


Limitation of excessive inflammation due to selective degradation of pro-inflammatory proteins is one of the cytoprotective functions attributed to autophagy. In the current study, we highlight that selective autophagy also plays a vital role in promoting the establishment of a robust inflammatory response. Under inflammatory conditions, here TLR3-activation by poly(I:C) treatment, the inflammation repressor TNIP1 (TNFAIP3 interacting protein 1) is phosphorylated by Tank-binding kinase 1 (TBK1) activating an LIR motif that leads to the selective autophagy-dependent degradation of TNIP1, supporting the expression of pro-inflammatory genes and proteins. This selective autophagy efficiently reduces TNIP1 protein levels early (0–4 h) upon poly(I:C) treatment to allow efficient initiation of the inflammatory response. At 6 h, TNIP1 levels are restored due to increased transcription avoiding sustained inflammation. Thus, similarly as in cancer, autophagy may play a dual role in controlling inflammation depending on the exact state and timing of the inflammatory response.
Introduction
Macroautophagy, hereafter referred to as autophagy, is a primary cytoprotective process that leads to the removal and lysosomal degradation of non-functional and/or superfluous cytoplasmic material. Autophagy is a constitutive process but may also be triggered by various forms of cell stresses (Mizushima and Levine, 2020). It involves the formation of a double membrane vesicle known as autophagosome that enwraps portions of the cytoplasm, including organelles. The content of the autophagosome is then targeted for degradation through the lysosome. Constitutive autophagy is regarded as a non-selective, bulk process, whereas stress-induced autophagy is often selective, aiming at removing the causes of stress, e.g., depolarized mitochondria in oxidative stress (Youle, 2019). Selective autophagy is carried out either by direct interactions between cargo and lipidated human ATG8 family members (MAP1LC3-A, -B, -C, GABARAP, GABARAPL1, GABARAPL2, commonly referred to as LC3s) that are anchored to autophagosomal membranes or by indirect interactions in which so-called selective autophagy receptors (SARs) tether cargo to LC3s (Johansen and Lamark, 2020). Cargo and receptors are then both degraded within lysosomes (Morishita and Mizushima, 2019). p62/SQSTM1, which is considered the founding member of the protein class of p62/SQSTM1-like receptors (SLRs), recognizes poly-ubiquitinated proteins/organelles destined for lysosomal degradation through its ubiquitin-associated (UBA) domain and interacts with lipidated LC3s through its LC3-interacting region (LIR) motif (Johansen and Lamark, 2020; Pankiv et al., 2007). SLRs are involved in the selective, autophagy-dependent degradation of a highly diverse set of substrates (Zellner et al., 2021).
The limitation of deleterious inflammatory responses is one of the cytoprotective functions attributed to autophagy (Deretic and Levine, 2018). The link between autophagy and tissue inflammation became obvious by genome-wide association studies that identified ATG16L1 as susceptibility locus for Crohn’s disease (Hampe et al., 2007). Since then several autophagy loci have been linked to inflammatory and autoimmune disorders (Mizushima and Levine, 2020). The selective degradation of inflammasome components, e.g., by p62/SQSTM1, is among the best-understood functions of autophagy in limiting tissue inflammation (Deretic and Levine, 2018; Samie et al., 2018). However, autophagy was also shown to support unconventional secretion of the pro-inflammatory cytokine IL1B (Dupont et al., 2011; Zhang et al., 2015), which argues for a fine-tuned and balanced role of autophagy in regulating inflammatory responses.
TNIP1 (also known as ABIN-1, Naf1, and VAN) is a ubiquitin-binding adaptor protein that has been implicated as a negative regulator of inflammatory signaling and cytokine-induced cell death (Dziedzic et al., 2018; Gao et al., 2011; Oshima et al., 2009; Su et al., 2019; Zhou et al., 2011). Interestingly, recent studies linked TNIP1, either as cargo or as receptor, to autophagy-dependent protein degradation (Shinkawa et al., 2022). Selective autophagy-dependent degradation of TNIP1 through interaction with the SLR optineurin (OPTN) was reported, supporting senescence-associated inflammation (Lee et al., 2021). While TNIP1 itself shows no catalytic activity, its ability to bind linear polyubiquitin chains through its Ub-binding domain in ABIN proteins and NEMO (UBAN) is important for its anti-inflammatory function (Nanda et al., 2011; Wagner et al., 2008). A number of studies suggest that TNIP1 exerts its negative function by recruiting the ubiqutitin-editing enzyme TNFAIP3 (also known as A20) to polyubiquitinated targets (Dziedzic et al., 2018; Gao et al., 2011; Mauro et al., 2006). However, the exact mechanism behind this negative regulation is not completely understood. Furthermore, TNIP1 shows activity independent of TNFAIP3 (Kattah et al., 2018; Oshima et al., 2009). The importance of TNIP1 as an anti-inflammatory signal transducer is highlighted by numerous studies implicating TNIP1 dysregulation in autoimmune disorders (Shamilov and Aneskievich, 2018). Uncovering the molecular function and dynamics of TNIP1 could, therefore, be valuable in understanding the mechanisms behind such complex disorders.
In the current study, we characterize TNIP1 as an autophagy substrate, which is selectively degraded at an early stage (0–4 h) under inflammatory conditions. We highlight that TNIP1 fulfills the structural characteristics of an autophagy receptor with an oligomerization domain, a ubiquitin-binding domain, and an LIR motif. Upon TLR3 activation, TNIP1 is phosphorylated by TBK1 on LIR proximal serine residues to increase binding to LC3s. Hence, TNIP1 is selectively degraded by autophagy in order to promote a competent initiation of pro-inflammatory signaling.
Results
TNIP1 is ubiquitinated and degraded within lysosomes
To identify new proteins potentially involved in autophagy regulation or autophagosomal targeting, we screened ubiquitination dynamics by quantitative mass spectrometry (MS)-based proteomics. U2OS and HeLa cells were differentially labeled by stable isotope labeling by amino acids in cell culture (SILAC), and autophagy was induced by inhibiting MTORC1 by rapamycin. In parallel, lysosomal degradation was blocked by concanamycin A (ConA), an inhibitor of lysosomal V-type ATPase (Klionsky et al., 2021). Respective cell lysates were mixed, proteins digested with the endoprotease LysC and ubiquitinated peptides were enriched using the UbiSite approach followed by MS-based identification and quantification (Akimov et al., 2018; Fig. 1 A). We identified more than 9,000 ubiquitination sites, of which more than 2,000 were quantified in minimally three biological replicates and could be localized clearly to specific amino acid residues (class I sites, Fig. 1 B and Table S1; Olsen et al., 2006). These sites were used for further analyses. Comparing abundance changes of ubiquitination sites of cells treated with rapamycin with cells treated with rapamycin and ConA, 148 sites were identified as significantly regulated; the majority being more abundant in cells in which lysosomal degradation was inhibited by ConA (paired t test, FDR < 0.05, Fig. 1 C). As anticipated, we identified many proteins involved in autophagosomal biogenesis and target recruitment. Importantly, four central SLRs, p62/SQSTM1, NBR1, CALCOCO2/NDP52, and TAX1BP1 (Johansen and Lamark, 2020), were identified as being increasingly ubiquitinated (Fig. 1 C), indicating that our experimental strategy was successful.
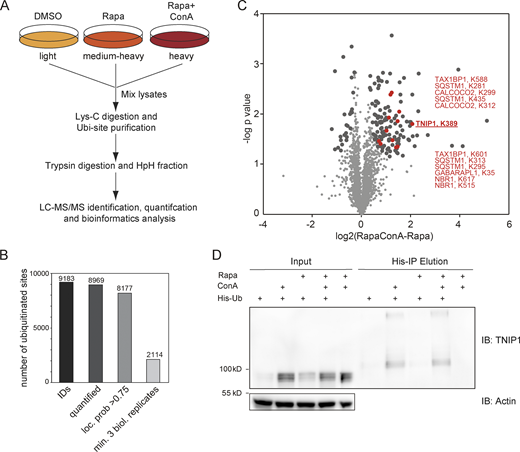
Ubiquitination and lysosomal degradation of TNIP1. (A) MS workflow to quantify ubiquitination sites potentially involved in autophagy-dependent lysosomal protein degradation. U2OS and HeLa cells were SILAC labeled and treated for 4 h with 100 nM rapamycin (Rapa), Rapa and 2 nM concanamycin A (ConA), or DMSO as control. After mixing of lysates, proteins were digested with Lys-C endoproteinase and ubiquitinated peptides were enriched using the UbiSite approach (Akimov et al., 2018). Enriched peptides were digested with trypsin, followed by high-pH reversed phase fractionation (Batth and Olsen, 2016) and shotgun LC-MS/MS analysis. (B) Detected ubiquitination sites. In three biological replicates, 9,183 ubiquitination sites were identified of which 8,969 were quantified. (C) Volcano plot highlighting significantly regulated ubiquitination sites. Significantly regulated sites comparing Rapa with Rapa + ConA treated cells are highlighted in dark grey (n = 3, paired two-sided t test, FDR < 0.05, S0 = 0.1, 148 sites in total; see Table S1). Data distribution was assumed to be normal but this was not formally tested. Non-regulated sites are colored in light gray. Sites identified on known autophagy receptors are colored in red exemplifying data quality. The newly identified site on TNIP1, K389, is highlighted in bold red. (D) TNIP1 gets ubiquitinated and degraded in the lysosome. U2-OS-StUbEx cells inducibly expressing His-FLAG-tagged ubiquitin at endogenous levels were used to enrich ubiquitinated proteins (Akimov et al., 2014). Under control conditions as well as under 4 h 100 nM rapamycin treatment TNIP1 got ubiquitinated as shown by anti-TNIP1 immunoblots. Ubiquitinated TNIP1 was stabilized by the addition of concanamycin A indicating its lysosomal degradation in treated and nontreated cells. The same was observed for starved cells (HBSS treatment; see Fig. S1). Actin was used as loading control. Note: Due to the design of the experiment and the used gels, the molecular weight marker (PageRuler Plus, #26619; Thermo Fisher Scientific) and the Rapa+/ConA+ sample were run in the same lane. In the respective source data, an additional replicate is shown in which samples ran separately. Source data are available for this figure: SourceData F1.
Ubiquitination and lysosomal degradation of TNIP1. (A) MS workflow to quantify ubiquitination sites potentially involved in autophagy-dependent lysosomal protein degradation. U2OS and HeLa cells were SILAC labeled and treated for 4 h with 100 nM rapamycin (Rapa), Rapa and 2 nM concanamycin A (ConA), or DMSO as control. After mixing of lysates, proteins were digested with Lys-C endoproteinase and ubiquitinated peptides were enriched using the UbiSite approach (Akimov et al., 2018). Enriched peptides were digested with trypsin, followed by high-pH reversed phase fractionation (Batth and Olsen, 2016) and shotgun LC-MS/MS analysis. (B) Detected ubiquitination sites. In three biological replicates, 9,183 ubiquitination sites were identified of which 8,969 were quantified. (C) Volcano plot highlighting significantly regulated ubiquitination sites. Significantly regulated sites comparing Rapa with Rapa + ConA treated cells are highlighted in dark grey (n = 3, paired two-sided t test, FDR < 0.05, S0 = 0.1, 148 sites in total; see Table S1). Data distribution was assumed to be normal but this was not formally tested. Non-regulated sites are colored in light gray. Sites identified on known autophagy receptors are colored in red exemplifying data quality. The newly identified site on TNIP1, K389, is highlighted in bold red. (D) TNIP1 gets ubiquitinated and degraded in the lysosome. U2-OS-StUbEx cells inducibly expressing His-FLAG-tagged ubiquitin at endogenous levels were used to enrich ubiquitinated proteins (Akimov et al., 2014). Under control conditions as well as under 4 h 100 nM rapamycin treatment TNIP1 got ubiquitinated as shown by anti-TNIP1 immunoblots. Ubiquitinated TNIP1 was stabilized by the addition of concanamycin A indicating its lysosomal degradation in treated and nontreated cells. The same was observed for starved cells (HBSS treatment; see Fig. S1). Actin was used as loading control. Note: Due to the design of the experiment and the used gels, the molecular weight marker (PageRuler Plus, #26619; Thermo Fisher Scientific) and the Rapa+/ConA+ sample were run in the same lane. In the respective source data, an additional replicate is shown in which samples ran separately. Source data are available for this figure: SourceData F1.
One protein that caught our attention was TNIP1/ABIN-1, which was identified as significantly ubiquitinated on the amino acid residue Lys389 (Fig. 1 C). TNIP1 is a key repressor of inflammatory signaling (Shamilov and Aneskievich, 2018), and posttranslational mechanisms regulating its protein abundance are largely unknown. In a reverse affinity purification (AP), we used U2OS-StUbEx cells inducibly expressing 6xHis-FLAG-tagged ubiquitin (Akimov et al., 2014), treated cells with rapamycin, or starved for amino acids with and without ConA and used Ni-NTA beads to enrich ubiquitinated proteins. Anti-TNIP1 immunoblotting validated the MS findings and characterized TNIP1 as increasingly ubiquitinated in cells in which lysosomal degradation was inhibited (Fig. 1 D and Fig. S1 A).
