. 3 F. Adenylate kinase 2 (AK2) served as control for an IMS protein with a disulfide bond. NDUFAF8-HA is mainly present in the oxidized state in YME1L knockout cells. This indicates that YME1L targets oxidized NDUFAF8 in the IMS. N = 3 replicates; error bars indicate SD. (I) Model for protease surveillance of NDUFAF8 import. In the wild-type situation, NDUFAF8 becomes at least in part oxidized by the mitochondrial disulfide relay. A large fraction of the oxidized protein is constantly degraded by YME1L, presumably because import mediated by the weak MTS of NDUFAF8 is slow and accumulation of oxidized NDUFAF8 in the IMS has to be prevented. If no oxidation of NDUFAF8 takes place, reduced NDUFAF8 is targeted by CLPP in the matrix, strongly decreasing the half-life of reduced NDUFAF8. Source data are available for this figure: SourceData F4.
Which protease detects and degrades NDUFAF8 in the matrix? The matrix protease CLPP has previously been shown to contribute to the quality control of misfolded proteins in the matrix, and we thus analyzed different NDUFAF8 variants in CLPP knockout cells. While wild-type NDUFAF8 levels were not affected at steady state, the levels of NDUFAF8-4CA and SU9MTS-NDUFAF8 were strongly stabilized (Fig. 4 D). This indicates that CLPP specifically recognizes and degrades reduced (potentially misfolded) NDUFAF8 in the matrix.
Intrigued by the recognition of reduced NDUFAF8 by CLPP, we next asked whether also in the IMS the levels of NDUFAF8 are monitored by a protease. We analyzed NDUFAF8 steady-state levels in cells deficient of the major IMS protease, YME1L (Fig. 4 E). We found that NDUFAF8 levels were strongly increased in YME1L knockout cells (fitting to proteomics data in MacVicar et al. [2019], where NDUFAF8 was also enriched in YME1L knockout cells). We also observed that NDUFAF8 was more stable in emetine chase experiments in YME1L knockout cells compared with corresponding wild-type cells (Fig. 4 F). Loss of YME1L only stabilized wild-type NDUFAF8 but not the SU9MTS-NDUFAF8 variant that became rapidly imported directly into the matrix (Fig. 4 G). Interestingly, also NDUFAF8-4CA was stabilized by loss of YME1L, indicating that transport across the IMM was rate-limiting for NDUFAF8 with its own weak MTS. YME1L thus does not appear to prefer NDUFAF8 in a specific redox state.
The redox state of the majority of cellular NDUFAF8 was reduced in wild-type cells (Fig. 3 F and Fig. 4 H). This changed dramatically in YME1L knockout cells, in which NDUFAF8 was almost completely oxidized (Fig. 4 H). This might implicate that despite the fact that YME1L does not directly sense the NDUFAF8 redox state, it prevents accumulation of excess oxidized NDUFAF8 in the IMS, presumably because MTS-dependent passage across the IMM is very slow and is the rate-limiting step for NDUFAF8 import.
Collectively, we demonstrated that YME1L and CLPP survey the import, levels, and redox state of NDUFAF8. We speculate that YME1L recognizes excess NDUFAF8 presumably to prevent its accumulation in the IMS and that CLPP efficiently removes reduced NDUFAF8 (Fig. 4 I). Thus, while the disulfide relay is not important for NDUFAF8 import, it sets the levels of the protein in the matrix by introducing stabilizing disulfide bonds.
NDUFAF8 is important for NDUFAF5 activity and complex I assembly
Next, we wanted to understand the role of the two-step import pathway of NDUFAF8 for its cellular function. NDUFAF8 serves in the assembly of the Q modules of complex I together with its partner NDUFAF5 (Alston et al., 2020; Floyd et al., 2016). We generated HEK293 knockout cells of NDUFAF8 and NDUFAF5 using the CRISPR-Cas9 system (Fig. S3, A and B). Using these cells, we confirmed that loss of NDUFAF5 or NDUFAF8 resulted in lowered complex I levels (Fig. 5 A; and Fig. S3, C and D) and that this phenotype could be complemented by introducing the respective wild-type protein (Fig. 5 A). Moreover, we confirmed the interaction of NDUFAF5 and NDUFAF8 with each other using immunoprecipitation approaches followed by immunoblot or mass spectrometric protein detection (Fig. 5, B and C). We also found that NDUFAF5 interacted with NDUFS7, in line with its role in the maturation of this subunit. Additionally, it coprecipitated other subunits of the Q-module (namely, NDUFA5, NDUFS2, and NDUFS3) suggesting an interaction of NDUFAF5 with the complete assembling Q module and not only NDUFS7. Intriguingly, both NDUFAF5 and NDUFAF8 also coprecipitated many subunits of the mitochondrial ribosome (Fig. S4). Based on these results, it is tempting to speculate that NDUFAF5 and NDUFAF8 closely coordinate their role in Q module assembly and synthesis of ND1 (the mitochondrially synthesized center of the Q module).
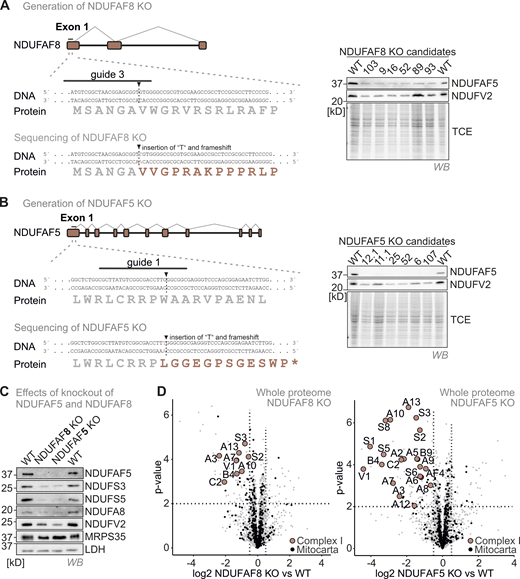
(Related toFig. 5 ). NDUFAF8 or NDUFAF5 loss results in an isolated complex I deficiency. (A) Strategy to generate NDUFAF8 knockout HEK293 cell lines using CRISPR Cas. A guide (#3) directed against the first exon of NDUFAF8 gave rise to multiple clones. Successful targeting of the gene was confirmed by immunoblotting against NDUAF5 (due to the lack of a suitable antibody against NDUFAF8) and by sequencing. (B) Strategy to generate NDUFAF5 knockout HEK293 cell lines using CRISPR Cas. A guide (#1) directed against the first exon of NDUFAF5 gave rise to multiple clones. Successful targeting of the gene was confirmed by immunoblotting against NDUAF5 and by sequencing. (C) Effects of NDUFAF5 and NDUFAF8 knockout cells. Levels of proteins were assessed using immunoblotting and quantitative label-free mass spectrometry. Subunits of complex I but not of other respiratory chain complexes are present in lowered amounts in both knockout cell lines indicating an isolated complex I deficiency. N = 4 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change > 2). Source data are available for this figure: SourceData FS3.
(Related toFig. 5 ). NDUFAF8 or NDUFAF5 loss results in an isolated complex I deficiency. (A) Strategy to generate NDUFAF8 knockout HEK293 cell lines using CRISPR Cas. A guide (#3) directed against the first exon of NDUFAF8 gave rise to multiple clones. Successful targeting of the gene was confirmed by immunoblotting against NDUAF5 (due to the lack of a suitable antibody against NDUFAF8) and by sequencing. (B) Strategy to generate NDUFAF5 knockout HEK293 cell lines using CRISPR Cas. A guide (#1) directed against the first exon of NDUFAF5 gave rise to multiple clones. Successful targeting of the gene was confirmed by immunoblotting against NDUAF5 and by sequencing. (C) Effects of NDUFAF5 and NDUFAF8 knockout cells. Levels of proteins were assessed using immunoblotting and quantitative label-free mass spectrometry. Subunits of complex I but not of other respiratory chain complexes are present in lowered amounts in both knockout cell lines indicating an isolated complex I deficiency. N = 4 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change > 2). Source data are available for this figure: SourceData FS3.