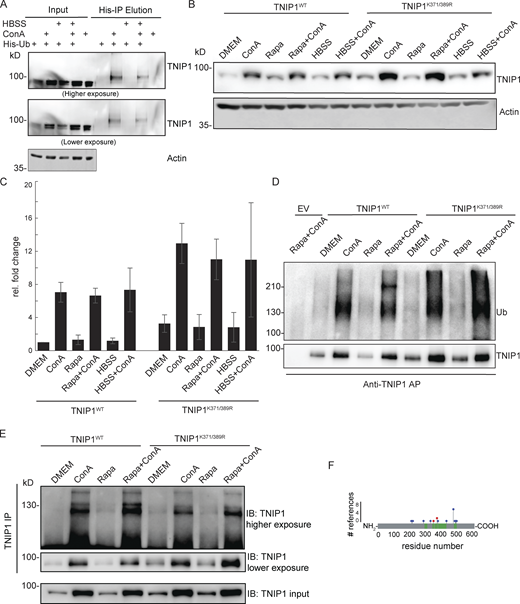
TNIP1 gets ubiquitinated and degraded in the lysosome. (A) U2-OS-StUbEx cells inducibly expressing His-FLAG-tagged ubiquitin at endogenous levels were used to enrich ubiquitinated proteins (Akimov et al., 2014). Under control conditions as well as under starvation treatment (HBSS), TNIP1 gets ubiquitinated as shown by anti-TNIP1 immunoblots. Ubiquitinated TNIP1 was stabilized by the addition of concanamycin A (ConA) indicating its lysosomal degradation in treated and nontreated cells. Actin was used as loading control. (B and C) Mutations of identified TNIP1 ubiquitination sites do not lead to reduced lysosomal degradation as indicated by stabilized protein amounts by ConA treatment. This is the case for fed control conditions (DMEM) as well as under active autophagy (Rapa and HBSS treatment). C shows quantification of blots exemplified in B (n = 3, error bars indicate SD). (D and E) Mutated TNIP1K371/389R is still getting ubiquitinated as indicated by anti-TNIP1 IP followed by anti-ubiquitin (D) and anti-TNIP1 (E) Western blot. The addition of ConA leads in all cases to a stabilization of non-ubiquitinated and polyubiquitinated protein variants. (F) Identified ubiquitination sites according to PhosphoSitePlus database and this study. Gray bar depicts the amino acid sequence of TNIP1. Sections in green mark tryptic peptides identified in this study, i.e., sequence coverage of TNIP1. Amino acids marked in blue highlight published ubiquitination sites, number of references shown on y-axis. Amino acids marked in red were identified in this study. Source data are available for this figure: SourceData FS1.
TNIP1 gets ubiquitinated and degraded in the lysosome. (A) U2-OS-StUbEx cells inducibly expressing His-FLAG-tagged ubiquitin at endogenous levels were used to enrich ubiquitinated proteins (Akimov et al., 2014). Under control conditions as well as under starvation treatment (HBSS), TNIP1 gets ubiquitinated as shown by anti-TNIP1 immunoblots. Ubiquitinated TNIP1 was stabilized by the addition of concanamycin A (ConA) indicating its lysosomal degradation in treated and nontreated cells. Actin was used as loading control. (B and C) Mutations of identified TNIP1 ubiquitination sites do not lead to reduced lysosomal degradation as indicated by stabilized protein amounts by ConA treatment. This is the case for fed control conditions (DMEM) as well as under active autophagy (Rapa and HBSS treatment). C shows quantification of blots exemplified in B (n = 3, error bars indicate SD). (D and E) Mutated TNIP1K371/389R is still getting ubiquitinated as indicated by anti-TNIP1 IP followed by anti-ubiquitin (D) and anti-TNIP1 (E) Western blot. The addition of ConA leads in all cases to a stabilization of non-ubiquitinated and polyubiquitinated protein variants. (F) Identified ubiquitination sites according to PhosphoSitePlus database and this study. Gray bar depicts the amino acid sequence of TNIP1. Sections in green mark tryptic peptides identified in this study, i.e., sequence coverage of TNIP1. Amino acids marked in blue highlight published ubiquitination sites, number of references shown on y-axis. Amino acids marked in red were identified in this study. Source data are available for this figure: SourceData FS1.
As only a fraction of TNIP1 appeared to be ubiquitinated, we investigated whether ubiquitination of Lys389 is necessary for lysosomal targeting of TNIP1. We performed site-directed mutagenesis and analyzed the stability and ubiquitination of wild-type (TNIP1WT) and respective TNIP1 variants. Next to Lys389, we also mutated the neighboring residue Lys371, which we identified in two out of the three SILAC experiments. The blockage of lysosomal degradation led to the accumulation of modified and non-modified variants of TNIP1, indicating that ubiquitination is not decisive for lysosomal degradation (Fig. S1 B). In addition, arginine-variants did neither exhibit alterations in their global ubiquitination pattern nor in their stability, indicating the presence of additional ubiquitination sites, which agrees with database entries (Fig. S1, B–F). Together, these results indicate that non-ubiquitinated and multi-ubiquitinated variants of TNIP1 accumulate upon the blockage of lysosomal acidification and that TNIP1 may be an autophagy substrate. Due to its importance in inflammation, we decided to study the regulation of TNIP1 protein abundance in more detail.
TNIP1 is degraded by autophagy
Because the blockage of lysosomal degradation by ConA led to an accumulation of non- and ubiquitinated TNIP1 variants, we examined whether TNIP1 is degraded by autophagy and whether proteasomal degradation also contributes to regulating TNIP1 protein abundance under basal conditions. TNIP1 behaved similarly as p62/SQSTM1 under basal and mTORC1 inhibited conditions using Torin-1, while the inhibition of lysosomal acidification by ConA led to a significant accumulation of TNIP1 protein, inhibition of the proteasome by MG132 did not (Fig. 2, A and B). Furthermore, confocal immunofluorescent imaging showed endogenous TNIP1 accumulation upon Bafilomycin A1 (BafA1), another V-type ATPase inhibitor, treatment (Fig. S2). Importantly, under these conditions TNIP1 colocalized with LC3-positive structures, which also strongly overlapped with p62/SQSTM1. Using airyscan super-resolution confocal microscopy, we found that TNIP1 is located inside LAMP1- and LC3-positive structures in untreated cells, and that TNIP1 is accumulated within these structures upon BafA1 treatment (Fig. 2 C). Another way to test for lysosomal degradation is using a tandem tag autophagy flux reporter system (Pankiv et al., 2007), in which TNIP1 is fused to a tandem mCherry-EYFP tag. While EYFP is sensitive to low pH and therefore quickly loses its fluorescence in acidic lysosomes, the mCherry tag is rather stable under these conditions. In neutral cytosol, both tags of mCherry-EYFP-TNIP1 will be visible, while in lysosomes, only the mCherry-tag will fluoresce and appear as red-only dots. Transient transfection of mCherry-EYFP-TNIP1 did indeed lead to the formation of many red-only dots per cell, supporting the notion that TNIP1 ends up in lysosomal structures (Fig. 2 D). Taken together, this suggests that TNIP1 is degraded by autophagy under basal conditions as well as upon Torin-1 treatment, with proteasomal protein degradation playing a negligible role.
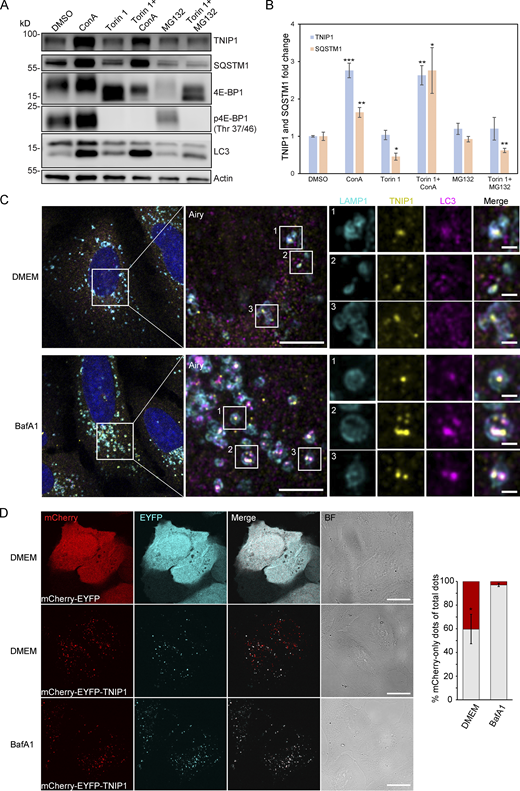
TNIP1 is degraded by autophagy. (A) U2OS cells were treated with 1 μM Torin-1 for 4 h; proteasomal or lysosomal degradation were inhibited by 10 μM MG132 or 2 nM ConA, respectively. Under fed conditions (DMSO) and in Torin-1 treated cells blockage of lysosomal acidification led to a significant increase of TNIP1 protein abundance. Shown are representative blots of three biological replicates. (B) Quantification of blots shown in A (n = 3). * = P < 0.05, ** = P < 0.01, *** = P < 0.001 unpaired, two-sided t test compared to DMSO treated samples. Error bars indicate SEM. (C) Confocal images showing colocalization between endogenous TNIP1, LAMP1 and LC3 in U2OS cells treated for 5 h with either BafA1 or vehicle DMSO. Cells were immunostained against endogenous TNIP1 (yellow), LAMP1 (cyan), and LC3 (purple) and imaged by Airyscan using the Zeiss LSM880 confocal microscope. Inserts highlight TNIP1 localized in LAMP1- and LC3-positive structures. Due to BafA1 treatment leading to accumulation of the immunostained proteins, signal intesities in the DMSO image have been increased relative to the BafA1 treated image during post-processing. Scale bars are 5 µm for the airyscan images and 1 µm for the inserts. (D) U2OS cells were transiently transfected with either mCherry-EYFP or mCherry-EYFP-TNIP1. 24 h after transfection, cells were either left untreated or treated with BafA1 for 4 h. BF = bright field. Scale bars, 20 µm. Quantification of red-only TNIP1 dots over total TNIP1 dots was done using Volocity software (PerkinElmer), with intensity cut-offs based on BafA1 intensity of red and green dots (n = 3). Around 2,000–3,000 dots were counted for each condition within each replicate. * = P < 0.05, unpaired, two-sided t test. Error bars indicates SD. In B and D, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F2.
TNIP1 is degraded by autophagy. (A) U2OS cells were treated with 1 μM Torin-1 for 4 h; proteasomal or lysosomal degradation were inhibited by 10 μM MG132 or 2 nM ConA, respectively. Under fed conditions (DMSO) and in Torin-1 treated cells blockage of lysosomal acidification led to a significant increase of TNIP1 protein abundance. Shown are representative blots of three biological replicates. (B) Quantification of blots shown in A (n = 3). * = P < 0.05, ** = P < 0.01, *** = P < 0.001 unpaired, two-sided t test compared to DMSO treated samples. Error bars indicate SEM. (C) Confocal images showing colocalization between endogenous TNIP1, LAMP1 and LC3 in U2OS cells treated for 5 h with either BafA1 or vehicle DMSO. Cells were immunostained against endogenous TNIP1 (yellow), LAMP1 (cyan), and LC3 (purple) and imaged by Airyscan using the Zeiss LSM880 confocal microscope. Inserts highlight TNIP1 localized in LAMP1- and LC3-positive structures. Due to BafA1 treatment leading to accumulation of the immunostained proteins, signal intesities in the DMSO image have been increased relative to the BafA1 treated image during post-processing. Scale bars are 5 µm for the airyscan images and 1 µm for the inserts. (D) U2OS cells were transiently transfected with either mCherry-EYFP or mCherry-EYFP-TNIP1. 24 h after transfection, cells were either left untreated or treated with BafA1 for 4 h. BF = bright field. Scale bars, 20 µm. Quantification of red-only TNIP1 dots over total TNIP1 dots was done using Volocity software (PerkinElmer), with intensity cut-offs based on BafA1 intensity of red and green dots (n = 3). Around 2,000–3,000 dots were counted for each condition within each replicate. * = P < 0.05, unpaired, two-sided t test. Error bars indicates SD. In B and D, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F2.

TNIP1 localizes to autophagosomes. U2OS cells kept in either fed or starved (HBSS) conditions and treated with either vehicle (DMSO) or BafA1 for 8 h. Cells were fixed and stained with antibodies against endogenous TNIP1 (green), p62 (orange), and LC3 (purple) and imaged using the Zeiss LSM800 confocal microscope. Line-profile co-localization plots were made using the line-profile quantification tool in the Zen blue imaging software (Zeiss). Vertical axis represents measurements of fluorescent intensity and the horizontal axis the drawn distances. Scale bar = 10 µm.
TNIP1 localizes to autophagosomes. U2OS cells kept in either fed or starved (HBSS) conditions and treated with either vehicle (DMSO) or BafA1 for 8 h. Cells were fixed and stained with antibodies against endogenous TNIP1 (green), p62 (orange), and LC3 (purple) and imaged using the Zeiss LSM800 confocal microscope. Line-profile co-localization plots were made using the line-profile quantification tool in the Zen blue imaging software (Zeiss). Vertical axis represents measurements of fluorescent intensity and the horizontal axis the drawn distances. Scale bar = 10 µm.