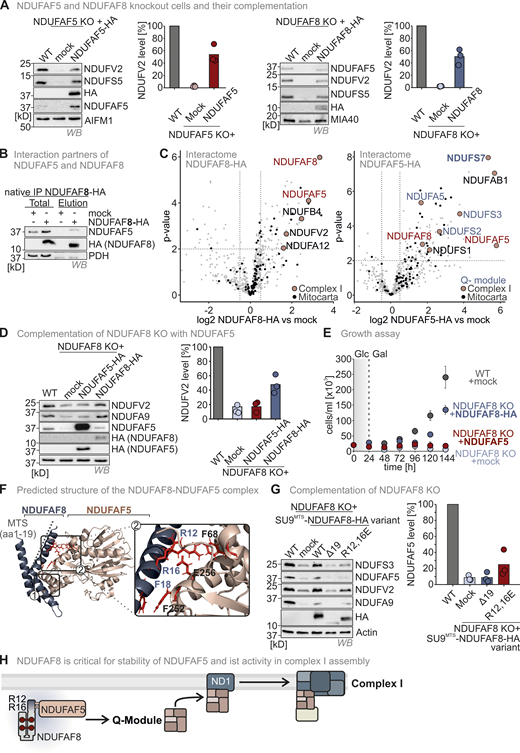
The NDUFAF8–NDUFAF5 interaction stabilizes and activates NDUFAF5 to allow NDUFAF5 to fulfill its function in complex I assembly. (A) Complex I subunit levels in NDUFAF5 and NDUFAF8 knockout cells. Levels of indicated proteins were assessed using immunoblotting. Subunits of complex I are present in lowered amounts in both knockout cell lines. This is in line with data from quantitative label-free mass spectrometry (Fig. S3, C and D). Moreover, NDUFS5 and NDUFV2 levels can be complemented by re-expressing NDUFAF5 and NDUFAF8 in the respective knockout cells. N = three biological replicates. (B) Assessment of NDUFAF5–NDUFAF8 interaction. NDUFAF8-HA was immunoprecipitated (IP) under native conditions. Precipitates were tested for NDUFAF5, HA (NDUFAF8), and, as negative control, the protein PDH by reducing SDS-PAGE and immunoblotting. 10% of the total lysate was loaded as input control for HA blot and 2.5% was loaded as input for the NDUFAF5 input control. NDUFAF8-HA coprecipitates NDUFAF5. (C) Proteomic analysis to assess the interactomes of NDUFAF5 and NDUFAF8. HEK293 cells expressing either NDUFAF5-HA or NDUFAF8-HA were lysed, proteins were immunoprecipitated using the HA-tag, and precipitates were analyzed using quantitative label-free proteomics. Both NDUFAF8 and NDUFAF5 coprecipitate the respective other partner as well as subunits of complex I but not of other complexes of the respiratory chain. Remarkably, NDUFAF5 precipitates almost all subunits of the Q-module which contains NDUFS7, the protein on which NDUFAF5 acts during complex I maturation (highlighted in blue). This might indicate that NDUFAF5 acts on the complete Q-module and not on monomeric NDUFS7 during complex I assembly (Guerrero-Castillo et al., 2017). N = 4–5 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change >2). (D) Protein levels in HEK293 wild-type cells, NDUFAF8 knockout cells, and NDUFAF8 knockout complemented with NDUFAF8 and NDUFAF5 (expression for 3 d). Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. The loss of complex I subunits in NDUFAF8 knockout cells cannot be complemented by re-expressing NDUFAF5-HA despite strongly increased NDUFAF5 levels. This indicates that the role of NDUFAF8 is not solely in NDUFAF5 stabilization but also in activation. N = 3 replicates. (E) Assay to assess growth of NDUFAF8 knockout cell lines in galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium was exchanged to a galactose-containing medium, and cell number was scored daily. Growth on galactose necessitates a functional respiratory chain. NDUFAF8 knockout cells and NDUFAF8 knockout cells complemented with NDUFAF5 were not capable to grow on galactose. N = 3 replicates; error bars indicate SD. (F) Predicted structure of the NDUFAF5-NDUFAF8 complex by Alpha Fold Multimer. For the prediction, ChimeraX software was run in AlphaFold 2. The complex structure was visualized by Pymol. NDUFAF8 and NDUFAF5 appeared to have a very extended interaction interface that spans one complete face of NDUFAF8. NDUFAF8 “snuggles” in an L-shaped conformation onto NDUFAF5. Two regions mediate the interaction between NDUFAF5 and NDUFAF8. In region 1, two perpendicular helices of NDUFAF5 and NDUFAF8 come in close proximity, and small aliphatic amino acids face each other in this region. Region 2 encompasses the MTS of NDUFAF8. Specifically, two arginine residues (R12 and R16 of NDUFAF8) are involved in contact with a glutamate residue and a phenylalanine in NDUFAF5 (E256 and F68 in NDUFAF5). Notably, NDUFAF8 and the other interaction partner of NDUFAF5, PYURF, appear to occupy distinct interaction sites on NDUFAF5 that are distal from each other excluding interference of these two proteins in their function (Pei et al., 2022). (G) Protein levels in NDUFAF8 knockout cells complemented with SU9MTS-NDUFAF8 and different MTS mutant variants of SU9MTS-NDUFAF8. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. Removing the MTS of NDUFAF8 or mutating the arginine residues important for interaction with NDUFAF5 impairs complementation. N = 3 replicates. (H) Model. NDUFAF8 is critical for NDUAF5 stability and activity. Interaction of NDUFAF8 and NDUFAF5 involving arginine residues in the MTS of NDUFAF8 is required for the functionality of NDUFAF5 and proper assembly of the Q module of complex I. Source data are available for this figure: SourceData F5.
The NDUFAF8–NDUFAF5 interaction stabilizes and activates NDUFAF5 to allow NDUFAF5 to fulfill its function in complex I assembly. (A) Complex I subunit levels in NDUFAF5 and NDUFAF8 knockout cells. Levels of indicated proteins were assessed using immunoblotting. Subunits of complex I are present in lowered amounts in both knockout cell lines. This is in line with data from quantitative label-free mass spectrometry (Fig. S3, C and D). Moreover, NDUFS5 and NDUFV2 levels can be complemented by re-expressing NDUFAF5 and NDUFAF8 in the respective knockout cells. N = three biological replicates. (B) Assessment of NDUFAF5–NDUFAF8 interaction. NDUFAF8-HA was immunoprecipitated (IP) under native conditions. Precipitates were tested for NDUFAF5, HA (NDUFAF8), and, as negative control, the protein PDH by reducing SDS-PAGE and immunoblotting. 10% of the total lysate was loaded as input control for HA blot and 2.5% was loaded as input for the NDUFAF5 input control. NDUFAF8-HA coprecipitates NDUFAF5. (C) Proteomic analysis to assess the interactomes of NDUFAF5 and NDUFAF8. HEK293 cells expressing either NDUFAF5-HA or NDUFAF8-HA were lysed, proteins were immunoprecipitated using the HA-tag, and precipitates were analyzed using quantitative label-free proteomics. Both NDUFAF8 and NDUFAF5 coprecipitate the respective other partner as well as subunits of complex I but not of other complexes of the respiratory chain. Remarkably, NDUFAF5 precipitates almost all subunits of the Q-module which contains NDUFS7, the protein on which NDUFAF5 acts during complex I maturation (highlighted in blue). This might indicate that NDUFAF5 acts on the complete Q-module and not on monomeric NDUFS7 during complex I assembly (Guerrero-Castillo et al., 2017). N = 4–5 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change >2). (D) Protein levels in HEK293 wild-type cells, NDUFAF8 knockout cells, and NDUFAF8 knockout complemented with NDUFAF8 and NDUFAF5 (expression for 3 d). Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. The loss of complex I subunits in NDUFAF8 knockout cells cannot be complemented by re-expressing NDUFAF5-HA despite strongly increased NDUFAF5 levels. This indicates that the role of NDUFAF8 is not solely in NDUFAF5 stabilization but also in activation. N = 3 replicates. (E) Assay to assess growth of NDUFAF8 knockout cell lines in galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium was exchanged to a galactose-containing medium, and cell number was scored daily. Growth on galactose necessitates a functional respiratory chain. NDUFAF8 knockout cells and NDUFAF8 knockout cells complemented with NDUFAF5 were not capable to grow on galactose. N = 3 replicates; error bars indicate SD. (F) Predicted structure of the NDUFAF5-NDUFAF8 complex by Alpha Fold Multimer. For the prediction, ChimeraX software was run in AlphaFold 2. The complex structure was visualized by Pymol. NDUFAF8 and NDUFAF5 appeared to have a very extended interaction interface that spans one complete face of NDUFAF8. NDUFAF8 “snuggles” in an L-shaped conformation onto NDUFAF5. Two regions mediate the interaction between NDUFAF5 and NDUFAF8. In region 1, two perpendicular helices of NDUFAF5 and NDUFAF8 come in close proximity, and small aliphatic amino acids face each other in this region. Region 2 encompasses the MTS of NDUFAF8. Specifically, two arginine residues (R12 and R16 of NDUFAF8) are involved in contact with a glutamate residue and a phenylalanine in NDUFAF5 (E256 and F68 in NDUFAF5). Notably, NDUFAF8 and the other interaction partner of NDUFAF5, PYURF, appear to occupy distinct interaction sites on NDUFAF5 that are distal from each other excluding interference of these two proteins in their function (Pei et al., 2022). (G) Protein levels in NDUFAF8 knockout cells complemented with SU9MTS-NDUFAF8 and different MTS mutant variants of SU9MTS-NDUFAF8. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. Removing the MTS of NDUFAF8 or mutating the arginine residues important for interaction with NDUFAF5 impairs complementation. N = 3 replicates. (H) Model. NDUFAF8 is critical for NDUAF5 stability and activity. Interaction of NDUFAF8 and NDUFAF5 involving arginine residues in the MTS of NDUFAF8 is required for the functionality of NDUFAF5 and proper assembly of the Q module of complex I. Source data are available for this figure: SourceData F5.

(Related to Fig. 5 ). NDUFAF8 or NDUFAF5 interact with subunits of the mitochondrial ribosome. Proteomic analysis to assess the interactomes of NDUFAF5 and NDUFAF8. HEK293 cells expressing either NDUFAF5-HA or NDUFAF8-HA were lysed, proteins were immunoprecipitated using the HA-tag, and precipitates were analyzed using quantitative label-free proteomics. Both NDUFAF8 and NDUFAF5 coprecipitate mitochondrial ribosomal subunits which might indicate a close coordination of their role in Q module assembly and ND1 synthesis. N = 4 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change > 2).
(Related to Fig. 5 ). NDUFAF8 or NDUFAF5 interact with subunits of the mitochondrial ribosome. Proteomic analysis to assess the interactomes of NDUFAF5 and NDUFAF8. HEK293 cells expressing either NDUFAF5-HA or NDUFAF8-HA were lysed, proteins were immunoprecipitated using the HA-tag, and precipitates were analyzed using quantitative label-free proteomics. Both NDUFAF8 and NDUFAF5 coprecipitate mitochondrial ribosomal subunits which might indicate a close coordination of their role in Q module assembly and ND1 synthesis. N = 4 biological replicates, an unpaired one-sample two-sided Student’s t test was applied (P < 0.05, fold change > 2).
If the main task of NDUFAF8 was to stabilize NDUFAF5, we reasoned that a strong overexpression of NDUFAF5 should also complement NDUFAF8 knockout cells. However, despite clearly increased levels of NDUFAF5 in this system, levels of the complex I subunits NDUFV2 and NDUFA9 did not return to normal levels (Fig. 5 D). In line, NDUFAF8 knockout cells overexpressing NDUFAF5 were, like the NDUFAF8 knockout cells by themselves, impaired in their growth on galactose, which requires a functional respiratory chain (Fig. 5 E). This points to a role of NDUFAF8 in stabilizing and activating NDUFAF5.
To obtain a better understanding of the NDUFAF5–NDUFAF8 interaction, we used Alphafold-guided biochemical experiments (Fig. 5 F). NDUFAF8 and NDUFAF5 share an extended interaction interface that spans one complete face of NDUFAF8. NDUFAF8 thereby “snuggles” in an L-shaped conformation onto NDUFAF5. In particular, two perpendicular helices of NDUFAF5 and NDUFAF8 come in close proximity and small aliphatic amino acids face each other in this region (Fig. 5 F, region 1). Intriguingly, a second interacting region encompasses the MTS of NDUFAF8. The arginine residues of this helix (R12, R16 in NDUFAF8) are involved in contacts with an arginine and a phenylalanine residue in NDUFAF5 (E256 and F68 in NDUFAF5), respectively (Fig. 5 F, region 2 and inset). To assess the validity of our structural model of the NDUFAF5–NDUFAF8 complex, we generated two NDUFAF8 mutants, one lacking the MTS and one in which we mutated the arginine residues in the MTS to glutamic acid residues. In both cases, we added the SU9MTS to guide these proteins into the matrix. Both mutants failed to complement the NDUFAF8 knockout, while SU9MTS-NDUFAF8 could, confirming the importance of the arginine residues and the (unprocessed) MTS in the function of NDUFAF8 toward NDUFAF5 (Fig. 5 G).
Taken together, we confirmed the role of NDUFAF5 and NDUFAF8 in complex I biogenesis. We found that NDUFAF8 stabilizes and activates NDUFAF5. We also demonstrated the importance of the non-processed NDUFAF8 MTS not only for slow mitochondrial import of NDUFAF8 allowing oxidative folding but also in establishing the interaction with NDUFAF5 (Fig. 5 H).
NDUFAF8 levels determine NDUFAF5-dependent complex I biogenesis
We next explored the contribution of the NDUFAF8 redox state and NDUFAF8 levels on complex I assembly by complementing NDUFAF8 knockout cells with two NDUFAF8 variants, SU9MTS-NDUFAF8 and NDUFAF8-4CA, that do not form disulfide bonds (see Fig. 3). Equipping NDUFAF8 with a SU9MTS led to NDUFAF8 levels similar to wild-type NDUFAF8. It also allowed NDUFAF8 to fulfill its function in activating NDUFAF5, restoring levels of the complex I subunit NDUFV2, allowing ND1 translation (Fig. 6 A, lane 4; Fig. S5), and restoring growth of NDUFAF8 knockout cells in galactose-containing medium (Fig. 6 B). Conversely, NDUFAF8-4CA was present at much lower levels in the matrix compared with SU9MTS-NDUFAF8 likely because it is imported slower than the SU9MTS-equipped NDUFAF8 and thus partially degraded en route (Fig. 6 A, lane 5). It could also not restore NDUFAF5 and NDUFV2 levels (Fig. 6 A), and it did not allow the growth of complemented NDUFAF8 knockout cells on galactose (Fig. 6 B). To exclude that the exchange of four cysteines in NDUFAF8 completely abolished its activity, we generated a SU9MTS-NDUFAF8-4CA mutant. This mutant was present at wild-type-like levels and could complement NDUFAF5 and complex I levels, ND1 translation as well as growth on galactose (Fig. 6, C and D; and Fig. S5).
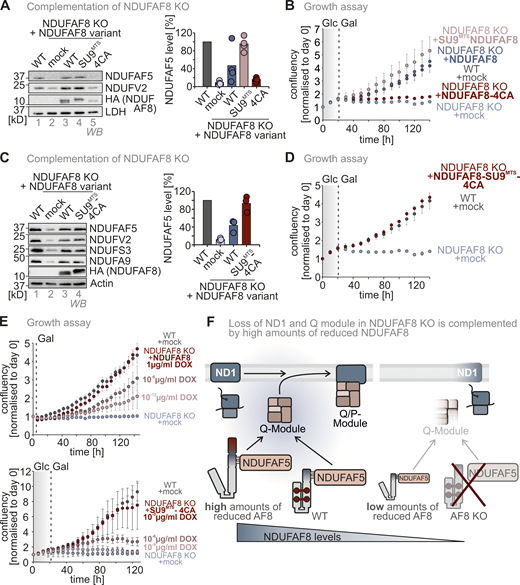
NDUFAF8 levels determine the efficiency of NDUFAF5 activation and complex I assembly. (A) Protein levels in NDUFAF8 knockout cells complemented with NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. Equipping NDUFAF8 with a SU9MTS allows complementation of NDUFAF8 knockout cells. Mutating the four conserved cysteines in NDUFAF8 (4CA) resulted in very low steady-state levels of NDUFAF8 and did not allow complementation of the NDUFAF8 knockout. N = 3 replicates; error bars indicate SD. (B) Assay to assess the growth of NDUFAF8 knockout cell lines in a galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium was exchanged to a galactose-containing medium and cell number was scored every 6 h NDUFAF8 knockout cells and NDUFAF8 knockout cells complemented with NDUFAF8-4CA were not capable to grow on galactose. N = 4 replicates; error bars indicate SD. (C) Protein levels in NDUFAF8 knockout cells complemented with NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. Equipping NDUFAF8-4CA with a SU9MTS allows complementation of NDUFAF8 knockout cells indicating that the cysteine of NDUFAF8 is dispensable for its function. Mutating the cysteines impacts the stability of NDUFAF8 in the matrix and only an efficient import of NDUFAF8-4CA by the SU9MTS ensures a sufficiently high supply of new NDUFAF8-4CA to overcome the high degradation rate of the protein in the matrix. N = 3 replicates; error bars indicate SD. (D) Assay to assess growth of complemented NDUFAF8 knockout cell lines in galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium were exchanged to galactose-containing medium and cell number was scored every 6 h NDUFAF8 knockout cells complemented with SU9MTS-NDUFAF8-4CA were able to grow on galactose. N = 4 replicates; error bars indicate SD. (E) Assay to assess the growth of complemented NDUFAF8 knockout cell lines in a galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium were exchanged to galactose-containing medium and cell number was scored every 6 h. Titration of NDUFAF8 variant levels was achieved by induction of their expression with different amounts of doxycycline. Upon reaching a certain threshold, knockout cells complemented with NDUFA8 or SU9MTS-NDUFAF8-4CA were able to grow on galactose. SU9MTS-NDUFAF8-4CA expressing cells required increased amounts of doxycycline to allow growth on galactose. N = 4 replicates; error bars indicate SD. (F) Model. The amounts of NDUFAF8 control NDUFAF5 levels and complex I assembly. Low amounts of NDUFAF8 as found in the NDUFAF8 knockout or in NDUFAF8 knockout cells expressing NDUFAF8-4CA do not activate and stabilize NDUFAF5 thereby preventing Q module assembly. High amounts of NDUFAF8 can complement NDUFAF8 knockout irrespective of whether they contain disulfide bonds or not. The formation of disulfide bonds in NUFAF8 is, however, important to stabilize the protein in the matrix. Source data are available for this figure: SourceData F6.
NDUFAF8 levels determine the efficiency of NDUFAF5 activation and complex I assembly. (A) Protein levels in NDUFAF8 knockout cells complemented with NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. Equipping NDUFAF8 with a SU9MTS allows complementation of NDUFAF8 knockout cells. Mutating the four conserved cysteines in NDUFAF8 (4CA) resulted in very low steady-state levels of NDUFAF8 and did not allow complementation of the NDUFAF8 knockout. N = 3 replicates; error bars indicate SD. (B) Assay to assess the growth of NDUFAF8 knockout cell lines in a galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium was exchanged to a galactose-containing medium and cell number was scored every 6 h NDUFAF8 knockout cells and NDUFAF8 knockout cells complemented with NDUFAF8-4CA were not capable to grow on galactose. N = 4 replicates; error bars indicate SD. (C) Protein levels in NDUFAF8 knockout cells complemented with NDUFAF8 variants. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab and the amount of protein was plotted. Equipping NDUFAF8-4CA with a SU9MTS allows complementation of NDUFAF8 knockout cells indicating that the cysteine of NDUFAF8 is dispensable for its function. Mutating the cysteines impacts the stability of NDUFAF8 in the matrix and only an efficient import of NDUFAF8-4CA by the SU9MTS ensures a sufficiently high supply of new NDUFAF8-4CA to overcome the high degradation rate of the protein in the matrix. N = 3 replicates; error bars indicate SD. (D) Assay to assess growth of complemented NDUFAF8 knockout cell lines in galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium were exchanged to galactose-containing medium and cell number was scored every 6 h NDUFAF8 knockout cells complemented with SU9MTS-NDUFAF8-4CA were able to grow on galactose. N = 4 replicates; error bars indicate SD. (E) Assay to assess the growth of complemented NDUFAF8 knockout cell lines in a galactose-containing medium. Cells were seeded in glucose-containing medium, and after 1 d, medium were exchanged to galactose-containing medium and cell number was scored every 6 h. Titration of NDUFAF8 variant levels was achieved by induction of their expression with different amounts of doxycycline. Upon reaching a certain threshold, knockout cells complemented with NDUFA8 or SU9MTS-NDUFAF8-4CA were able to grow on galactose. SU9MTS-NDUFAF8-4CA expressing cells required increased amounts of doxycycline to allow growth on galactose. N = 4 replicates; error bars indicate SD. (F) Model. The amounts of NDUFAF8 control NDUFAF5 levels and complex I assembly. Low amounts of NDUFAF8 as found in the NDUFAF8 knockout or in NDUFAF8 knockout cells expressing NDUFAF8-4CA do not activate and stabilize NDUFAF5 thereby preventing Q module assembly. High amounts of NDUFAF8 can complement NDUFAF8 knockout irrespective of whether they contain disulfide bonds or not. The formation of disulfide bonds in NUFAF8 is, however, important to stabilize the protein in the matrix. Source data are available for this figure: SourceData F6.
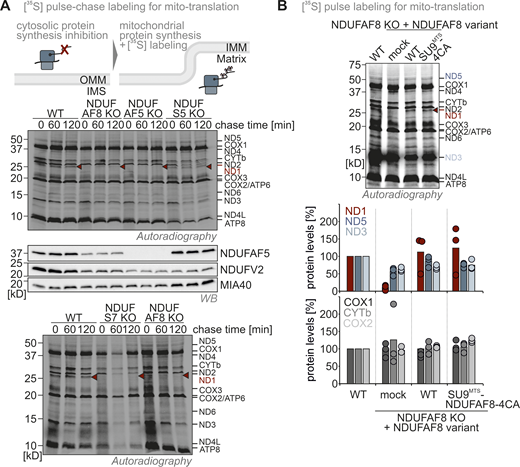
(Related toFig. 5 ). Translation of ND1 is impaired in NDUFAF5 and NDUFAF8 knockout cells. (A) Assessment of mitochondrial translation in different knockout cell lines. Cells were treated with emetine to block cytosolic translation. Under these conditions, only the 13 proteins encoded in the mitochondrial genome are synthesized. Their synthesis was assessed by radioactive pulse-labeling. Subsequently, stability of the mitochondrial translation products was assessed using chase times of 1 and 2 h, respectively. Cells were lysed and proteins were analyzed by reducing SDS-PAGE and autoradiography. Except for ND1 no other mitochondrial translation product was affected by the loss of NDUFAF5 (AF5 KO) or NDUFAF8 (AF8 KO). ND1 was completely missing even directly after the pulse period. This is in line with earlier findings that Q module assembly is directly linked to the biogenesis of the mitochondria-encoded subunit ND1 which is rapidly degraded if Q module assembly is impaired (Zurita Rendon and Shoubridge, 2012), and is also fitting to our findings with an NDUFS7 knockout (S7 KO) cell line. ND1 was not lost in NDUFS5 knockout (S5 KO) cells in which complex I is also lost, but assembly is affected at a later stage (Salscheider et al., 2022). (B) Assessment of mitochondrial translation in NDUFAF8 knockout cell lines complemented with NDUFAF8 variants. Experiment was performed as described in A. Both NDUFAF8 and SU9MTS-NDUFAF8-4CA could complement the loss of NDUFAF8 with respect to ND1 synthesis indicating again their full functionality despite the absence of disulfide bonds. Signals were quantified using Image Lab and the amount of imported protein was plotted. N = 3 replicates. Source data are available for this figure: SourceData FS5.