Next, we aimed to characterize the molecular events that led to autophagosomal recruitment of TNIP1. We immunoprecipitated HA-tagged TNIP1 and performed an MS analysis of its interactome. This screen identified a number of autophagy-related proteins as possible TNIP1 interactors, including p62/SQSTM1, TAX1BP1, OPTN, and TBK1 (Fig. 3, A and B and Table S2). The former two, p62/SQSTM1 and TAX1BP1, were also identified by MS as enriched in IPs of endogenous TNIP1 under basal conditions (Fig. S3 A). The interaction with p62/SQSTM1 was also observed by immunoprecipitation followed by Western blotting (Fig. 3 C). In ATG101 KO, FIP200 KO, and pentaKO HeLa cells (which do not express the SLRs p62/SQSTM1, NBR1, NDP52, TAX1BP1 and OPTN [Sarraf et al., 2020]) TNIP1 no longer showed stabilization upon lysosomal blockage (Fig. 3, D and E). This was also true for single KOs of p62 and OPTN (but not TAX1BP1), which also showed a reduction in TNIP1 abundance upon lysosomal blockage (Fig. S3, B and C). Taken together, this suggests that more than one SLR is mediating the basal autophagic turnover of TNIP1. The staining of endogenous TNIP1 in U2OS cells showed an increase in the number of TNIP1 puncta (Fig. 3, F and G) and increased colocalization between TNIP1 and the SLRs p62/SQSTM1, TAX1BP1, and NDP52 upon BafA1 treatment (Fig. 3 H). Thus, under basal conditions, TNIP1 interacts with SLRs, which target TNIP1 to autophagosomes for lysosomal degradation. To further investigate the basal turnover, we analyzed the autophagic flux using mCherry-EYFP-TNIP1 in U2OS cells KO for FIP200, ATG9, ATG7, and ATG16L1. As quantified in Fig. 3 I, FIP200 and ATG9 KO cells showed little to no red-only dots. This is consistent with the importance of these proteins in both ATG7-dependent and -independent autophagy (Goodwin et al., 2017). ATG7 and ATG16L1 KOs showed reduced formation of red-only dots, but not a complete loss, indicating that there may also be ATG7-independent degradation of TNIP1 under basal conditions. Altogether, these results suggest that autophagic degradation of TNIP1 under basal conditions is aided by SLRs, and that this turnover may be both ATG7-dependent and -independent.
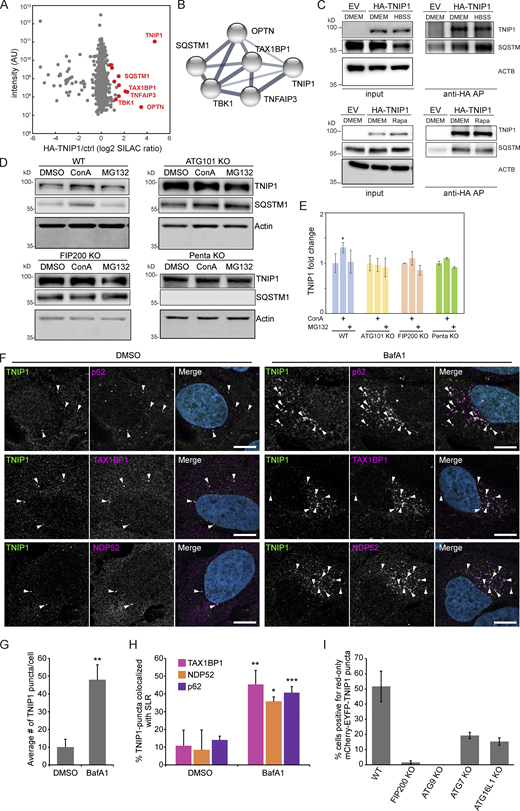
TNIP1 localizes to p62 bodies. (A) HA-TNIP1 affinity purification (AP)-MS highlights its interaction with autophagy receptors. HeLa cells expressing HA-TNIP1 and vector control cells (ctrl.) were differentially SILAC-labeled and anti-HA APs were performed under basal conditions, followed by quantitative MS analyses (n = 3). Proteins that were significantly enriched in minimum two out of three replicates are highlighted in red (P < 0.05, BH corrected). Proteins with known functions in autophagy and inflammation are annotated. (B) TNIP1 interactome. STRING DB was used to highlight the TNIP1 interactome identified in A (Szklarczyk et al., 2019). Thickness of edges indicate confidence of interaction. (C) TNIP1 interacts with p62/SQSTM1. Anti-HA affinity purifications followed by Western blot analyses were performed to test for HA-TNIP1-p62/SQSTM1 interactions as identified in A in basal (DMEM), stress conditions (amino acid starvation, HBSS) and after rapamycin (Rapa) treatment each for 4 h. EV, empty vector. (D and E) TNIP1 is degraded in an autophagy- and SLR-dependent manner. Shown are representative blots of three biological replicates. HeLa WT cells, ATG101 KO cells, FIP200 KO cells and PentaKO cells were treated with 10 μM MG132 or 2 nM ConA for 4 h. TNIP1 abundance was significantly increased in ConA-treated HeLa WT cells, while treatment had no effect in HeLa ATG101 KO, FIP200 KO, and pentaKO cells. E shows quantifications of D (n = 3). * = P < 0.05, unpaired, two-sided t test. Error bars indicates SEM. (F) U2OS cells were treated with either vehicle (DMSO) or BafA1 for 5 h and stained for endogenous TNIP1 (green) together with either endogenous p62, NDP52 or TAX1BP1. Representative images are shown. Colocalization between TNIP1 and respective SLRs are indicated by arrowheads. Scale bars, 10 µm. (G) Quantification of the average number of TNIP1 puncta per cell imaged in F (>40 cells analyzed for each condition within each replicate [n = 3]). ** = P < 0.01, unpaired two-sided t test. Error bars indicate SD. (H) Quantification of percent TNIP1 puncta colocalizing with the indicated SLRs in F (>40 cells analyzed for each condition within each replicate [n = 3]). *** = P < 0.001, ** = P < 0.01, * = P < 0.05, unpaired, two-sided t test. Error bars indicate SD. (I) Transient transfection of mCherry-EGFP-TNIP1 in WT U2OS cells and indicated KO cell lines. The graph bars indicate the percentage of transfected cells containing >5 red-only puncta indicative of autophagic degradation. Each graph bar shows the mean value from three separate transfections (n = 3, >100 cells counted per transfection, unpaired, two-sided t test). Error bars indicate SD. In E and G–I, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F3.
TNIP1 localizes to p62 bodies. (A) HA-TNIP1 affinity purification (AP)-MS highlights its interaction with autophagy receptors. HeLa cells expressing HA-TNIP1 and vector control cells (ctrl.) were differentially SILAC-labeled and anti-HA APs were performed under basal conditions, followed by quantitative MS analyses (n = 3). Proteins that were significantly enriched in minimum two out of three replicates are highlighted in red (P < 0.05, BH corrected). Proteins with known functions in autophagy and inflammation are annotated. (B) TNIP1 interactome. STRING DB was used to highlight the TNIP1 interactome identified in A (Szklarczyk et al., 2019). Thickness of edges indicate confidence of interaction. (C) TNIP1 interacts with p62/SQSTM1. Anti-HA affinity purifications followed by Western blot analyses were performed to test for HA-TNIP1-p62/SQSTM1 interactions as identified in A in basal (DMEM), stress conditions (amino acid starvation, HBSS) and after rapamycin (Rapa) treatment each for 4 h. EV, empty vector. (D and E) TNIP1 is degraded in an autophagy- and SLR-dependent manner. Shown are representative blots of three biological replicates. HeLa WT cells, ATG101 KO cells, FIP200 KO cells and PentaKO cells were treated with 10 μM MG132 or 2 nM ConA for 4 h. TNIP1 abundance was significantly increased in ConA-treated HeLa WT cells, while treatment had no effect in HeLa ATG101 KO, FIP200 KO, and pentaKO cells. E shows quantifications of D (n = 3). * = P < 0.05, unpaired, two-sided t test. Error bars indicates SEM. (F) U2OS cells were treated with either vehicle (DMSO) or BafA1 for 5 h and stained for endogenous TNIP1 (green) together with either endogenous p62, NDP52 or TAX1BP1. Representative images are shown. Colocalization between TNIP1 and respective SLRs are indicated by arrowheads. Scale bars, 10 µm. (G) Quantification of the average number of TNIP1 puncta per cell imaged in F (>40 cells analyzed for each condition within each replicate [n = 3]). ** = P < 0.01, unpaired two-sided t test. Error bars indicate SD. (H) Quantification of percent TNIP1 puncta colocalizing with the indicated SLRs in F (>40 cells analyzed for each condition within each replicate [n = 3]). *** = P < 0.001, ** = P < 0.01, * = P < 0.05, unpaired, two-sided t test. Error bars indicate SD. (I) Transient transfection of mCherry-EGFP-TNIP1 in WT U2OS cells and indicated KO cell lines. The graph bars indicate the percentage of transfected cells containing >5 red-only puncta indicative of autophagic degradation. Each graph bar shows the mean value from three separate transfections (n = 3, >100 cells counted per transfection, unpaired, two-sided t test). Error bars indicate SD. In E and G–I, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F3.
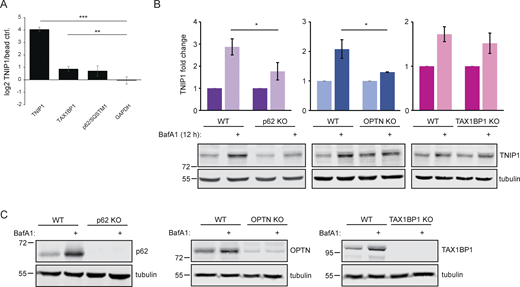
Endogenous TNIP1 interacts with TAX1BP1 and p62/SQSTM1 under basal conditions. (A) SILAC-based, IP-MS analyses of anti-TNIP1 immunoprecipitations identified TAX1BP1 and p62/SQSTM1 as enriched compared to negative control IPs using beads only. GAPDH is shown as negative control. Shown are average values of three biological replicates (n = 3). Error bars: SD, ** = P < 0.01, *** = P < 0.001, unpaired, two-sided t test. (B and C) Lysosomal turnover of TNIP1 is mediated by several SLRs. HeLa WT and HeLa p62, OPTN, and TAX1BP1 KO cells were either left untreated or treated with 200 nM BafA1 for 12 h. Shown are average values of three biological replicates. Error bars: SD, * = P < 0.05, unpaired, two-sided t test. Source data are available for this figure: SourceData FS3.
Endogenous TNIP1 interacts with TAX1BP1 and p62/SQSTM1 under basal conditions. (A) SILAC-based, IP-MS analyses of anti-TNIP1 immunoprecipitations identified TAX1BP1 and p62/SQSTM1 as enriched compared to negative control IPs using beads only. GAPDH is shown as negative control. Shown are average values of three biological replicates (n = 3). Error bars: SD, ** = P < 0.01, *** = P < 0.001, unpaired, two-sided t test. (B and C) Lysosomal turnover of TNIP1 is mediated by several SLRs. HeLa WT and HeLa p62, OPTN, and TAX1BP1 KO cells were either left untreated or treated with 200 nM BafA1 for 12 h. Shown are average values of three biological replicates. Error bars: SD, * = P < 0.05, unpaired, two-sided t test. Source data are available for this figure: SourceData FS3.
TNIP1 interacts with human ATG8 family proteins through LIR motifs
Having established that TNIP1 colocalizes with several SLRs and is degraded by autophagy, we aimed to examine whether TNIP1 may also function independently of SLRs in autophagy. TNIP1 contains coiled-coil domains for oligomerization and a ubiquitin-binding UBAN domain similar to that of OPTN, which preferentially binds K63- and M1-linked polyubiquitin chains (Fig. 4 A; Herhaus et al., 2019; Wagner et al., 2008). TNIP1 only needs an LIR motif interacting with ATG8 proteins to be able to function as a SAR itself. Hence, we performed a peptide array screen for potential LIR motifs in human TNIP1. The peptide array, containing overlapping 20-mer peptides of TNIP1 moved by increments of three amino acids to cover the 636 amino acids full-length sequence, was probed with GST-GABARAP and revealed two potential LIRs in TNIP1, with core sequences 83-FDPL-86 and 125-FEVV-128, respectively (Fig. 4 B). The N-terminal region harboring these LIR motifs is missing in the two other TNIP family members, TNIP2 and -3. Of these two candidates, only LIR2 is conserved in the evolution of vertebrates down to cartilagous fishes, while LIR1 is only conserved down to marsupials and is not found in platypus (Fig. 4 C). LIR2 also has acidic- and phosphorylatable residues flanking the core LIR motif, making it a very strong candidate for a functional LIR that could be positively regulated by phosphorylation (Johansen and Lamark, 2020; Wirth et al., 2019). LIR1 has a proline within the core motif which is usually inhibitory to binding to the ATG8s (Alemu et al., 2012; Johansen and Lamark, 2020). We then further tested the interaction between TNIP1 and the six human ATG8 family proteins by GST-pulldown assays. GST and GST-tagged human LC3 and GABARAP proteins were used to pull down in vitro translated wild-type TNIP1. Here, we observed that TNIP1 bound very well to several of the human ATG8s, with the strongest interaction being with LC3A, LC3B, GABARAP, and GABARAPL1, and weak binding to LC3C and GABARAPL2 (Fig. 4, D and E). To test whether any of the two potential LIRs identified in the peptide array scan were responsible for this interaction, we mutated the conserved aromatic- and hydrophobic residues in each core LIR sequence to alanine, namely, F83A/L86A and F125A/V128A (Fig. 4 C). GST-pulldown assays with these mutants revealed that the F125A/V128A mutations in LIR2 strongly reduced the interaction between TNIP1 and the GST-ATG8s, while the F83A/L86A mutations in LIR1 had either no or a very minor effect compared to mutating LIR2 alone (Fig. 4, D and E). We also tested the influence of LIRs for in vivo interactions of TNIP1 with LC3s using HeLa cells expressing HA-TNIP1 variants by performing anti-HA affinity purifications (APs) followed by anti-LC3A/B Western blots (Fig. 4, F and G). In agreement with the in vitro observations, TNIP1 with both LIR motifs mutated (mLIR1+2) interacted significantly weaker with LC3A/B in vivo. Thus, TNIP1 fulfills all structural characteristics of being an SLR, with LIR2 being the motif mainly responsible for binding to ATG8s.