(Related toFig. 5 ). Translation of ND1 is impaired in NDUFAF5 and NDUFAF8 knockout cells. (A) Assessment of mitochondrial translation in different knockout cell lines. Cells were treated with emetine to block cytosolic translation. Under these conditions, only the 13 proteins encoded in the mitochondrial genome are synthesized. Their synthesis was assessed by radioactive pulse-labeling. Subsequently, stability of the mitochondrial translation products was assessed using chase times of 1 and 2 h, respectively. Cells were lysed and proteins were analyzed by reducing SDS-PAGE and autoradiography. Except for ND1 no other mitochondrial translation product was affected by the loss of NDUFAF5 (AF5 KO) or NDUFAF8 (AF8 KO). ND1 was completely missing even directly after the pulse period. This is in line with earlier findings that Q module assembly is directly linked to the biogenesis of the mitochondria-encoded subunit ND1 which is rapidly degraded if Q module assembly is impaired (Zurita Rendon and Shoubridge, 2012), and is also fitting to our findings with an NDUFS7 knockout (S7 KO) cell line. ND1 was not lost in NDUFS5 knockout (S5 KO) cells in which complex I is also lost, but assembly is affected at a later stage (Salscheider et al., 2022). (B) Assessment of mitochondrial translation in NDUFAF8 knockout cell lines complemented with NDUFAF8 variants. Experiment was performed as described in A. Both NDUFAF8 and SU9MTS-NDUFAF8-4CA could complement the loss of NDUFAF8 with respect to ND1 synthesis indicating again their full functionality despite the absence of disulfide bonds. Signals were quantified using Image Lab and the amount of imported protein was plotted. N = 3 replicates. Source data are available for this figure: SourceData FS5.
Thus, the disulfide bonds in NDUFAF8 are apparently not important for its function in activating NDUFAF5. They are, however, critical to allow the accumulation of sufficient NDUFAF8 amounts in the matrix because they stabilize the protein and prevent CLPP-mediated degradation. To test the correlation between NDUFAF8 levels and its activity, we made use of our cell system that allows doxycycline-titratable expression of NDUFAF8 variants in the NDUFAF8 KO background. Indeed, titrating not only the amounts of NDUFAF8-WT but also of the SU9MTS-NDUFAF8-4CA variant resulted in an according growth on galactose (Fig. 6 E).
Collectively, we demonstrated that the disulfide bonds in NDUFAF8 are dispensable for function. However, since reduced NDUFAF8 is more rapidly degraded in the matrix, improved import rates and thus increased initial levels of the protein are required for this reduced protein to fulfill its function (Fig. 6 F). In principle, this could also be solved by equipping NDUFAF8 with a strong endogenous MTS, raising the question of whether the detour of NDUFAF8 through the IMS serves regulatory purposes.
The activity of the mitochondria disulfide relay links IMS redox conditions to levels and activity of the matrix protein NDUFAF5
NDUFAF8 levels are critical for NDUFAF5 activation. These levels are linked to its stability, which depends on the presence of disulfide bonds and thus likely on the functionality of the mitochondrial disulfide relay. We thus next assessed how the loss of AIFM1 and MIA40/CHCHD4, the core components of the disulfide relay, affected NDUFAF5 levels (as a proxy for endogenous NDUFAF8, which we cannot detect). Notably, we cannot use complex I as a readout for this assay as AIFM1 and MIA40/CHCHD4 directly affect the import of four complex I subunits including NDUFS5 (Erdogan et al., 2018; Hangen et al., 2015; Meyer et al., 2015; Salscheider et al., 2022). Instead, we followed NDUFAF5 levels as even the knockout of NDUFS5, which results in the loss of complex I, did not affect NDUFAF5 levels (Fig. 7 A). This implies that any impact that AIFM1 and MIA40/CHCHD4 have directly on complex I will not impact NDUFAF5, and thus all effects that we would observe on NDUFAF5 would be via NDUFAF8.
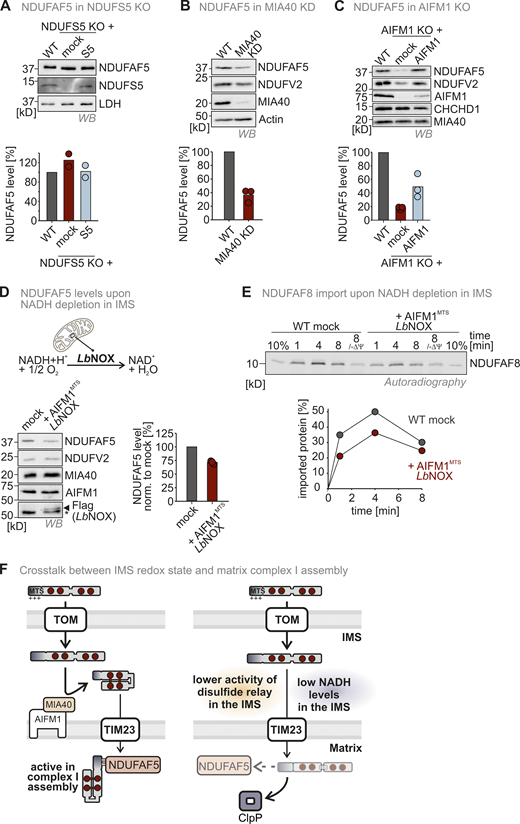
Functionality of the mitochondrial disulfide relay and redox conditions in the IMS determine the efficiency of NDUFAF5 activation and stability. (A) Protein levels in NDUFS5 knockout cells. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. Loss of NDUFS5 does not affect NDUFAF5 levels. Since NDUFS5 is a structural subunit of complex I, its loss results in complex I loss. Thus, complex I loss does not affect NDUFAF5 levels. N = 3 replicates; error bars indicate SD. (B) Protein levels in MIA40/CHCHD4 knockdown cells. Experiment was performed as described in A. Depletion of MIA40/CHCHD4 results in depletion of NDUFAF5. This effect is independent of complex I levels and most likely mediated by the depletion of NDUFAF8 in these cells. N = 3 replicates; error bars indicate SD. (C) Protein levels in AIFM1 knockout cells. Experiment was performed as described in A. Loss of AIFM1 results in depletion of NDUFAF5. Like for MIA40/CHCHD4, this effect is independent from complex I levels and most likely mediated by the depletion of NDUFAF8 in these cells. N = 3 replicates; error bars indicate SD. (D) Protein levels in HEK293 cells expressing IMS-targeted LbNOX. Experiment was performed as described in A. Shifting the NADH/NAD+ ratio toward NAD+ results in the depletion of NDUFAF5. N = 3 replicates; error bars indicate SD. (E) In organello import assay with NDUFAF8 to test for its dependence on IMS NADH levels. Experiment was performed as described in Fig. 2 A. Import was performed into mitochondria isolated from wild-type or AIFM1MTS-LbNOX expressing cells. NDUFAF8 import into mitochondria with low NADH levels was reduced. N = 1 replicate. (F) Model. The two-step import pathway of NDUFAF8 combined with the close surveillance of its levels in IMS and matrix allows control of NDUFAF5 levels and activity and efficiency of complex I assembly dependent on the activity of IMS redox processes. Both impairment of the mitochondrial disulfide relay as well as NADH depletion directly has an impact on NDUFAF5 levels in the matrix. Source data are available for this figure: SourceData F7.
Functionality of the mitochondrial disulfide relay and redox conditions in the IMS determine the efficiency of NDUFAF5 activation and stability. (A) Protein levels in NDUFS5 knockout cells. Lysates from different cells were analyzed by reducing SDS-PAGE and immunoblotting. Signals were quantified using Image Lab, and the amount of protein was plotted. Loss of NDUFS5 does not affect NDUFAF5 levels. Since NDUFS5 is a structural subunit of complex I, its loss results in complex I loss. Thus, complex I loss does not affect NDUFAF5 levels. N = 3 replicates; error bars indicate SD. (B) Protein levels in MIA40/CHCHD4 knockdown cells. Experiment was performed as described in A. Depletion of MIA40/CHCHD4 results in depletion of NDUFAF5. This effect is independent of complex I levels and most likely mediated by the depletion of NDUFAF8 in these cells. N = 3 replicates; error bars indicate SD. (C) Protein levels in AIFM1 knockout cells. Experiment was performed as described in A. Loss of AIFM1 results in depletion of NDUFAF5. Like for MIA40/CHCHD4, this effect is independent from complex I levels and most likely mediated by the depletion of NDUFAF8 in these cells. N = 3 replicates; error bars indicate SD. (D) Protein levels in HEK293 cells expressing IMS-targeted LbNOX. Experiment was performed as described in A. Shifting the NADH/NAD+ ratio toward NAD+ results in the depletion of NDUFAF5. N = 3 replicates; error bars indicate SD. (E) In organello import assay with NDUFAF8 to test for its dependence on IMS NADH levels. Experiment was performed as described in Fig. 2 A. Import was performed into mitochondria isolated from wild-type or AIFM1MTS-LbNOX expressing cells. NDUFAF8 import into mitochondria with low NADH levels was reduced. N = 1 replicate. (F) Model. The two-step import pathway of NDUFAF8 combined with the close surveillance of its levels in IMS and matrix allows control of NDUFAF5 levels and activity and efficiency of complex I assembly dependent on the activity of IMS redox processes. Both impairment of the mitochondrial disulfide relay as well as NADH depletion directly has an impact on NDUFAF5 levels in the matrix. Source data are available for this figure: SourceData F7.
Excitingly, depletion of MIA40/CHCHD4 or loss of AIFM1 did lower NDUFAF5 levels in line with their role in NDUFAF8 import (Fig. 7, B and C). Thus, impairment of the mitochondrial disulfide relay results in NDUFAF5 depletion via loss of NDUFAF8. AIFM1 is an NADH-dependent enzyme, and its activity in the mitochondrial disulfide relay critically requires NADH binding (Hangen et al., 2015; Salscheider et al., 2022). Thus, we lastly tested whether shifting the ratio of the NADH/NAD+ couple toward lowered NADH levels by expressing the water-producing NADH oxidase from Lactobacillus brevis (LbNOX) in the IMS would impact NDUFAF5 levels (Titov et al., 2016). Also, this perturbation lowered NDUFAF5 levels (Fig. 7 D) and attenuated NDUFAF8 import (Fig. 7 E).
In summary, our data are in line with a model in which NDUFAF8 takes a two-step import pathway (Fig. 7 F). After traversing the OMM, it interacts with the disulfide relay to receive two disulfide bonds. This interaction is enabled because its weak MTS prevents rapid import into the matrix. In its oxidized state, NDUFAF8 translocates through TIM23 to the matrix where it activates NDUFAF5. For this, it relies on its MTS which is not cleaved during import. NDUFAF5 in turn facilitates efficient complex I assembly at an early stage. The maturation of NDUFAF8 is closely surveyed by the proteases CLPP and YME1L. CLPP rapidly removes reduced NDUFAF8 in the matrix. Thus, the redox state of NDUFAF8 is directly linked to matrix NDUFAF8 levels and its activity in complex I assembly. Consequently, perturbations of the IMS disulfide relay and changes in the IMS NADH/NAD+ ratio can directly impact this activity. Thus, the here-described import pathway essentially allows matrix processes to “read-out” the functionality of IMS and matrix import and folding pathways at the same time.
Additional substrates of the mitochondrial disulfide relay carry weak N-terminal targeting sequences that drive matrix import
Having characterized the import pathway of NDUFAF8, we next wondered whether additional proteins follow this pathway. To identify these proteins, we screened the known substrates of the human disulfide relay system (Edwards et al., 2021; Habich et al., 2019b) for (weak) N-terminal MTS. For most substrates, the TargetP prediction did only show very low scores for MTS; however, for CHCHD1, CHCHD2/9, CHCHD10, and COA6 isoform 2, the sliding iMTS approach revealed potential MTS (Fig. 8 A). These proteins all carry a positive net charge in their first 20 amino acids. To test whether these potential MTS can drive import into mitochondria, we fused them individually to DHFRmut and investigated import into isolated mitochondria (Fig. 8 B). Compared to an MTS-less DHFR, all MTS could confer mitochondrial import of DHFRmut albeit with low efficiency. To complement this approach, we performed the split GFP approach with the full-length proteins (Fig. 8 C and Fig. S1). Using this approach, we confirmed matrix localization of at least a share of all of these proteins. Using IMS-targeted LbNOX, we next tested whether modulating NADH levels in the IMS also influenced the levels of these proteins (Fig. 8 D). Indeed, we found that CHCHD1 and COA6 levels were lowered upon depletion of IMS NADH, as was the case with NDUFAF8.
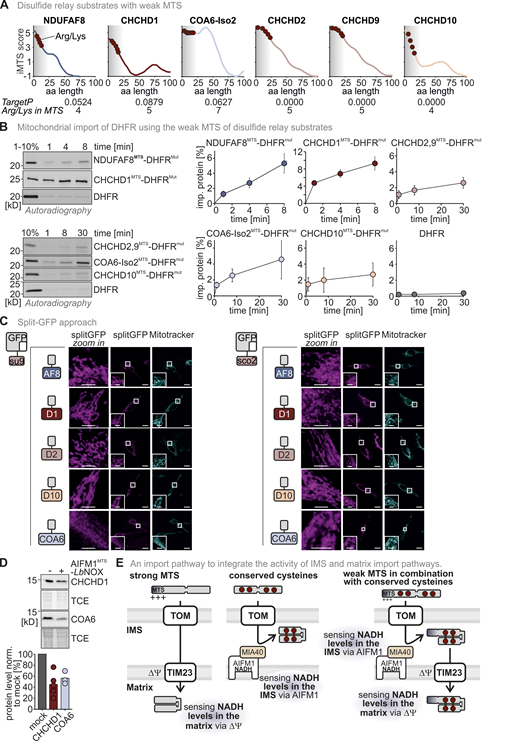
Additional substrates of the mitochondrial disulfide relay carry weak N-terminal targeting sequences that can drive matrix import. (A) iMTS prediction of different disulfide relay substrates. NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2 all have very low TargetP scores, but a positive iMTS score in their first 19 amino acids. All proteins contain in their N-terminal amino acid stretch arginine and lysine residues but not negatively charged amino acid residues. MTS, mitochondrial targeting signal. (B) In organello import assay with disulfide relay substrate-DHFR fusion constructs. The experiment was performed as described in Fig. 2 A. To test the capacity of the N-terminal amino acids of NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2 to serve as MTS, these amino acids (usually the first 19–24 amino acids) were fused to the cytosolic protein DHFRmut. All N-terminal stretches facilitated the import of DHFRmut albeit with different efficiencies. DHFR alone was not imported into mitochondria. N = 3 replicates; error bars indicate SD. (C) Split-GFP assay to detect the localization of full-length NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2. The experiment was performed as described in Fig. 1 C. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, or COA6 isoform 2 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS respectively (see “split GFP” signal). DHFR-GFP11 co-expression did not result in the reconstitution of GFP with any of the mitochondria localized GFP1-10 s. In the case of NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, or COA6 isoform 2, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10 indicating for all proteins that at least a small fraction localized also to the matrix. Bar corresponds to 20 µm. (D) Protein levels in HEK293 cells expressing IMS-targeted LbNOX. Experiment was performed as described in Fig. 7 D. Shifting the NADH/NAD+ ratio towards NAD+ results in depletion of CHCHD1 and COA6. N = 3–5 replicates; error bars indicate SD. (E) Model. The import pathway combining the TIM23 and mitochondrial disulfide relay pathways allows the simultaneous readout of the functionality of both pathways and thereby potentially the energy state of matrix and IMS, respectively. In both cases, NADH levels influence import, either through establishing the mitochondrial membrane potential or by allowing the reconstitution of the MIA40/CHCHD4-AIFM import receptor complex. Source data are available for this figure: SourceData F8.
Additional substrates of the mitochondrial disulfide relay carry weak N-terminal targeting sequences that can drive matrix import. (A) iMTS prediction of different disulfide relay substrates. NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2 all have very low TargetP scores, but a positive iMTS score in their first 19 amino acids. All proteins contain in their N-terminal amino acid stretch arginine and lysine residues but not negatively charged amino acid residues. MTS, mitochondrial targeting signal. (B) In organello import assay with disulfide relay substrate-DHFR fusion constructs. The experiment was performed as described in Fig. 2 A. To test the capacity of the N-terminal amino acids of NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2 to serve as MTS, these amino acids (usually the first 19–24 amino acids) were fused to the cytosolic protein DHFRmut. All N-terminal stretches facilitated the import of DHFRmut albeit with different efficiencies. DHFR alone was not imported into mitochondria. N = 3 replicates; error bars indicate SD. (C) Split-GFP assay to detect the localization of full-length NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, and COA6 isoform 2. The experiment was performed as described in Fig. 1 C. GFP1-10 was equipped either with an MTS for the matrix (SU9MTS) or for the IMS (SCO2MTS). GFP11 was C-terminally fused to full-length NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, or COA6 isoform 2 and as controls for matrix, IMS, and cytosol to SOD2, MIA40/CHCHD4, and DHFR, respectively. For SOD2 and MIA40/CHCHD4, fluorescence could only be observed for matrix and IMS respectively (see “split GFP” signal). DHFR-GFP11 co-expression did not result in the reconstitution of GFP with any of the mitochondria localized GFP1-10 s. In the case of NDUFAF8, CHCHD1, CHCHD2, CHCHD9, CHCHD10, or COA6 isoform 2, GFP reassembled for both SU9MTS-GFP1-10 and SCO2MTS-GFP1-10 indicating for all proteins that at least a small fraction localized also to the matrix. Bar corresponds to 20 µm. (D) Protein levels in HEK293 cells expressing IMS-targeted LbNOX. Experiment was performed as described in Fig. 7 D. Shifting the NADH/NAD+ ratio towards NAD+ results in depletion of CHCHD1 and COA6. N = 3–5 replicates; error bars indicate SD. (E) Model. The import pathway combining the TIM23 and mitochondrial disulfide relay pathways allows the simultaneous readout of the functionality of both pathways and thereby potentially the energy state of matrix and IMS, respectively. In both cases, NADH levels influence import, either through establishing the mitochondrial membrane potential or by allowing the reconstitution of the MIA40/CHCHD4-AIFM import receptor complex. Source data are available for this figure: SourceData F8.
Collectively, we found that employing an unconventional two-step import pathway that uses the disulfide relay system for oxidative folding to stabilize proteins that are subsequently imported into the matrix might be followed by a number of proteins to allow integration of the functionality of the disulfide relay with the TIM23 pathway (Fig. 8 E).
Discussion
An unconventional matrix import pathway for substrates of the mitochondrial disulfide relay
Here, we report an unusual two-step import pathway for the matrix protein NDUFAF8 that sequentially employs the mitochondrial disulfide relay as well as the TIM23 pathway for its import into the mitochondrial matrix. To this end, NDUFAF8 combines in its primary structure four highly conserved cysteine residues and a very weak MTS. While a strong MTS guides a precursor protein efficiently across the OMM and IMM (as the SU9MTS does), the weak MTS of NDUFAF8 enables the protein to interact with the mitochondrial disulfide relay in the IMS. This oxidative machinery introduces two disulfide bonds into NDUFAF8 that stabilize a helix-loop-helix motif. Oxidized NDUFAF8 then traverses the IMM to bind and activate in the matrix its partner protein NDUFAF5, which in turn serves in modifying NDUFS7 and in the assembly of the Q module of complex I. This also indicates that the TIM23 import channel can accommodate the helix-loop-helix motif of NDUFAF8 (i.e., a partially folded protein). This had also been observed before, e.g., for the ribosomal protein Mrp10 in yeast (Huang et al., 2002; Longen et al., 2014). Moreover, our data also support that very stable disulfide bonds as they are present in NDUFAF8 can withstand the highly reducing milieu of the matrix. The MTS of NDUFAF8 is not cleaved after import; instead, it is critical to establish the interaction with NDUFAF5.