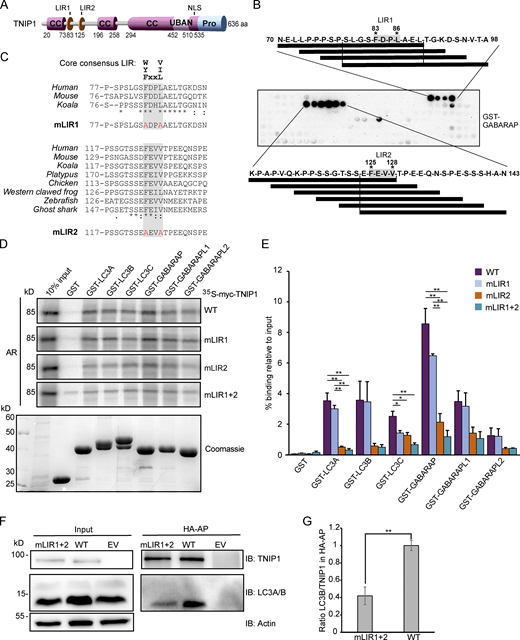
TNIP1 interacts with human ATG8 family proteins through a LIR motif. (A) Schematic drawing of the domain architecture of TNIP1, showing possible LIRs. (B) Peptide array of 20-mer peptides covering full length TNIP1 was used to probe for possible LIRs, using GST-GABARAP. (C) Amino-acid sequence alignment showing conservation of the core consenesus LIRs in TNIP1 across species. TNIP1 amino acid sequences were collected from UniProt, and multiple sequence alignment performed with Clustal Omega. Asterisk (*) indicates fully conserved residues; colon (:) indicates conservation between groups of strongly similar properties (>0.5 Gonnet PAM 250 matrix); and a period (.) indicates conservation between groups of weakly similar properties (0–0.5 Gonnet PAM 250 matrix). Mutated residues for the LIR mutants (mLIR1 and mLIR2) are shown in red. (D) In vitro GST-pulldown assay using 35S-labeled myc-TNIP1, myc-TNIP1-F83A/L86A (mLIR1), myc-TNIP1-F125A/V128A (mLIR2) and myc-TNIP1-F83A/L86A/F125A/V128A (mLIR1+2) against recombinant GST and GST-tagged human ATG8s. Bound myc-TNIP1 WT and LIR mutants were detected by autoradiography (AR). (E) Quantification of GST-pulldown from D. Relative % binding was quantified against 10% input (n = 3). * = P < 0.05, ** = P < 0.01, based on one-way ANOVA (post hoc Tukey test). Error bars indicate SEM. (F) LIR mutation impacts in vivo interaction with LC3A/B. Anti-HA AP of cells expressing HA-TNIP1WT and HA-TNIP1mLIR1+2 were performed followed by Western blot against indicated proteins. LIR mutation reduced the interaction between TNIP1 and LC3A/B. (G) Quantification of blots exemplified in panel F (n = 3). Error bars indicate SEM. ** = P < 0.01, unpaired, two-sided t test. In E and G, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F4.
TNIP1 interacts with human ATG8 family proteins through a LIR motif. (A) Schematic drawing of the domain architecture of TNIP1, showing possible LIRs. (B) Peptide array of 20-mer peptides covering full length TNIP1 was used to probe for possible LIRs, using GST-GABARAP. (C) Amino-acid sequence alignment showing conservation of the core consenesus LIRs in TNIP1 across species. TNIP1 amino acid sequences were collected from UniProt, and multiple sequence alignment performed with Clustal Omega. Asterisk (*) indicates fully conserved residues; colon (:) indicates conservation between groups of strongly similar properties (>0.5 Gonnet PAM 250 matrix); and a period (.) indicates conservation between groups of weakly similar properties (0–0.5 Gonnet PAM 250 matrix). Mutated residues for the LIR mutants (mLIR1 and mLIR2) are shown in red. (D) In vitro GST-pulldown assay using 35S-labeled myc-TNIP1, myc-TNIP1-F83A/L86A (mLIR1), myc-TNIP1-F125A/V128A (mLIR2) and myc-TNIP1-F83A/L86A/F125A/V128A (mLIR1+2) against recombinant GST and GST-tagged human ATG8s. Bound myc-TNIP1 WT and LIR mutants were detected by autoradiography (AR). (E) Quantification of GST-pulldown from D. Relative % binding was quantified against 10% input (n = 3). * = P < 0.05, ** = P < 0.01, based on one-way ANOVA (post hoc Tukey test). Error bars indicate SEM. (F) LIR mutation impacts in vivo interaction with LC3A/B. Anti-HA AP of cells expressing HA-TNIP1WT and HA-TNIP1mLIR1+2 were performed followed by Western blot against indicated proteins. LIR mutation reduced the interaction between TNIP1 and LC3A/B. (G) Quantification of blots exemplified in panel F (n = 3). Error bars indicate SEM. ** = P < 0.01, unpaired, two-sided t test. In E and G, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F4.
TNIP1 does not affect basal autophagy flux
We next asked whether TNIP1 was capable of regulating autophagy flux. For this, we compared the levels of several known SLRs as well as the ratio of LC3-I and II under basal and starvation conditions in WT HeLa and two TNIP1 KO clones. The ratio of LC3-I and LC3-II was not affected by TNIP1 KO, and neither was the conversion upon starvation by HBSS, suggesting that TNIP1 does not affect autophagic flux under the tested conditions (Fig. 5, A and B). With the exception of OPTN, the basal protein levels of several SLRs and their degradation upon starvation were also unaffected by TNIP1 KO. However, the basal protein levels of OPTN were elevated in both TNIP1 KO clones, indicating that OPTN might be involved in a compensatory response. Indeed, we identified an upregulation of OPTN mRNA in TNIP1 KO clones (Table S3).
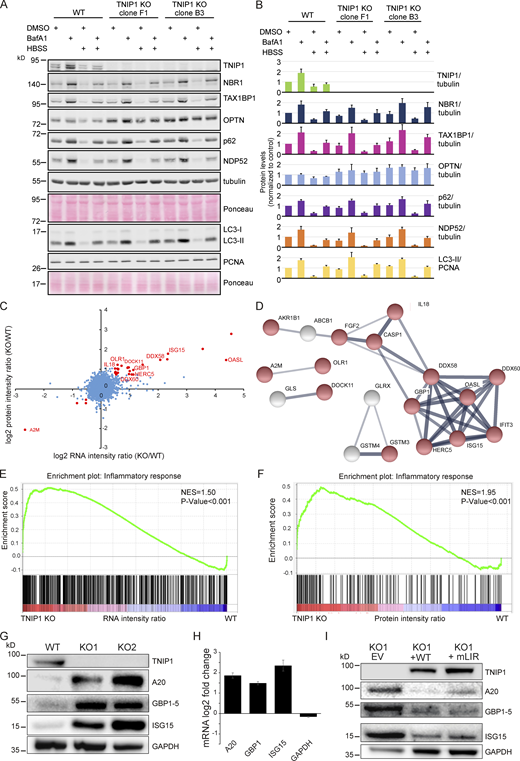
Loss of TNIP1 leads to an increase in inflammatory proteins. (A) Loss of TNIP1 does not alter autophagy flux under basal and starvation conditions. TNIP1 knock out HeLa cells generated by CRISPR/Cas9 were used to study its effect on autophagy. Wild-type HeLa cells and the two TNIP1 knockout clones denoted KO1 and KO2 were kept in either fed or starved (HBSS) conditions and treated with either vehicle (DMSO) or BafA1 for 8 h. Blots were probed for several known SLRs as well as LC3. (B) Quantification of blots shown in A (n = 3). Error bars indicate SD. (C) Loss of TNIP1 leads to an increased transcription of inflammatory genes. Fold changes of RNA and protein intensities of TNIP1 KO and WT cells were compared. Shown are average values of two KO clones compared to WT cells (n = 3 per cell type for RNAseq; n = 5 per cell type for proteomics). Genes that were significantly regulated on RNA and protein level are highlighted in red (P < 0.01). Genes linked to immune effector processes and interferon-stimulated genes are annotated. (D) Protein-protein interactions of significantly regulated proteins. Proteins highlighted in red in C were analyzed on known interactions using STRING DB (Szklarczyk et al., 2019). Interactions between 17 proteins were identified, of which 13 are linked to stress response (marked in red). Thickness of edges indicate confidence of interaction. (E and F) Gene set enrichment analysis of significantly dysregulated mRNAs and proteins identifies an increased transcription and translation of genes involved in inflammation. NES denotes normalized enrichment score. (G–I) TNIP1 represses translation of pro-inflammatory gene products. Whereas knockout of TNIP1 led to an increased abundance of indicated inflammatory proteins (G), which is likely due to transcriptional changes (H, n = 3, error bars indicate SEM), re-expression of TNIP1WT or TNIP1mLIR1+2 blunted this phenotype (I). Source data are available for this figure: SourceData F5.
Loss of TNIP1 leads to an increase in inflammatory proteins. (A) Loss of TNIP1 does not alter autophagy flux under basal and starvation conditions. TNIP1 knock out HeLa cells generated by CRISPR/Cas9 were used to study its effect on autophagy. Wild-type HeLa cells and the two TNIP1 knockout clones denoted KO1 and KO2 were kept in either fed or starved (HBSS) conditions and treated with either vehicle (DMSO) or BafA1 for 8 h. Blots were probed for several known SLRs as well as LC3. (B) Quantification of blots shown in A (n = 3). Error bars indicate SD. (C) Loss of TNIP1 leads to an increased transcription of inflammatory genes. Fold changes of RNA and protein intensities of TNIP1 KO and WT cells were compared. Shown are average values of two KO clones compared to WT cells (n = 3 per cell type for RNAseq; n = 5 per cell type for proteomics). Genes that were significantly regulated on RNA and protein level are highlighted in red (P < 0.01). Genes linked to immune effector processes and interferon-stimulated genes are annotated. (D) Protein-protein interactions of significantly regulated proteins. Proteins highlighted in red in C were analyzed on known interactions using STRING DB (Szklarczyk et al., 2019). Interactions between 17 proteins were identified, of which 13 are linked to stress response (marked in red). Thickness of edges indicate confidence of interaction. (E and F) Gene set enrichment analysis of significantly dysregulated mRNAs and proteins identifies an increased transcription and translation of genes involved in inflammation. NES denotes normalized enrichment score. (G–I) TNIP1 represses translation of pro-inflammatory gene products. Whereas knockout of TNIP1 led to an increased abundance of indicated inflammatory proteins (G), which is likely due to transcriptional changes (H, n = 3, error bars indicate SEM), re-expression of TNIP1WT or TNIP1mLIR1+2 blunted this phenotype (I). Source data are available for this figure: SourceData F5.
As SLRs are themselves autophagy substrates, and as loss of TNIP1 was shown to lead to increased inflammatory signaling (Shamilov and Aneskievich, 2018), we next asked if loss of TNIP1 in our cell systems also led to an increase of pro-inflammatory proteins. Comparing gene expression by RNAseq and protein abundance by SILAC-based proteomics between WT and TNIP1 KO HeLa cells, we indeed observed an upregulation of a number of genes involved in inflammatory signaling in TNIP1 KO cells on mRNA and protein level, including TNFAIP3, ISG15, and GBP1 (Fig. 5, C and D; and Table S3). This was confirmed by gene set enrichment analyses (GSEA), which highlighted an activation of the inflammatory response in TNIP1 KO cells at both mRNA and protein level (Fig. 5, E and F). At the mRNA level, we identified several chemokines which were more abundant in TNIP1 KO cells, CCL5 being the most differentially regulated gene (Table S3 B). The upregulation of single proteins was also observed by Western blot analysis and was at least in part due to changes at the transcriptional level (Fig. 5, G and H). Importantly, re-expression of TNIP1 blunted this effect confirming that it was indeed the absence of TNIP1 that led to the observed changes of the respective proteins. The LIR motifs did not affect this response under basal conditions as expression of TNIP1-mLIR1+2 led to the same consequences as TNIP1-WT (Fig. 5 I). Taken together, whereas TNIP1 seemed not to affect autophagy flux under basal conditions, its loss led to an increased abundance of pro-inflammatory proteins, indicating that selective autophagy may contribute to the tuning of inflammatory signaling by regulating TNIP1 protein levels. Thus, in this context, TNIP1 is a bona fide autophagy substrate whose protein level is critical for the regulation of inflammatory signaling.