We demonstrate that this two-step import pathway is presumably also taken by a number of structurally similar proteins including CHCHD1 (MRPS37, yeast Mrp10), a subunit of the mitochondrial ribosome and the isoform 2 of COA6, as well as CHCHD2, CHCHD9, and CHCHD10. CHCHD2 and CHCHD10 had already been identified as interaction partners of the matrix protein p32, further supporting at least a partial matrix localization (Burstein et al., 2018).
At steady state, NDUFAF8 appears to be a dually localized protein. However, it does not appear that NDUFAF8 has a function in the IMS. It might thus be that the IMS fraction of NDUFAF8 at steady state is a consequence of a slow TIM23-dependent part of the import pathway and relatively low stability of (even folded and oxidized) NDUFAF8 in the matrix that results then in a relatively high fraction of NDUFAF8 that is en route to the matrix but still in the IMS. For other substrates of this two-step-import pathway, this might be different and they might perform functions in both compartments. For example, CHCHD2 and CHCHD10 interact with the matrix protein p32 but also play roles in respiratory chain regulation and regulation of OPA1 stability in the IMS (Burstein et al., 2018; Liu et al., 2020). For those proteins, the import pathway might allow dual localization and balance their levels and functions in IMS and matrix.
Coordination of disulfide bond formation with different matrix processes
Interestingly, many matrix proteins contain very weak MTS (∼20% of all matrix proteins have a TargetP score between 0 and 0.1). The resulting inefficient TIM23-dependent import might, for example, be important for increasing the sensitivity to fluctuations in the mitochondrial membrane potential, for enabling posttranslational modifications of precursors along the way, or for enabling other modes of dual localization (Haucke et al., 1997; Nargund et al., 2012).
In the case of NDUFAF8, the weak MTS leaves sufficient time to enable interaction with the mitochondrial disulfide relay and NDUFAF8 oxidation before IMM translocation. The presence of disulfide bonds in NDUFAF8 is not critical for matrix import and is not a prerequisite for its activity in stabilizing and activating NDUFAF5. However, since the matrix protease CLPP closely monitors the NDUFAF8 folding and redox state, reduced NDUFAF8 is rapidly degraded. Thus, disulfide formation strongly influences NDUFAF8 stability and thereby levels in the matrix. In line, we found that the absence of the cysteines in NDUFAF8 or interference with the disulfide bond formation in the IMS destabilizes NDUFAF8. This can only be complemented by strong overexpression of the protein.
Under physiological conditions, the activity of the IMS disulfide relay might be influenced by different factors including the availability of oxidized cytochrome c, the availability of oxygen, and the activity of the respiratory chain. In addition, recent work has demonstrated that the disulfide relay is sensitive to changes in IMS NADH levels through the oxidoreductase function of its core component AIFM1 (Hangen et al., 2015; Romero-Tamayo et al., 2021; Salscheider et al., 2022). AIFM1 requires NADH binding to undergo dimerization. AIFM1 dimerization is a prerequisite to bind MIA40/CHCHD4, and only this thereby-formed trimeric complex is efficient in disulfide formation (Salscheider et al., 2022). It was thus interesting to observe that disturbing the NADH/NAD+ ratio in the IMS by using LbNOX also influenced the here-described two-step import pathway. Levels of CHCHD1 and COA6 were reduced as well. Moreover, NDUFAF5 levels were decreased presumably because NDUFAF8 failed to acquire disulfide bonds. It is tempting to speculate that NADH/NAD+ ratios can be sensed by the mitochondrial disulfide relay thus allowing the matrix to sense NADH/NAD+ ratios in the IMS through the stability of disulfide relay-dependent proteins; high NADH levels would hereby facilitate disulfide formation, and low levels would prevent it.
In conclusion, we propose that the here-described two-step import pathway might serve as a mechanism to sense and integrate the activity of the disulfide relay in the IMS with matrix processes, and thereby constitute an additional regulatory layer that allows the tight coordination of assembly of complexes from proteins of dual genetic origin.
Materials and methods
Plasmids, cell lines, and chemical treatments of cells
For plasmids and cell lines used in this study, see Tables S1 and S2. Cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS) and penicillin/streptomycin (Pen/Strep) at 37°C under 5% CO2.
For the MitoBlock (MB)-10 treatment during import, isolated mitochondria were incubated for 30 min with 100 µM MB-10 (dissolved in DMSO) prior to import assay. MB-10 was also added to the lysate mix (100 µM).
For the emetine and the cycloheximide (CHX) chase experiments, cells were treated for the indicated times with 100 µg/ml emetine (dissolved in water) or 100 µg/ml CHX (dissolved in DMSO). The cells were washed once with 0.5 ml DMEM containing 100 µg/ml emetine or CHX and another 0.5 ml DMEM with 100 µg/ml emetine or CHX was added. After each time point, the cells were washed with 0.5 ml ice-cold PBS and harvested in reducing Laemmli buffer. For the generation of stable, inducible cell lines the HEK293 cell line–based Flp-In T-REx-293 cell line was used with the Flp-In T-REx system (Invitrogen).
CRISPR-Cas9-based generation of NDUFAF8 and NDUFAF5 HEK293 knockout cell lines
For the generation of NDUFAF5 and NDUFAF8 knockout cells, guide RNA sequences targeting human NDUFAF5 or NDUFAF8 were cloned into the pSpCas9(BB)-2A-GFP (PX458) vector, which was a gift from Feng Zhang (Broad Institute of the Massachusetts Institute of Technology and Harvard, Cambridge, MA, USA; plasmid #48138; Addgene; Ran et al., 2013). For NDUFAF8, the guide RNA sequence (Guide #3: 5′-TCGGCTAACGGAGCGGTGTG-3′), and for NDUFAF5, the guide RNA sequence (Guide #1: 5′-CTTATGTCGGCGACCTTGG-3′) were used. HEK Flp-In T-REx-293 cells were transfected with the plasmid using polyethylenimine (PEI; Thermo Fisher Scientific). After 24 h, GFP-positive cells were collected via FACS and single-cell clones were seeded onto 96-well plates (1 cell/well). Clonal cell lines were screened for NDUFAF5 protein expression by Western blotting and sequenced to verify indel mutations (by Eurofins GATC Biotech).
Complementation of NDUFAF8, NDUFAF5, YME1L, and CLPP CRISPR clones
For the complementation of CRISPR clones with different NDUFAF8 variants, the inducible Flp-In T-REx System was used. All NDUFAF8 constructs were cloned into the pcDNA5 FRT-TO vector and cotransfected with the pOG44 vector into the different CRISPR clones by using the transfection reagent FuGene, according to the manufacturer’s guideline. Positive clones were selected with glucose-containing medium (DMEM supplemented with 1 mM sodium pyruvate, 1 × nonessential amino acids, 10% FCS, and 500 mg/ml Pen/Strep, 50 µg/ml Uridine) containing 10 µg/ml Blasticidin and 100 µg/ml Hygromycin.
Cell proliferation assay to test for growth on different carbon sources
For the cell proliferation assay, 20,000 cells were seeded on a 24-well dish and incubated at 37°C. After several hours, the complementing cell lines were treated with doxycycline (1 µg/ml). After 16–18 h, the cells were washed with 500 µl PBS and trypsinized with 50 µl trypsin. Then, 200 µl DMEM was added, and the cells were harvested for cell counting. The medium was exchanged with DMEM containing galactose (DMEM supplemented with 4.5 g/l galactose, 1 mM sodium pyruvate, 1 × nonessential amino acids, 10% FCS, and 500 mg/ml Pen/Strep). Every day the cells were counted for 6 d using the cell counter (Luna). Triplicates were measured for each day, and the standard deviation was calculated.
For the cell proliferation assay recorded with the cytosmart omni, 15,000 cells were seeded on a poly-L-coated 48-well dish and incubated at 37°C. After several hours, the complementing cell lines were treated with doxycycline. Next day, the medium was exchanged with DMEM containing galactose (DMEM supplemented with 4.5 g/l galactose, 1 mM sodium pyruvate, 1 × nonessential amino acids, 10% FCS, and 500 mg/ml Pen/Strep). Every day, the medium was exchanged by removing 250 µl and adding 250 µl fresh DMEM containing galactose. Every 6 h, the coverage of each well was scanned for 6 d using the cytosmart omni.
Split-GFP assay
For the split-GFP assay, 200,000 Hela cells were seeded onto 12-well plates. 1 d after seeding, cells were transfected with 1 µg of split-GFP-10- and split-GFP-11-containing plasmids/well (see Table S1) and 4 µl Fugene HD/well (Promega) following the manufacturer’s protocol. The cells were incubated overnight, and the next day, 25,000 of these cells were seeded on 12-well plates containing poly-L-lysine-coated coverslips. The cells were treated with 1 µg/ml doxycycline. Next day, the media was exchanged with DMEM containing mitotracker (1:100,000; Invitrogen), and the cells were incubated for 1 h. The mitotracker was removed by exchanging the media, and the cells were incubated for 30 min. After a washing step with PBS, 1 ml of fixation buffer (4% paraformaldehyde in PBS) was added and incubated for 15 min at room temperature (RT). The cells were washed 3 × with 1 ml PBS and 0.5 ml of 4′,6-diamidino-2-phenylindole (DAPI) solution (1 µg/ml, Roth) was added for 15 min at RT. The cells were washed with 1 ml PBS. The coverslips were mounted on microscopy slides with Mowiol (Sigma-Aldrich) and DABCO (Roth) and were dried for 1 d at 4°C. The cells were analyzed by immunofluorescence microscopy.
Image acquisition
Microscope image acquisition
For the image acquisition, the microscope LSM 980 with Airyscan 2 and multiplex from Carl Zeiss Microscopy was used with a Plan-Apochromat 63×/1,4 Oil DIC objective and the GaAsP-PMT, Multi-Alkali-PMT detector. The cells were imaged at room temperature with oil as the imaging medium. The following fluorochromes were used: GFP, Mitotracker CMXRos, and Alexa Fluor488. Images were displayed using the acquisition software ZEN 3.3. and were processed using the software OMERO.insight. The inset magnification ranges from 4.5–13.5 fold magnification.
Western blot image acquisition
The immunoblotting images were detected using the ChemiDoc Touch Imaging system (Bio-Rad). The autoradiography images were detected using the image analyzer Typhoon FLA 7000.
Native and denaturing immunoprecipitation
To detect protein–protein interactions, native or denaturing immunoprecipitation (IP) was performed. Corresponding cell lines were seeded on a 10-cm dish and grown until a confluency of 90%. Before IP was performed, inducible cell lines were induced with doxycycline overnight. Then the cells were washed with 5 ml ice-cold PBS containing 20 mM N-Ethylmaleimide (NEM) and incubated with another 5 ml ice-cold PBS containing 20 mM NEM for 15 min on ice. The cells were scratched off and pelleted at 700 × g for 3 min at 4°C. For native IP, the pellet was resuspended in 500 µl native IP buffer (100 mM Na-Pi, pH 8.1, 1% TX-100) and was incubated for at least 30 min on ice. For denaturing IP, the pellet was resuspended in 210 µl denaturing IP buffer (100 mM Tris, pH 8.1, 150 mM NaCl, 1 mM EDTA, 2.5% TX-100) and 40 µl 10% SDS was added. The denaturing IP samples were sonified at maximum amplitude. After cooling down, 750 µl renaturing buffer (100 mM Tris, pH 8.1, 150 mM NaCl, 1 mM EDTA, 1% TX-100) was added and incubated for at least 30 min on ice. After that, the samples were centrifuged at 21,817 × g for 1 h at 4°C. The supernatant was collected and incubated with 10 µl equilibrated HA beads (monoclonal anti-HA-Agarose produced in mouse) overnight at 4°C. Then, the beads were washed four times with 1 ml washing buffer containing TX-100 (100 mM Na-Pi, pH 8.1, 1% TX-100, 250 mM NaCl for native IP; 100 mM Tris, pH 8.1, 150 mM NaCl, 1 mM EDTA, 1% TX-100 for denaturing IP) and 1 × with washing buffer without TX-100 with a centrifugation step at 2,000 × g for 1 min at 4°C. After the last centrifugation step, the beads were dried completely, 20 µl reducing Laemmli was added, and the samples were boiled for 10 min. The samples were analyzed by SDS-PAGE and Western blot.
Isolation of crude mitochondria from HEK293 cells
Isolation of crude mitochondria from HEK293 cells was performed as described in Murschall et al. (2021). In short, cells were cultivated for 4 d on 15 cm dishes. For harvesting, the cells were washed two times with 10 ml ice-cold 1 × PBS and scraped off using a cell scraper. Afterward, the cells were centrifuged at 500 × g for 5 min at 4°C. The pellets were resuspended in 5 ml 1× M buffer (220 mM mannitol, 70 mM Sucrose 5 mM HEPES-KOH, pH 7.4, 1 mM EGTA-KOH, pH 7.4) containing 1 × Complete TM Protease Inhibitor Cocktail. The cells were homogenized using a precooled potter homogenizer (1,000 rpm, 15 strokes). The homogenized cells were pelleted at 600 × g for 5 min at 4°C. The supernatant containing the crude mitochondria was centrifuged at 8,000 × g for 10 min at 4°C. The pellet was washed with 2 ml ice-cold 1× M buffer (without the complete protease inhibitor cocktail). The crude mitochondria were pelleted at 6,000 × g for 10 min at 4°C, and the supernatant was carefully removed. The pellet was resuspended in 400 µl, and the concentration was measured using the BCA Reagent ROTI Quant Assay according to the manufacturer’s instructions.
Submitochondrial fractionation of isolated mitochondria
For the subcellular fraction of NDUFAF8, a stable inducible cell line was used. These cells were treated with doxycycline for 18 h and 10 min with 100 µg/ml emetine prior to isolation of mitochondria. After the isolation of crude mitochondria, 40 µg mitochondria was centrifuged at 10,000 × g for 5 min at 4°C. Each pellet was resuspended in 95 µl of corresponding buffer (isotonic buffer: 1 × M buffer [220 mM mannitol, 70 mM sucrose, 5 mM HEPES-KOH, pH 7.4, 1 mM EGTA-KOH, pH 7.4]; hypotonic buffer: 10 mM HEPES, pH 7.4; and TX-100 buffer: 1× M buffer, 1% TX-100; each buffer containing either 40 µg/ml PK or no PK) by pipetting up and down using a 200-µl pipette tip that was cut. The samples were incubated for 5 min on ice, and after the incubation time, 2.5 µl of 0.2 M PMSF (final concentration 5 mM) was added to all six samples, and incubation was allowed for a further 5 min on ice. Then, the samples were centrifuged at 10,000 × g for 5 min, and the pellets were resuspended with 100 µl of each buffer (isotonic or hypotonic) containing 1 mM PMSF. 4× Laemmli buffer and DTT were added to a final concentration of 50 mM DTT. All samples were boiled at 95°C for 5 min. The samples were analyzed by SDS-PAGE and Western blot.
Protein import assay into isolated mitochondria
The import assay was performed as described in Murschall et al. (2021). After isolation, 20 µg of mitochondria per import reaction was centrifuged at 8,000 × g for 5 min at 4°C. The resulting pellet was carefully mixed and resuspended with 4 µl [35S]-labeled precursor lysate (prepared according to the manufacturer’s protocol and Murschall et al. [2021]) using a cut 10-µl pipette tip. For the CCCP control, 1 µl of 20 mM CCCP solution was added and incubated on ice prior to the import reaction. The import reactions were incubated at 30°C while shaking at 600 rpm and stopped at different time points by putting on ice. The samples were directly centrifuged at 8,000 × g 5 min at 4°C. Then, the samples were treated with proteinase K (PK) by resuspending mitochondria in 400 µl of 1 × M buffer (220 mM mannitol, 70 mM Sucrose, 5 mM HEPES-KOH, pH 7.4, 1 mM EGTA-KOH, pH 7.4) containing 20 µg/ml PK. The samples were incubated for 20 min on ice, and the digestion was stopped by adding 2 µl of 200 mM PMSF (1 mM final). After another centrifugation step at 10,000 × g for 5 min at 4°C, the pellets were washed with 400 µl buffer M containing 1 mM PMSF. The resulting pellets were resuspended in 30 µl reducing Laemmli buffer. The samples were boiled at 96°C for 5 min. In case the samples turned yellow, the samples were pH adjusted with a 1 M Tris solution (unbuffered). The samples were analyzed by SDS-PAGE and autoradiography.
Proteinase K-shaved mitochondria
Prior to the import assay, 20 µg mitochondria was resuspended in 100 µl buffer M containing 20 µg/ml PK. After 5 min, the digest was stopped with 1 mM PMSF and incubated for 5 min on ice. After one centrifugation step at 10,000 × g for 5 min at 4°C, the pellets were resuspended in 400 µl buffer M containing 1 mM PMSF. After another centrifugation step, the pellets were used for the import assay as described in section Protein import assay into isolated mitochondria.
Preoxidized NDUFAF8 import into mitoplasts
Mitochondria was isolated as described in section Isolation of crude mitochondria from HEK293 cells. 20 µg mitochondria was resuspended in hypotonic buffer (10 mM HEPES, pH 7.4) and centrifuged at 8,000 × g for 5 min at 4°C. The resulting pellet was carefully mixed and resuspended with pre-oxidized NDUFAF8 [35S]-labeled precursor lysate which was incubated with 1 mM Diamide for 5 min at 30°C prior to use. The import assay and PK treatment were performed as described in section Protein import assay into isolated mitochondria.
Submitochondrial fractionation after organelle import
The import assay was performed as described in section Protein import assay into isolated mitochondria. After the PK treatment, the pellets were washed with 400 µl buffer M containing 1 mM PMSF. The resulting pellets were washed another time with 400 µl buffer M without PMSF and were centrifuged at 10,000 × g for 5 min at 4°C. The pellets were resuspended 95 µl of corresponding buffer (isotonic buffer: 1 × M buffer [220 mM mannitol, 70 mM Sucrose, 5 mM HEPES-KOH, pH 7.4, 1 mM EGTA-KOH, pH 7.4]; hypotonic buffer: 10 mM HEPES, pH 7.4; TX-100 buffer: 1× M buffer, 1% TX-100; each buffer containing either 40 µg/ml PK or no PK) by pipetting up and down using a 200 µl pipet tip that was cut. The samples were treated as described in the section Submitochondrial fractionation of isolated mitochondria.
Protein import assay into mitochondria coupled to native immunoprecipitation
To follow the interaction between two proteins during mitochondrial import, the protein import assay can be coupled to a native IP to coprecipitate possible interaction partners during import. For this assay, respective cell lines were treated with doxycycline for 16–18 h. Crude mitochondria were isolated as described in the section Isolation of crude mitochondria from HEK293 cells. 80 µg of isolated mitochondria was centrifuged at 10,000 × g for 5 min at 4°C. The resulting pellet was resuspended with 4 µl [35S]-labeled precursor lysate and incubated for 30 min at 30°C while shaking at 600 rpm. The reaction was stopped on ice, and the sample was centrifuged at 10,000 × g for 5 min at 4°C. The pellet was resuspended in 400 µl of 1× M buffer (220 mM mannitol, 70 mM sucrose, 5 mM HEPES-KOH, pH 7.4, 1 mM EGTA-KOH, pH 7.4) containing 20 µg/ml proteinase K (PK). The samples were incubated for 5 min on ice. PK digest was stopped by adding 2 µl of 200 mM PMSF (1 mM final). After another centrifugation step at 10,000 × g for 5 min at 4°C, the pellets were washed with 400 µl of buffer M containing 1 mM PMSF. This step was repeated another time and the resulting pellets were resuspended in 250 µl native IP buffer (100 mM Na-Pi, pH 8.1, 1% TX-100). After 30 min incubation time on ice, a native IP with Streptactin beads (directed against MIA40/CHCHD4-Strep) was performed.
Assay to assess the TOM-TIM23 import tethering complex
The baker’s yeast Saccharomyces cerevisiae wild-type strain YPH499 (Chacinska et al., 2004) was grown at 30°C on YPD medium (1% [wt/vol] yeast extract, 2% [wt/vol] peptone, 2% [vol/vol] glucose). Mitochondria were isolated by differential centrifugation following published procedures (Priesnitz et al., 2020). Radiolabeled precursor proteins were generated by cell-free translation in the presence of [35S] methionine (Priesnitz et al., 2020). 35S-labeled precursor proteins were preincubated with 5 μM methotrexate in import buffer (1% [wt/vol] BSA, 250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 10 mM MOPS/KOH, pH 7.2, 5 mM methionine, 2.5 mM KH2PO4, pH 7.2, 2 mM ATP, 120 μg/ml creatine kinase, and 12 mM creatine phosphate) for 15 min on ice. The addition of methotrexate induces stable folding of the DHFR domain. Afterward, isolated mitochondria and 2 mM NADH were added to start the import reaction. Samples were incubated at 24°C for 25 min. The import reaction was stopped by the addition of 1 µM valinomycin. Mitochondria were reisolated (13,000 g, 4°C, 10 min) and washed with ice-cold SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS-KOH, pH 7.2). The imported proteins were analyzed by blue native electrophoresis followed by autoradiography using a Typhoon Biomolecular Imager (GE Healthcare).