Pro-inflammatory signaling induces LIR-dependent, autophagic degradation of TNIP1
So far, we characterized TNIP1 as a constitutive autophagic cargo as we did not observe changes in autophagosomal recruitment and lysosomal degradation based on the metabolic status of cells. To test whether inflammatory signaling could lead to specific effects, we treated cells with the double-stranded RNA mimic poly(I:C), which mimics viral infection and elicits a TLR3 signaling response (Glavan and Pavelic, 2014). After 4 h of poly(I:C) treatment, we observed a significant decrease in endogenous TNIP1 levels, followed by an increase after 6 h (Fig. 6, A and B). Parallel to the observed decrease of TNIP1, an increase in pro-inflammatory proteins like ISG15 and the chemokine CCL5 was observed (Fig. 6 A and Fig. S4 A). The decrease of TNIP1 was dependent on canonical autophagy, as loss of ATG7, ATG101, and FIP200 inhibited the response, but independent of SLRs, as loss of the SLRs p62/SQSTM1, NBR1, NDP52, TAX1BP1, and OPTN in a pentaKO cell line did not inhibit the response (Fig. 6, A and B). In agreement, lysosomal inhibition by ConA could block this decrease in WT and pentaKO cells but had no effect on the autophagy incompetent cell lines (Fig. S4 B). Proteasomal inhibition by MG132 did not stabilize TNIP1 levels in any of the cell lines (Fig. S4 B). The observed increase at 6 h of treatment is due to transcriptional upregulation of TNIP1 (Fig. 6 C). Hence, TNIP1 is itself specifically targeted to autophagosomes under poly(I:C) treatment. Confocal imaging showed that upon poly(I:C) treatment in WT cells, endogenous TNIP1 changes from being mostly diffuse to forming dots in the cytoplasm (Fig. 6, D and E). The formation of TNIP1 dots in response to poly(I:C) was also observed in ATG7- and pentaKO cells (Fig. 6, D and E). In ATG7 KO cells, there was an increased amount of TNIP1 dots already at the basal level, which increased even further upon poly(I:C) treatment. In pentaKO cells, we observed an increased diffuse TNIP1 staining of cells at the basal level likely reflecting an increase in basal protein levels. Upon 4 h stimulation with poly(I:C) TNIP1 puncta increased also in pentaKO cells similarly as in ATG7 KO cells. Taken together, our data suggest that neither ATG7 nor SLRs are required for poly(I:C)-induced TNIP1 aggregation into dots. However, ATG7, but not SLRs, is necessary for TNIP1 degradation upon poly(I:C) stimulation.
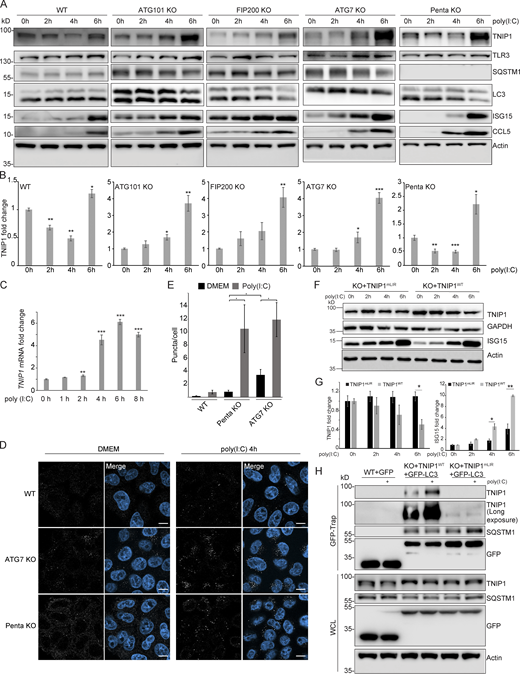
Poly(I:C) stimulation induces LIR-dependent, specific degradation of TNIP1 by autophagy. (A) Poly(I:C) treatment leads to time-dependent changes in TNIP1 abundance. Poly(I:C) stimulation leads to an autophagy dependent and SLR independent decrease of TNIP1 abundance within the first 4 h as indicated by a block of degradation in ATG101, FIP200 and ATG7 KO cells. Autophagy receptors appear to have a minor influence as degradation still occurs in pentaKO cells. (B) Quantification of blots shown in A (n = 3). Error bars indicate SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001 unpaired, two-sided t test compared to 0 h values of respective cell lines. (C) After 2–4 h of poly(I:C) treatment TNIP1 transcription is significantly upregulated. Bar diagrams show quantification of three biological replicates (n = 3), error bars: SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired, two-sided t test. (D) Representative immunofluorescent images showing endogenous TNIP1 response to poly(I:C) in WT, ATG7 KO, and pentaKO. Cells were either left untreated or treated with 5 µg/ml poly(I:C) for 4 h. Scale bar = 10 µm. (E) Quantification of images shown in D, error bars indicate SEM. * = P < 0.05, unpaired two-sided t test. (F and G) The degradation of TNIP1 depends on functional LIR motifs. TNIP1 WT is degraded in a time-dependent fashion after poly(I:C) stimulation. The double LIR mutant TNIP1 (LIR1+2, TNIP1mLIR) is spared from degradation. Note: Protein amounts of TNIP1 and ISG15 correlate inversely. Due to ectopic expression of TNIP1 variants regulation based on transcriptional/translational control as shown in A is lost. E shows quantification of blots exemplified in D (n = 3). Error bars indicate SEM. * = P < 0.05, unpaired, two-sided t test. KO1 cells were used for reconstitution. (H) Poly(I:C) induces a LIR-dependent interaction with LC3. Indicated HeLa cells expressing GFP-LC3 were used for anti-GFP AP. Cells expressing TNIP1mLIR do not exhibit an increased interaction between TNIP1 and GFP-LC3 after poly(I:C) treatment, in contrast to cells expressing TNIP1WT. KO1 cells were used for reconstitution. In B, C, E, and G, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F6.
Poly(I:C) stimulation induces LIR-dependent, specific degradation of TNIP1 by autophagy. (A) Poly(I:C) treatment leads to time-dependent changes in TNIP1 abundance. Poly(I:C) stimulation leads to an autophagy dependent and SLR independent decrease of TNIP1 abundance within the first 4 h as indicated by a block of degradation in ATG101, FIP200 and ATG7 KO cells. Autophagy receptors appear to have a minor influence as degradation still occurs in pentaKO cells. (B) Quantification of blots shown in A (n = 3). Error bars indicate SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001 unpaired, two-sided t test compared to 0 h values of respective cell lines. (C) After 2–4 h of poly(I:C) treatment TNIP1 transcription is significantly upregulated. Bar diagrams show quantification of three biological replicates (n = 3), error bars: SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired, two-sided t test. (D) Representative immunofluorescent images showing endogenous TNIP1 response to poly(I:C) in WT, ATG7 KO, and pentaKO. Cells were either left untreated or treated with 5 µg/ml poly(I:C) for 4 h. Scale bar = 10 µm. (E) Quantification of images shown in D, error bars indicate SEM. * = P < 0.05, unpaired two-sided t test. (F and G) The degradation of TNIP1 depends on functional LIR motifs. TNIP1 WT is degraded in a time-dependent fashion after poly(I:C) stimulation. The double LIR mutant TNIP1 (LIR1+2, TNIP1mLIR) is spared from degradation. Note: Protein amounts of TNIP1 and ISG15 correlate inversely. Due to ectopic expression of TNIP1 variants regulation based on transcriptional/translational control as shown in A is lost. E shows quantification of blots exemplified in D (n = 3). Error bars indicate SEM. * = P < 0.05, unpaired, two-sided t test. KO1 cells were used for reconstitution. (H) Poly(I:C) induces a LIR-dependent interaction with LC3. Indicated HeLa cells expressing GFP-LC3 were used for anti-GFP AP. Cells expressing TNIP1mLIR do not exhibit an increased interaction between TNIP1 and GFP-LC3 after poly(I:C) treatment, in contrast to cells expressing TNIP1WT. KO1 cells were used for reconstitution. In B, C, E, and G, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F6.
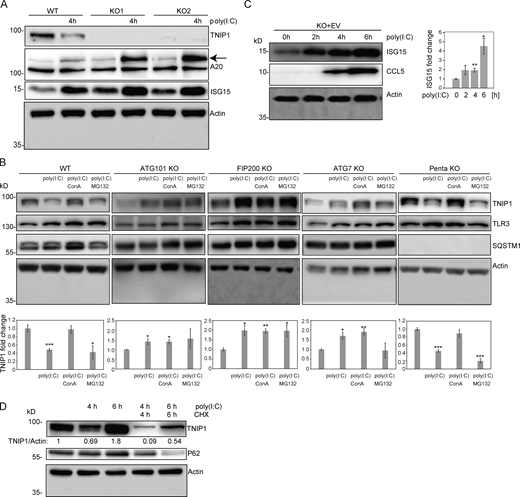
Regulation of TNIP1 protein abundance. (A) Reduction of TNIP1 correlates with an increase of ISG15 and TNFAIP3/A20 in HeLa cells. The increase of the TNIP1 interaction partner TNFAIP3 under poly(I:C) treatment indicates the existence of distinct TNIP1 pools, i.e., free and bound to TNFAIP3. Arrow marks A20 band. (B) Poly(I:C) treatment leads to an autophagy-dependent and SLR-independent lysosomal degradation of TNIP1. Whereas lysosomal inhibition by ConA treatment leads to a significant block of TNIP1 degradation in WT and pentaKO cells, proteasomal inhibition by MG132 treatment has no effect. In ATG101 KO, FIP200 KO and ATG7 KO, autophagy incompetent cell lines poly(I:C) does not lead to TNIP1 degradation. TLR3 and SQSTM1 are monitored as positive controls, actin as loading control. Shown are representative blots of three biological replicates each. Bar diagram shows quantification, error bars: SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, unpaired, two-sided t test. (C) TNIP1 KO cells transfected with an empty control vector (EV) do also respond to poly(I:C) treatment by an upregulation of ISG15 and CCL5 similar to KO cells transfected with a TNIP1 expression construct. This indicates that ISG15 and CCL5 abundances are not only regulated by TNIP1. Shown are representative blots of three biological replicates. Bar diagram shows quantification. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired, two-sided t test. (D) Blockage of protein translation by cycloheximide (CHX) treatment reduced the time- and poly(I:C)-dependent increase of TNIP1 after 6 h of treatment indicating a regulation on translational level. Source data are available for this figure: SourceData FS4.
Regulation of TNIP1 protein abundance. (A) Reduction of TNIP1 correlates with an increase of ISG15 and TNFAIP3/A20 in HeLa cells. The increase of the TNIP1 interaction partner TNFAIP3 under poly(I:C) treatment indicates the existence of distinct TNIP1 pools, i.e., free and bound to TNFAIP3. Arrow marks A20 band. (B) Poly(I:C) treatment leads to an autophagy-dependent and SLR-independent lysosomal degradation of TNIP1. Whereas lysosomal inhibition by ConA treatment leads to a significant block of TNIP1 degradation in WT and pentaKO cells, proteasomal inhibition by MG132 treatment has no effect. In ATG101 KO, FIP200 KO and ATG7 KO, autophagy incompetent cell lines poly(I:C) does not lead to TNIP1 degradation. TLR3 and SQSTM1 are monitored as positive controls, actin as loading control. Shown are representative blots of three biological replicates each. Bar diagram shows quantification, error bars: SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, unpaired, two-sided t test. (C) TNIP1 KO cells transfected with an empty control vector (EV) do also respond to poly(I:C) treatment by an upregulation of ISG15 and CCL5 similar to KO cells transfected with a TNIP1 expression construct. This indicates that ISG15 and CCL5 abundances are not only regulated by TNIP1. Shown are representative blots of three biological replicates. Bar diagram shows quantification. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired, two-sided t test. (D) Blockage of protein translation by cycloheximide (CHX) treatment reduced the time- and poly(I:C)-dependent increase of TNIP1 after 6 h of treatment indicating a regulation on translational level. Source data are available for this figure: SourceData FS4.
Because our data suggested that TNIP1 is degraded specifically by autophagy upon poly(I:C) exposure, we investigated whether this was LIR-dependent. To this end, we observed that in cells reconstituted with HA-TNIP1-WT, 6 h of poly(I:C) treatment led to a significant decrease in TNIP1 levels, while we did not observe this decrease in cells expressing HA-TNIP1-mLIR1+2 (Fig. 6, F and G; and Fig. S4 C). ISG15 levels anticorrelated with HA-TNIP1 levels confirming the inhibitory role of TNIP1 in regulating ISG15 expression (Fig. 6, F and G; and Fig. S4 A). Importantly, the relative increase of ISG15 was significantly higher in HA-TNIP1-WT compared to HA-TNIP1-mLIR1+2 expressing cells, supporting the interpretation that LIR-dependent degradation of TNIP1 is critical for a stimulus- and time-dependent expression of pro-inflammatory genes. Note that the time-dependent increase of endogenous and exogenous TNIP1 after prolonged poly(I:C) treatment differed, indicating a long-term transcriptional and/or translational regulation next to the observed short-term autophagy-dependent effects (Fig. 6, F and G; and Fig. S4 D). TNIP1 expression has previously been shown to be transcriptionally regulated by NF-κB, making it likely that prolonged poly(I:C) treatment can lead to the increased expression of TNIP1 (Tian et al., 2005). To further test the LIR-dependent recruitment of TNIP1 to autophagosomes under poly(I:C) treatment, we performed anti-GFP-LC3B IPs followed by anti-TNIP1 Western blotting. In these IP experiments, we observed a poly(I:C)-dependent increase in interaction of TNIP1 with LC3B in TNIP1 KO cells reconstituted with WT TNIP1, which was significantly reduced in KO cells reconstituted with TNIP1-mLIR1+2 (Fig. 6 H). Thus, pro-inflammatory signaling as exemplified by poly(I:C) treatment appears to lead to the specific, LIR-dependent degradation of TNIP1 by autophagy.