Assessment of mitochondrial translation
To analyze the translation of proteins at mitochondrial ribosomes and to assess their stability, radioactive pulse and pulse-chase assays were performed. Cells were seeded on a poly-L-lysine-coated 3.5-cm dish and were grown until 80–90% confluence was reached. For the pulse assay, the cells were preincubated for 30 min in a starvation medium (DMEM without methionine and FCS). Then the cells were treated for 5 min in starvation medium containing 100 μg/ml Emetine. For the pulse treatment, newly synthesized proteins were pulse-labeled with EasyTag EXPRESS [35S]-Protein Labeling Mix (Perkin Elmer) at a concentration of 42 mCi/ml incubated at 37°C. For the pulse-chase assay, pulse-labeling was stopped after 2 h, and DMEM with methionine was added for 1 and 2 h. Before harvesting cells with a cell scraper, the cells were incubated for 5 min in DMEM. The cells were washed twice with PBS and were harvested. After a centrifugation step for 15 min at 16,000 × g at 4°C, the pellets were resuspended in 50 µl reducing Laemmli buffer (containing 50 mM DTT). The samples were incubated for 10 min at 45°C. After a short spin at 21,817 g, the samples were sonified to break DNA. The samples were analyzed by SDS-PAGE and autoradiography.
Determination of cellular protein levels by quantitative label-free proteomics
For quantitative label-free proteomics, the experiments were performed as described in Habich et al. (2019a). Respective cells were seeded on a 6-well dish. The next day, the cells were washed with PBS and harvested by cell scraper and centrifugation at 500 × g for 5 min. The pellets were resuspended in 20 µl lysis buffer (4% SDS in PBS containing protease inhibitor) and were sonified. Afterward, the samples were boiled for 5 min at 96°C. After cooling down, 160 µl ice-cold acetone was added and stored at −80°C overnight. The next day, TCA precipitations were thawed and the samples were centrifuged for 15 min at 16,000 × g. The resulting pellets were washed with 500 µl acetone and then air-dried. The pellets were then resuspended in 50 µl 8 M urea in TEAB buffer supplemented with protease inhibitor cocktail and sonified. The samples were centrifuged for 15 min at 20,000 × g. The supernatant was transferred into a new tube and the protein concentration of the samples was determined by Pierce Protein Assay Reagent. The assay was performed according to the manufacturer’s protocol, and the concentration was measured at 600 nm. 50 µg of each sample was transferred to a new reaction tube and filled up to 40 µl with the Urea/TEAB buffer. Then, DTT with a final concentration of 5 mM was added, and the samples were incubated for 1 h at 37°C. Next, chloroacetamide (CAA) with a final concentration of 40 mM was added to the samples and then incubated for 30 min in the dark. For the digestion of the peptides, first Lysyl Endopeptidase with an enzyme-to-substrate ratio of 1:75 was used, and the samples were incubated for 4 h at 25°C. For the trypsin digest, the samples were first diluted with TEAB buffer to reach a urea concentration below 2 M and then trypsin with an enzyme-to-substrate ratio of 1:75 was added. The samples were incubated at 25°C overnight. In the last step, the samples were purified by STAGE tips, which were equilibrated with methanol and buffers containing 0.1% formic acid and 80% acetonitrile. The samples were loaded on the STAGE tips and were washed with buffers containing 0.1% formic acid and 80% acetonitrile. The STAGE tips were completely dried and until measurement stored at 4°C. The mass spectrometry was performed and analyzed by the proteomics core facility Cologne.
Determination of protein interaction partners by proteomics (interactome analysis)
For the interactome data, an in-gel digest was performed. The native IP was performed as described in the respective section except that no NEM was used in the initial washing steps. After the native IP was performed, the beads were dried and boiled in 20 µl reducing Laemmli buffer (without bromphenolblue) for 10 min. The samples were reduced by the addition of DTT with a final concentration of 5 mM and incubated at 56°C for 30 min. Free cysteine thiols were alkylated by the addition of CAA to a final concentration of 40 mM to the samples which were then incubated for 30 min at room temperature in the dark. The samples were run on SDS-PAGE until the samples migrated for 1 cm into the separation gel. Then the gels were fixed for 1 h in a fixing solution (10% Acetic acid/20% Methanol in water). The gel bands were cut into smaller pieces and transferred to individual tubes. 100 µl of 50 mM ABC/50% Acetonitrile was added to the gel pieces and were incubated for 20 min at room temperature. The solution was exchanged with fresh 50 mM ABC/50% Acetonitrile and the remaining solution were discarded after 20 min incubation. The gel pieces were covered with 100 µl acetonitrile and incubated for 10 min. The gel pieces were then dried in a speedvac for ∼5 min. A digestion solution of 10 ng/μl of 90% trypsin and 10% LysC in 50 mM ammonium bicarbonate (ABC) was added to the gel pieces until they were fully covered. The gel pieces were incubated for 30 min at 4°C with the digest solution. After the incubation time, the excessive digest solution was removed, and 50 mM ABC buffer was used to cover the gel pieces. The samples were incubated overnight at 37°C while shaking at 750 rpm. The next day, the supernatant of the gel pieces was transferred into new tubes. The gel pieces were covered with 100 µl 30% ACN/3% TFA and incubated for 20 min at room temperature. The extract was combined with the supernatant of the previous step. The gel pieces were covered with 100 µl 100% acetonitrile and again incubated for 20 min at room temperature. The extract was also combined with the supernatant from the previous step and the organic solvents of the samples were reduced in the speedvac until a remaining volume of 50 µl was reached. The samples were acidified by the addition of formic acid to a final concentration of 1%, and the STAGE tip purification protocol as it is described in the section Determination of cellular protein levels by quantitative label-free proteomics was performed. The STAGE tips were stored at 4°C. The mass spectrometry was performed and analyzed by the proteomics core facility Cologne.
Assay to detect redox states of protein thiols (+emetine)
To assess the redox state of NDUFAF8 in different cell lines, 60,000 cells were seeded on a 24-well dish. The cells were cultivated for 2 d. Before the cells were harvested, they were treated for 19 h with doxycycline (1 µg/ml) to induce NDUFAF8 expression. If the assay was coupled to an emetine treatment, the cells were treated with 100 µg/ml emetine before they were harvested and modified. For harvesting, the cells were washed with 500 µl ice-cold PBS and the non-reducing Laemmli buffer (2% SDS, 60 mM Tris, pH 6.8, 10% glycerol, 0.0025% bromphenol blue) containing 15 mM mmPEG12 was added to obtain “steady state” samples. The “maximum reduced” and “unmodified” samples were harvested after washing with PBS in Laemmli buffer containing 10 mM Tris(2-carboxyethyl) phosphine (TCEP). The maximum reduced samples were boiled for 15 min at 96°C. All samples were sonicated and analyzed by SDS-PAGE and Western blot.
Structure modeling using Alphafold-Multimer
To model the structure of the NDUFAF5/NDUFAF8 complex, AlphaFold multimer was used (Jumper et al., 2021). For the prediction, the sequences of both proteins were run in AlphaFold using ChimeraX software (version 1.4). Predicted best model structure was visualized with PyMol.
Determination of iMTS score
For the prediction of the iMTS score, iMTS-L prediction tool was used following the general workflow on the homepage (http://iomiqsweb1.bio.uni-kl.de/; Boos et al., 2018).
Quantification and statistical analysis
The intensity of autoradiography and immunoblot signals were quantified using ImageQuant and Image Lab (Biorad), respectively. Microscope images were processed with Fiji. Error bars in the figures represent standard deviation. The number of experiments is reported in the figure legend.
Online supplemental material
Fig. S1 confirms that tagged and untagged NDUFAF8 and CHCHD1 behave the same and provide the full data set for the split-GFP experiment. Fig. S2 provides additional insights into the import mechanism of NDUFAF8. Fig. S3 presents the characterization of the NDUFAF5 and NDUFAF8 knockout cell lines. Fig. S4 highlights the interaction of mitoribosomal subunits with NDUFAF8 and NDUFAF5. Fig. S5 shows that translation of the mitochondria-encoded ND1 is disturbed in NDUFAF5 and NDUFAF8 knockout cell lines. Table S1 contains the cell lines used in this study. Table S2 lists primers and plasmids used in this study. Table S3 contains the antibodies used in this study. Table S4 contains all oligonucleotides and siRNAs used in this study. Table S5 lists all further equipment and software employed in this study.
Data availability
The datasets generated and/or analyzed during the current study are available via ProteomeXchange with identifiers PXD040495, PXD040453, and PXD040441, and the Dryad Database https://doi.org/10.5061/dryad.0zpc8672t.
Acknowledgments
We thank Kathrin Ulrich for her critical reading of the manuscript. We thank Anja Wittmann for technical support throughout the project and the CECAD Proteome and Imaging Facilities for the provision of instrumentation, training, and technical support.
The Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) provided funds for research in the Laboratory of J. Riemer through the grants RI2150/5-1 project number 435235019, RI2150/2-2 project number 251546152, RTG2550/1 project number 411422114, and CRC1218 project number 269925409. Research in the lab of T. Becker is funded by DFG grants BE4679/2-2 project number 269424439 and CRC1218 project number 269925409. Research in the lab of A. Trifunovic is funded by the DFG including CRC1218 project number 269925409. Research in the lab of T. Langer is funded by the DFG including CRC1218 project number 269925409. Mike Ryan (Monash University) provided the HEK293 knockout cell for NDUFS7.
Author contributions: J. Riemer and E. Peker designed the study and planned experiments. K. Weiss and E. Peker designed, cloned the constructs, and generated cell lines. V. Boehm and N.H. Gehring helped to verify cell lines. S. Gerlich performed bioinformatical analyses. A. Trifunovic and T. Langer contributed cell lines. E. Peker and CECAD proteomics facility carried out the mass spectrometry-based proteomics and performed the bioinformatical analysis of the mass spectrometry data. J. Song and T. Becker carried out and analyzed the BN-PAGE crosslinking experiments. E. Peker, C. Zarges, and S. Gerlich carried out the biochemical experiments. K. Weiss carried out the fluorescence microscopy experiments and the image analysis. J. Riemer, E. Peker, and K. Weiss analyzed the data. J. Riemer and E. Peker wrote the manuscript with the help and input of all authors.