TBK1 positively regulates LIR-dependent interaction of OPTN with LC3B by phosphorylating the LIR motif of OPTN (Wild et al., 2011). In addition, TBK1 is an important mediator of TLR3-inducd antiviral signaling (Louis et al., 2018). Since we identified TBK1 as a possible interaction partner of TNIP1 (Fig. 3 A), we tested whether TBK1 also plays a role in selective TNIP1 degradation. Indeed, confocal imaging showed that upon poly(I:C) treatment in WT cells, several of the observed endogenous TNIP1 dots colocalized with phosphorylated (Ser172), i.e., active, TBK1 (Fig. 7 A). Poly(I:C) treatment also led to a time-dependent activation of TBK1 as indicated by phosphorylation of Ser172 (Fig. 7 B). Interestingly, loss of TNIP1 led to an increased activation of TBK1 indicating a feedback regulatory mechanism.
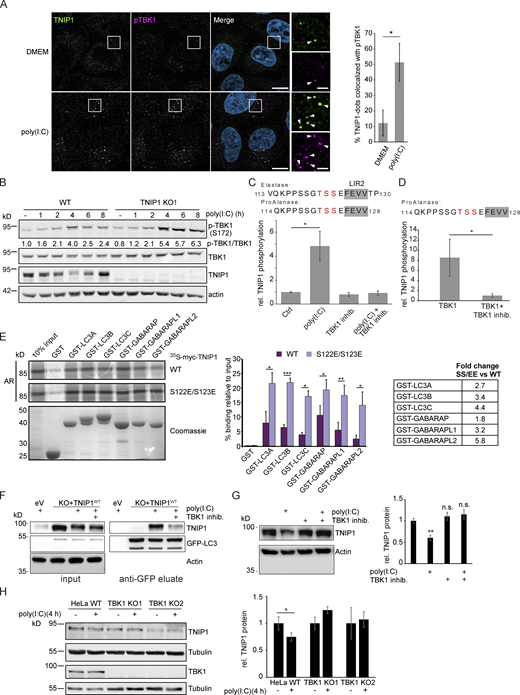
Poly(I:C) stimulation induces TBK1-dependent, specific degradation of TNIP1 by autophagy. (A) Immunofluorescence images showing colocalization between TNIP1 and pTBK1 upon poly(I:C) treatment. Cells were either left untreated or treated with 5 µg/ml poly(I:C) for 4 h, and subsequently stained for endogenous TNIP1 and pTBK1. Colocalization between TNIP1 and pTBK1 is indicated by arrowheads. Quantification of TNIP1 dots colocalizing with pTBK1 was done using Volocity software (PerkinElmer). Around 160–220 cells were counted for each condition in each independent experiment (n = 3). * = P < 0.05, unpaired two-sided t test. Error bars indicate SD. Scale bar in overview image is 10 µm, and scale bar in insert is 2 µm. (B) Time-course effect of poly(I:C) treatment on TBK1 activation and TNIP1. Representative blot and the corresponding quantification of the relative pTBK1 over total TBK1 levels are shown. (C and D) TBK1 phopshorylates TNIP1 N-terminal of LIR2. (C) In vivo phosphoproteomics using Elastase or ProAlanase as proteolytic enzymes identified indicated phosphopeptides. The single phosphorylation site could not be unambiguously localized to one of the three amino acid residues highlighted in red. Inhibition of TBK1 blocked the respective phosphorylation event (n ≥ 3). (D) In vitro kinase assay using purified TBK1 and TNIP1 coupled to phosphoproteomics indicates that TBK1 directly phosphorylates TNIP1 on one of the amino acid residues highlighted in red. * = P < 0.05, unpaired two-sided t test. Error bars indicate SEM. (E) In vitro GST-pulldown assay using 35S-labeled myc-TNIP1 and myc-TNIP1-S122E/S123E against recombinant GST and GST-tagged human ATG8s. Bound myc-TNIP1 WT and S122E/S123E was detected using autoradiography (AR). Quantification and fold change of n = 3, * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired two-sided t test. Error bars indicate SD. (F) The interaction between TNIP1 and LC3B is regulated by TBK1. GFP-LC3B is purified using GFP trap beads. Bound TNIP1 is deteced by Western blot. Inhibition of TBK1 by MRT67307 negatively regulates the poly(I:C)-dependent interaction of TNIP1 with LC3. KO1 cells were used for reconstitution. (G) Inhibition of TBK1 negatively interferes with poly(I:C)-dependent degradation of TNIP1. Western blots of whole cell lysate indicate TNIP1 stabilization by TBK1 inhibition. Actin was used as loading control (n = 3). Error bars indicate SEM. ** = P < 0.01, unpaired, two-sided t test. (H) TBK1 KO negatively interferes with poly(I:C)-dependent degradation of TNIP1. Western blots of whole cell lysate indicate TNIP1 stabilization by TBK1 KO in two independent cell lines. Tubulin was used as loading control (n = 3). Error bars indicate SD. ** = P < 0.01, unpaired, two-sided t test. In A, C, D, E, G, and H, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F7.
Poly(I:C) stimulation induces TBK1-dependent, specific degradation of TNIP1 by autophagy. (A) Immunofluorescence images showing colocalization between TNIP1 and pTBK1 upon poly(I:C) treatment. Cells were either left untreated or treated with 5 µg/ml poly(I:C) for 4 h, and subsequently stained for endogenous TNIP1 and pTBK1. Colocalization between TNIP1 and pTBK1 is indicated by arrowheads. Quantification of TNIP1 dots colocalizing with pTBK1 was done using Volocity software (PerkinElmer). Around 160–220 cells were counted for each condition in each independent experiment (n = 3). * = P < 0.05, unpaired two-sided t test. Error bars indicate SD. Scale bar in overview image is 10 µm, and scale bar in insert is 2 µm. (B) Time-course effect of poly(I:C) treatment on TBK1 activation and TNIP1. Representative blot and the corresponding quantification of the relative pTBK1 over total TBK1 levels are shown. (C and D) TBK1 phopshorylates TNIP1 N-terminal of LIR2. (C) In vivo phosphoproteomics using Elastase or ProAlanase as proteolytic enzymes identified indicated phosphopeptides. The single phosphorylation site could not be unambiguously localized to one of the three amino acid residues highlighted in red. Inhibition of TBK1 blocked the respective phosphorylation event (n ≥ 3). (D) In vitro kinase assay using purified TBK1 and TNIP1 coupled to phosphoproteomics indicates that TBK1 directly phosphorylates TNIP1 on one of the amino acid residues highlighted in red. * = P < 0.05, unpaired two-sided t test. Error bars indicate SEM. (E) In vitro GST-pulldown assay using 35S-labeled myc-TNIP1 and myc-TNIP1-S122E/S123E against recombinant GST and GST-tagged human ATG8s. Bound myc-TNIP1 WT and S122E/S123E was detected using autoradiography (AR). Quantification and fold change of n = 3, * = P < 0.05, ** = P < 0.01, *** = P < 0.001; unpaired two-sided t test. Error bars indicate SD. (F) The interaction between TNIP1 and LC3B is regulated by TBK1. GFP-LC3B is purified using GFP trap beads. Bound TNIP1 is deteced by Western blot. Inhibition of TBK1 by MRT67307 negatively regulates the poly(I:C)-dependent interaction of TNIP1 with LC3. KO1 cells were used for reconstitution. (G) Inhibition of TBK1 negatively interferes with poly(I:C)-dependent degradation of TNIP1. Western blots of whole cell lysate indicate TNIP1 stabilization by TBK1 inhibition. Actin was used as loading control (n = 3). Error bars indicate SEM. ** = P < 0.01, unpaired, two-sided t test. (H) TBK1 KO negatively interferes with poly(I:C)-dependent degradation of TNIP1. Western blots of whole cell lysate indicate TNIP1 stabilization by TBK1 KO in two independent cell lines. Tubulin was used as loading control (n = 3). Error bars indicate SD. ** = P < 0.01, unpaired, two-sided t test. In A, C, D, E, G, and H, data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData F7.
With the TBK1-mediated regulation of the OPTN LIR in mind, we used MS-based phosphoproteomics to study whether TBK1 can phosphorylate serine and threonine residues located N-terminal to the core LIR sequence FEVV of LIR2 in TNIP1 (Fig. 4 C). Due to the surrounding amino acid sequence, we could not follow the standard workflow of bottom-up proteomics experiments using trypsin to generate respective peptides. Instead, we performed a multi-protease digestion protocol using Elastase or ProAlanase to generate optimal sequence coverage (Eisenhardt et al., 2016; Samodova et al., 2020). We were able to identify and quantify two phosphopeptide species which were phosphorylated within the TSS motif just in front of LIR2 (Fig. 7 C). Comparing cells treated or not with poly(I:C) and/or the TBK1 inhibitor MRT67307 clearly indicated that TBK1 phosphorylates TNIP1 in a stimulus-dependent manner (Fig. 7 C). To determine whether TNIP1 is a direct target of TBK1, we performed in vitro kinase assays and could recapitulate the in vivo observations characterizing TNIP1 as a bona fide TBK1 substrate (Fig. 7 D). To test the effect of phosphorylations of the evolutionary conserved S122 and S123 residues N-terminal to the core LIR motif (Fig. 4 C), we analyzed the binding of the phosphomimicking S122E/S123E TNIP1 mutant to ATG8 family proteins by GST pulldown. Strikingly, the phosphomimicking mutant displayed strongly increased binding to all ATG8 proteins. The binding increase was particularly evident for LC3B (3.4-fold), LC3C (4.4-fold) and GABARAPL2 (5.8-fold; Fig. 7 E). Finally, we tested the effects of poly(I:C) treatment and TBK1 inhibition on TNIP1-LC3B interaction by GFP-LC3B IP. Inhibition of TBK1 led to a decreased interaction of TNIP1-WT with GFP-LC3B indicating that TBK1-dependent phosphorylation of the TNIP1 LIR motif was responsible for the observed increased interaction between LC3 and TNIP1 (Fig. 7 F). In agreement, pharmacological and genetic inhibition of TBK1 led to a stabilization of TNIP1 under poly(I:C) treatment, i.e., interfered with its stimulus-dependent degradation (Fig. 7, G and H). Thus, whereas SLRs appear to support autophagy-dependent, basal turnover of TNIP1, activation of its LIR motif by TBK1 induces its selective, autophagy-dependent removal, supporting the mounting of a transcriptional program to induce a robust inflammation response (Fig. 8).

Model of TNIP1 regulation. Under basal, unstimulated conditions (left panel), TNIP1 functions as a negative regulator of inflammatory signaling and is subject to constitutive autophagic degradation through interaction with autophagy receptors such as p62/SQSTM1. Upon poly(I:C)-induced activation of TLR3 (right panel), activated TBK1 phosphorylates TNIP1 in the vicinity of its LIR, increasing TNIP1 affinity for human LC3 and GABARAP proteins. This, in turn, leads to a LIR-dependent increase in TNIP1 degradation through selective autophagy. The removal of TNIP1 relieves the negative effect on inflammatory signaling, allowing the establishment of a robust inflammatory response upon antiviral signaling.
Model of TNIP1 regulation. Under basal, unstimulated conditions (left panel), TNIP1 functions as a negative regulator of inflammatory signaling and is subject to constitutive autophagic degradation through interaction with autophagy receptors such as p62/SQSTM1. Upon poly(I:C)-induced activation of TLR3 (right panel), activated TBK1 phosphorylates TNIP1 in the vicinity of its LIR, increasing TNIP1 affinity for human LC3 and GABARAP proteins. This, in turn, leads to a LIR-dependent increase in TNIP1 degradation through selective autophagy. The removal of TNIP1 relieves the negative effect on inflammatory signaling, allowing the establishment of a robust inflammatory response upon antiviral signaling.
Discussion
Autophagy is largely regarded as a cytoprotective, anti-inflammatory response ensuring cell and organismal homeostasis (Deretic, 2021; Deretic and Levine, 2018). Several autophagy loci have been linked to genetic predispositions for chronic inflammatory and autoimmune diseases. This indicates that the lack of removal of damaged organelles may indirectly promote tissue destabilizing pro-inflammatory signaling. Autophagy may also contribute on a direct molecular level to limit excessive inflammatory signaling, e.g., by the selective degradation of inflammasome components by SARs, such as p62/SQSTM1 (Shi et al., 2012). Furthermore, by removing other endogenous pro-inflammatory sources including damaged organelles and components from viral and bacterial infections, SARs play an important role in anti-inflammatory responses (Deretic, 2021). The pro-inflammatory functions of autophagy were so far limited to its contribution to interleukin secretion and to senescence-associated inflammation (Dupont et al., 2011; Lee et al., 2021; Zhang et al., 2015). In the current article, we highlight that the role of autophagy in inflammation appears to be more complex than anticipated and that both processes appear to be more intimately intertwined on a molecular level.
As TNIP1 has no reported enzymatic activity itself, it was believed that its functions in inflammation were directly linked to its interaction with TNFAIP3 (also known as A20), a ubiquitin-editing enzyme that contains both ubiquitin ligase and deubiquitinase activities and that was shown to negatively interfere with NF-κB signaling (Shamilov and Aneskievich, 2018; Song et al., 1996). Several recent studies, however, indicate that TNIP1 itself may function as a key repressor of inflammatory signaling, its dysregulation being linked to hyperinflammatory diseases like psoriasis (Nair et al., 2009), systemic lupus erythematosus (Gateva et al., 2009), systemic sclerosis (Allanore et al., 2011), and senescence (Lee et al., 2021). TNIP1 is thus a potential target for the design of anti-inflammatory therapeutics. It has also been suggested that TNIP1 can function by out-competing other pro-inflammatory mediators for polyubiquitin binding, thereby negatively affecting inflammatory signaling (Shamilov and Aneskievich, 2018).
Whereas single nucleotide polymorphisms were shown to alter TNIP1 expression and microRNAs to decrease TNIP1 mRNA levels (Shamilov and Aneskievich, 2018), posttranslational mechanisms regulating TNIP1 protein abundance are largely unknown. We and others localized TNIP1 to autophagosomes, but did so far not address underlying mechanisms and phenotypical consequences (Mejlvang et al., 2018; Shinkawa et al., 2022; Zellner et al., 2021). In the current study, we corroborate observations that TNIP1 can be regarded as a substrate for constitutive autophagosomal degradation. We did not identify changes in autophagy-dependent lysosomal degradation under fed and starved conditions, as well as under blocked mTORC1 signaling. The association of TNIP1 with SLRs and its colocalization with p62 bodies and LC3 puncta further supports this interpretation, indicating that ubiquitination of TNIP1 and direct protein-protein interactions lead to SLR-dependent lysosomal degradation (Fig. 8). Indeed, we could show that TNIP1 was still ubiquitinated even after mutating the two ubiquitination sites identified in this study. This is in agreement with www.phosphosite.org, which lists 11 ubiquitination sites of TNIP1, Lys389 being one of them (Akimov et al., 2018). In addition, also non-modified TNIP1 could be stabilized by lysosomal inhibition. Thus, TNIP1 appears to be constitutively degraded by autophagy, the role of ubiquitination being not entirely clear.
Interestingly, this changes under inflammatory conditions. Poly(I:C) treatment led to SLR-independent, selective removal of TNIP1 by autophagy dependent on its LIR motif. This suggests the existence of distinct TNIP1 pools within cells: non-modified TNIP1, phosphorylated and/or ubiquitinated TNIP1 (Fig. 8). This interpretation is supported by the observation that TNFAIP3 appeared to not be degraded by autophagy in a poly(I:C)-dependent manner, indicating that the pool of TNIP1 which interacts with TNFAIP3 under these conditions is also spared from degradation (see Fig. S4 A). The time-dependent regulation of TNIP1 protein levels indicates that autophagy-dependent degradation within the first 4 h of poly(I:C) treatment contributes to the establishment of a robust inflammatory response. At 6 h, TNIP1 levels rise again due to transcriptional upregulation starting already at 4 h. This regulation occurs to prevent excessive inflammatory signaling, which may lead to cell and tissue damage. In light of the model suggesting that TNIP1 can compete with pro-inflammatory proteins for ubiquitin binding (Shamilov and Aneskievich, 2018), it is possible that TNIP1 is degraded during the early stages of TLR3-activation to prevent this competition. The subsequent increase in TNIP1 at later stages of the signaling response could then be to outcompete other pro-inflammatory mediators in order to limit excessive signaling. In a recent study, OPTN was characterized as a direct, ubiquitination-independent binding partner of TNIP1 contributing to the specific, autophagy-dependent degradation of TNIP1 in senescent cells (Lee et al., 2021). The pathway characterized by us is different as TNIP1 degradation under inflammation conditions appears as SLR-independent (Fig. 8). OPTN and TNIP1 appear to function in parallel and to be positively linked on the transcriptional level since TNIP1 KO led to an upregulation of OPTN mRNA and protein levels, potentially in a compensatory fashion.
The canonical core LIR motif has the consensus sequence W/F/Y-X1-X2-L/I/V. The W/F/Y and the L/I/V occupy two hydrophobic pockets of the LIR docking site (LDS) of ATG8 family proteins. Additionally, acidic residues N-terminal, within and C-terminal, to the core motif, as found in the main LIR2 of TNIP1, increase binding affinity to the LDS which has a largely basic surface surrounding the two hydrophobic pockets (reviewed in Johansen and Lamark, 2020). The presence of serine or threonine residues N-terminal to the core motif which can be phosphorylated to increase the acidic nature of the LIR is a neat strategy for a switchable LIR-LDS interaction. It has been shown for a number of SARs, including OPTN and several mitophagy receptors, that phosphorylation of N-terminal residues flanking the core LIR enhances the LIR-LDS interaction (Di Rita et al., 2018; Rogov et al., 2017; Wild et al., 2011; Wu et al., 2014; Zhu et al., 2013). This is also found for Beclin1, VPS34, and SCOC LIR-ATG8 interactions (Birgisdottir et al., 2019; Wirth et al., 2021). In the case of OPTN, the LIR-proximal phosphorylation is mediated by TBK1, leading to the enhanced binding of OPTN to LC3 (Wild et al., 2011). In this study, we show that upon TLR3-activation, TNIP1 is phosphorylated at residues N-terminal to its main LIR-motif. Similar to OPTN, TNIP1 phosphorylation N-terminal of LIR2 enhances the interaction of TNIP1 with human LC3B. In vitro, phosphomimic S122E/S123E mutations of TNIP1 led to a strong increase in the binding to all of the human LC3 and GABARAP proteins, further supporting that LIR2 of TNIP1 is regulated by phosphorylation. Whether phosphorylation also influences the ubiquitination status of TNIP1 is currently not known, and a potential crosstalk between the two PTMs will have to be addressed in future studies.
During revision of our article, TNIP1 was reported as a potential SLR acting in the lipopolysaccharide (LPS)- and TLR1/TLR2-dependent degradation of MYD88 and IRAK1 (Shinkawa et al., 2022). In contrast to our data, this group reported LIR1 of murine TNIP1 as being responsive to TLR1/TLR2 activation by LPS (Shinkawa et al., 2022). In another very recent study, TNIP1 was shown to negatively interfere with mitophagy by interfering with ULK1 complex dynamics (Le Guerroue et al., 2022 Preprint). In accordance with our results, these authors reported that LIR2 mutation abolished all binding to ATG8 proteins, while LIR1 mutations had no effect on binding. As we noted, LIR2 is conserved in vertebrate evolution down to cartilagous fishes, while LIR1 is only conserved down to marsupials (Fig. 4 A).
Our results strongly suggest that TBK1 is responsible for phosphorylation of the LIR2 proximal TSS motif. TBK1 is an important kinase in response to innate antiviral signaling. Upon TLR3-activation, TBK1 gets activated through TRIF and TRAF3 leading to the phosphorylation and activation of IRF3 and induction of type I interferons (Louis et al., 2018). TRIF itself may undergo autophagy-dependent degradation (Gentle et al., 2017; Inomata et al., 2012; Lim et al., 2019; Samie et al., 2018). How this anti-inflammatory effect of autophagy is coordinated with the described pro-inflammatory acting degradation of TNIP1 will be an interesting question to address in the future studies. TBK1 is also implicated in the regulation of autophagy and is known to phosphorylate several autophagy proteins (Oakes et al., 2017). It is possible that TBK1 promotes TNIP1 degradation in order to ensure the efficient activation of downstream interferon signaling. Our results indicate that TNIP1 may rely on the other SLRs for its basal turnover in unstimulated cells, but upon TBK1 activation in response to innate immune stimuli, the selective degradation of TNIP1 is promoted (Fig. 8). Interestingly, TNIP1 fulfills all characteristics of a SAR, having a UBD, LIR motif, and coiled-coil regions supporting multimerization. TLR1/2-induced degradation of TNIP1 was shown to mediate the degradation of MYD88 and IRAK1 (Shinkawa et al., 2022). Whether TLR3-activation leads to selective TNIP1-dependent degradation of specific cargo proteins will have to be addressed in future studies. The knockout of TNIP1 resulted in a basal increase in the expression and protein levels of several pro-inflammatory genes, even in the absence of pro-inflammatory signaling. This suggests that TNIP1 may have a role in preventing the induction of an inflammatory response even in untreated cells. Whether this is caused by TNIP1-mediated selective degradation of pro-inflammatory-mediators or -complexes will also be important to address in future studies.
Taken together, we identified TBK1-dependent phosphorylation sites immediately N-terminal to the core LIR motif of TNIP1 that lead to its selective degradation by autophagy under inflammatory conditions. Autophagy may contribute to the establishment of a potent inflammatory response before it limits excessive cytotoxic inflammatory signaling. Thus in addition to cancer, inflammation is another condition in which autophagy may have a dual role either supporting or inhibiting underlying processes depending on the exact cell state and timing.
Materials and methods
Cell culture and cell treatments
HeLa cells and HEK293T cells were obtained from ATCC. U2OS cells were obtained from ECACC. StUbEx U2OS cells were a gift from Blagoy Blagoev (Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark; Akimov et al., 2014). HeLa CCL2.2, HeLa CCL2.2 ATG7 KO cells, and HeLa CCL2.2 pentaKO cells were a gift from Richard J. Youle (National Institutes of Health, Bethesda, MD; Sarraf et al., 2020). HeLa p62 KOs have been described previously (Abudu et al., 2021). All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin. For starvation experiments, cells were incubated with Hanks’ Balanced Salt solution (HBSS) for the indicated times. Cells were treated with 100 nM rapamycin, 200 nM bafilomycin A1, 5 µg/ml Concanamycin A, 20 µg/ml cycloheximide, or 10 μM MG132, for the indicated time periods. For SILAC, cells were grown for >14 d in SILAC DMEM supplemented with dialyzed fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, and “light” (12C614N2-Lysine and 12C614N4-Arginine), “medium” (D4-Lysine and 13C6-Arginine), or “heavy” (13C615N2-Lysine and 13C615N4-Arginine) stable isotope-labeled amino acids. For transient DNA transfection, subconfluent cells were transfected using TransIT LT1 transfection reagent according to the manufacturer’s instructions.
Plasmid constructs
Plasmids used in this study are listed in Table S4. Cloning into pDest-vectors was performed using the Gateway cloning system (Invitrogen). QuickChange site-directed mutagenesis kit (Stratagene) was used to create desired point mutations, which were verified by DNA sequencing (BigDye Sequencing kits, Applied Biosystems). Oligonucleotides for mutagenesis and sequencing were from Invitrogen. TNIP1 cDNA was obtained from Genscript (NM_001252390), TNIP1 LIR mutation fragment was synthesized by IDT gBlocks Fragments (Integrated DNA Technologies). TNIP1 and TNIP1_mLIR were cloned into pDONR201 and pLenti CMV Blast DEST (706-1) by Gateway recombination cloning according to manufacturer’s instructions. All plasmid constructs were verified by sequencing (BigDye; Applied Biosystems) and/or restriction digestion.
Generation of knockout (KO) and stable cell lines
The TNIP1 KO cells were generated by CRISPR/Cas9-mediated genome-editing tools provided by the F. Zhang laboratory (Broad Institute, MIT, Boston, MA). A target sequence in exon twenty of human TNIP1 was selected (sgTNIP1-1, 5′-CGTACCGGATCTACGACCCT-3′; sgTNIP1-2, 5′-GGCCCTGGAGTTCAACCGAC-3′; sgTNIP1-3, 5′-CACCCGACAGCGTGAGTACC-3′). The sgRNA oligos were cloned into the Bbs1 site of hSpCas9 plasmid. Lipofectamine LTX and Plus reagent (15338100; Invitrogen) were used to transfect hSpCas9-sgRNA into HeLa cells according to the manufacturer’s instructions. Cells were selected with 2 μg/ml puromycin for 2 d and single cells were isolated by serial dilutions. TNIP1 deficiency was screened by immunoblotting.
CRISPR knockout of FIP200 and ATG101 in HeLa cells was performed using sgRNAs cloned into the plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (#42230; Addgene): ATG101, 5′- ACCAGAAGAAGAAGTCTCGC-3′ and 5′- GTTATCCACCTCCGACTGTG-3′; FIP200, 5′-GTAGTTTTAGGAATAGCAGG-3′. CRISPR plasmids and pEGFP-puromycin plasmid were co-transfected in HeLa cells using lipofectamine LTX reagent (15338030; Thermo Fisher Scientific), according to manufacturer’s instructions. 24-h post-transfection, co-transfected cells were selected in culture medium supplemented with 3 μg/ml puromycin (Invivogen, ant-pr-1). Efficiently transfected cells were isolated to generate clonal lineages by single-cell cloning in 96-well plates; colonies were all evaluated for KO efficiency by Western blotting against the targeted protein. The sgRNAs were designed using CHOPCHOP CRISPR/gRNA algorithm (Labun et al., 2019).
Generation of HeLa CCL2.2 cells KO for OPTN and TAX1BP1 and HeLa KO for TBK1 was carried out as described previously (Abudu et al., 2021). The following gRNAs were used: sgOPTN: 5′-CCCACGAGAACAGTCTCCAC-3′, sgTAX1BP1: 5′-AGACCTGCATACTGCACGCT-3′, sgTBK1: 5′- AACGTGGATGTACTTTAGGG-3′.
For generation of stable cell lines, lentiviral vectors were co-transfected with packaging (psPax2) and envelope plasmids (pMD2.G) in HEK293T cells using jetPRIME reagent (Polyplus transfection). Medium was changed to fresh DMEM 15 h after transfection. After 24 h incubation, the supernatant was harvested by filtration through 0.22-μm filter. Polybrene (8 µg/ml) was added before infection of recipient cells. To obtain stable cell lines, infected cells were cultured in the presence of 5 μg/ml blasticidin for 1 wk and monitored by immunoblotting.
Enrichment of ubiquitinated peptides
Ubiquitinated peptides were purified according to Akimov et al. (2018). Triple SILAC labeled U2OS and HeLa cells were treated with DMSO, Rapa, and Rapa + ConA. Cells were washed twice with cold PBS and lysed with 12 ml lysis buffer (8 M guanidine-HCl, 25 mM ammonium bicarbonate [ABC], pH 8.5). The lysates were sonicated to reduce viscosity and cleared by centrifugation at 15,000 RCF for 30 min. Protein concentration was determined by BCA protein assay kit (23225 and 23227 Pierce), and the same amount of proteins of each label were mixed. Proteins were reduced with 2 mM DTT for 30 min at room temperature and alkylated with 11 mM chloro acet amide in the dark for 30 min at room temperature. The concentration of guanidine-HCl was diluted to 2 M with 25 mM ammonium bicarbonate and filtrated with low binding 0.45-µm PVDF filters (Millipore). The Lys-C endopeptidase was added at a 1:100 enzyme-to-protein ratio to digest proteins at room temperature overnight. The peptide mixture was cleaned and purified using C18 cartridges (WATERS) and lyophilized for 24–36 h. The lyophilized peptides were dissolved in 12 ml of IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl, pH 7.5–8.0) plus 0.1% Triton X-100. Dissolved peptides were spun down and passed through low binging 0.45-µm PVDF filters and incubated with 500 μl of UbiSite conjugated matrix for 5 h at 4°C. The Ubisite matrix was washed three times with IAP buffer without detergent and three times with 150 mM NaCl. After the third wash, the matrix was transferred to a small Poly-Prep column (BioRAD). Enriched peptides were eluted with 250 μl 0.1% TFA three times, and each time incubated for 5 min. The eluted peptides were pooled and neutralized with 1 M ABC buffer to a final concentration at 25 mM, followed by trypsin digestion overnight at 37°C. The fractionation of tryptic peptides was performed as described previously (Akimov et al., 2018). The resulting peptides were lyophilized and cleaned by STAGE-tips.
Enrichment of ubiquitinated proteins by StUbEx
StUbEx U2OS cells were induced with doxycycline for 48 h and treated with Rapa and lysosomal degradation was blocked with ConA. The cells were washed with cold PBS and lysed with binding buffer (6 M Guanidinium-HCl, 50 mM Na2HPO4/NaH2PO4, pH 8.0, 500 mM NaCl, 5 mM imidazole). Sample viscosity was reduced by sonication followed by centrifugation at 11,000 RCF for 30 min at room temperature. Protein concentration was determined by BCA assay. Ubiquitin conjugates were purified by cOmplete His-tag purification beads (Roth) for 4 h at room temperature, followed by washes with binding buffer and washing buffers 1–3 (WB1—8 M Urea, 50 mM Na2HPO4/NaH2PO4, pH 8.0, 500 mM NaCl, 0.1% Triton-X-100; WB2—same as WB1 but with 0.2% Triton X-100; WB3 -8 M Urea, 25 mM Tris, pH 8.0, 150 mM NaCl). The ubiquitin conjugates were finally eluted with three times elution buffer (150 mM NaCl, 50 mM Tris, pH 8.0, and 300 mM imidazole).
Immunoblotting
For immunoblot analysis, treated cells were harvested and lysed in modified RIPA buffer supplemented with 2% SDS and 0.1% benzonase Nuclease or 1× SDS buffer (50 mM Tris, pH 6.8, 2% SDS, and 10% glycerol). For cells harvested in modified RIPA buffer, lysates were centrifuged for 15 min at 11,000 RCF. For cells harvested in 1× SDS buffer, lysates were incubated at 100°C for 10 min. Protein concentration was measured using Pierce BCA Protein Assay Kit. Quantified cell lysates or AP elutes were either reduced with 2 mM DTT at 75°C for 10 min or 100 mM DTT at 100°C for 10 min and resolved on SDS-PAGE gels. Proteins were transferred to a PVDF or nitrocellulose membrane, and subsequently prepared for either fluorescent or chemiluminescent detection. For fluorescent detection, membranes were blocked in Intercept (PBS or TBS) Blocking Buffer, followed by overnight incubation at 4°C with primary antibody. Membranes were subsequently washed 4× in PBS or TBS containing 0.1% Tween (PBS-T/TBS-T), followed by incubation with secondary antibody diluted in Intercept Blocking Buffer. After 4× wash in PBS-T/TBS-T, a final wash was carried out in PBS/TBS without Tween. Fluorescent signal was detected using LiCOR Odyssey CLx imaging system. Chemiluminescent detection: membranes were blocked in either 5% milk or 5% bovine serum albumin in PBS-T or TBS-T, followed by incubation with primary antibody. Membranes were washed 3–4× in PBS-T/TBS-T followed by incubation with secondary antibody. After 3–4× washes in PBS-T/TBS-T, membranes were developed using SuperSignal West Pico Chemiluminescent Substrate on GE Fujifilm LAS4000 Luminescent image analyzer or Odyssey Fc reader (LI-COR Biosciences-GmbH). The densitometry of immunoblotting was performed by either ImageJ (National Institutes of Health) or ImageStudio software (LI-COR Biosciences-GmbH).
Immunofluorescence staining and confocal fluorescence microscopy
For imaging, cells were grown on #1.5 round 12-mm coverslips (#631-0150; VWR) for immunofluorescence staining or 8-well Lab-Tek chamber coverglass for double-tag analysis (#155411; Thermo Fisher Scientific), and fixed with 4% formaldehyde in PBS for 30 min. For immunofluorescence staining, cells were washed 3× with PBS and then permeabilized with either 0.1% Triton X-100 in PBS for 10 min, or ice-cold methanol for 10 min. Next, cells were washed 5× with PBS or TBS, and blocked for 1 h in 5% BSA in PBS or TBS. Subsequently, cells were incubated with primary antibodies diluted in 1% BSA in PBS or TBS for 1–2 h at room temperature. After 5× wash in PBS or TBS, cells were incubated with Alexa Fluor secondary antibodies diluted in 1% BSA in PBS or TBS for 1 h at room temperature. After 5× wash in PBS or TBS, cell nuclei were stained with 1 µg/ml DAPI diluted in PBS for 5 min, followed by 2× final washes in PBS or TBS. Coverslips were mounted using ProLong Gold or ProLong Glass Antifade Mountant. Cells were imaged using Zeiss LSM800 or LSM880 (Carl Zeiss Microscopy) using a 63 × NA1.4 oil immersion lens for coverslips or a 40× NA1.2 water immersion lens for chambered coverglass. Images were collected in ZEN software (Zeiss). For Airyscan super-resolution images, optimal pixel size and z spacing as suggested by ZEN was used. Optimal excitation and emission settings were determined using the Smart Setup function. All fluorescence channels were recorded at non-saturating levels, and settings were kept identical between all samples within replicates used for comparisons or quantifications.
Image analysis
For quantification of red-only dots of mCherry-EYFP-TNIP1, transiently transfected cells with low expression of mCherry-EYFP-TNIP1 were visually selected and imaged. Cells with high levels of overexpression were excluded, as this resulted in the formation of large aggregates. All images within each replicate were taken using identical settings. For quantification of relative amounts of red-only dots versus total dots, mCherry and EYFP dot detection was performed using a custom-made measurement protocol using intensity thresholding, size exclusion, and noise filtering, based on signal intensity of the BafA1 control in Volocity software (PerkinElmer) ver. 6.3. The number of mCherry-only positive dots was counted by subtracting total EYFP dots from total mCherry dots for each experiment. For quantification of the number of cells positive for red-only mCherry-EYFP-TNIP1 dots, the images were analyzed manually by a subject that was blind to the analyzed conditions.
To quantify the number of TNIP1 dots and the colocalization between TNIP1 and SLRs/pTBK1, populations of objects representing fluorescent puncta in each channel were segmented using a custom-made protocol in Volocity ver. 6.3 (PerkinElmer). Detection of TNIP1, SLRs, and pTBK1 puncta was performed by intensity thresholding, size exclusion, and noise reduction. Overlap between TNIP1 and pTBK1 was identified by excluding TNIP1 objects not touching SLRs/pTBK1. The percentage of TNIP1 dots colocalizing with SLRs/pTBK1 was calculated by dividing the number of TNIP1 dots overlapping with SLRs/pTBK1 with the total number of TNIP1 dots. For line-profiles, the line-profile tool in Zen Blue software (Zeiss) was used to measure signal intensity across indicated lines.
SPOT synthesis and peptide array
TNIP1 peptide array was synthesized on cellulose membranes using MultiPrep peptide synthesizer (INTAVIS Bioanalytical Instruments AG). Membranes were blocked with 5% non-fat milk in TBS-T and peptide interactions were tested using GST-GABARAP by overlaying the membrane with 1 µg/ml recombinant protein for 2 h at room temperature. Membranes were washed three times in TBS-T. Bound GST-GABARAP was visualized with HRP-conjugated anti-GST antibody (Johansen et al., 2017). Putative LIR motifs in 20, 3 arrays (20-mer peptides moved a window of 3 residues along the protein sequence) were identified as 4–6 consecutive strong spots containing the core LIR consensus (W/F/Y)XX(L/I/V).
Recombinant protein production and GST-pulldown analysis
GST and GST-fusion proteins were expressed in Escherichia coli strain SoluBL21 (DE3; #C700200;Genlantis). Protein expression was induced by adding 50 μg/ml isopropyl β-D-1-thiogalactopyranoside (IPTG). The bacterial cells were sonicated in lysis buffer (20 mM Tris-HCl, pH 7.5, 10 mM EDTA, 5 mM EGTA, 150 mM NaCl) and GST-fused proteins were immobilized on Glutathione Sepharose 4 Fast Flow beads (#17-5132-01; GE Healthcare) by incubating in a rotator at 4°C for 1 h. The beads containing GST-fusion proteins were subsequently used for pulling down in vitro translated proteins or cell lysates. For in vitro translated proteins, a pDest-myc-vector containing the protein of interest and a T7 promoter was used. In vitro translation was performed using the TNT T7 Reticolucyte Lysate System (Promega Corp.), in the presence of radioactive 35S-methionine. In vitro translated protein was then precleared by incubation with empty Glutathione Sepharose beads in 100 μl of NETN buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40) supplemented with cOmplete Mini EDTA-free protease inhibitor (Merck) for 30 min at 4°C. Precleared lysates were then incubated with GST-fusion protein bound beads for 1–2 h on a rotator at 4°C. Beads were then washed 5× with NETN-buffer, and resuspended in 2 × SDS-PAGE gel loading buffer (125 mM Tris, pH 7.5, 4% SDS, 0.04% bromophenol blue, 20% glycerol, 100 mM dithiothreitol), boiled for 10 min, and resolved by SDS-PAGE. Gels were stained with Coomassie Brilliant Blue for protein visualization and then vacuum-dried. The radioactive signal was then detected on imaging plates with Fujifilm BAS-5000 (Fujifilm). Signals from 35S-labeled proteins were measured in terms of unit of photostimulated luminescent (PSL) and quantified in comparison with 10% of the in vitro translated lysate (input) using the Image Gauge software (Fuji; Johansen et al., 2017).
Affinity purification
Cells were lysed in ice-cold modified RIPA buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% Sodium deoxycholate) containing complete protease inhibitor cocktail and phosphatase inhibitor cocktail. Lysates were centrifugated for 15 min at 13,000 RCF and protein concentration was determined by BCA assay. GFP-tagged proteins were affinity purified by GFP-trap (ChromoTek) according to the manufacturer’s instructions. HA-TNIP1 was affinity purified by anti-HA magnetic beads (Pierce). TNIP1 polyclonal antibody-coupled protein G dynabeads were used for affinity purification of endogenous TNIP1. For denaturing purification, the lysate was supplemented with 2% SDS and incubated for 30 min at room temperature to break protein-protein interaction, then diluted to 0.5% SDS with lysis buffer and incubated with HA or anti-TNIP1 antibody for affinity purification.
For TNIP1 interactome analysis by MS, HA-TNIP1 cells and empty vector cells were cultured in “heavy” and “light” SILAC medium, respectively. After 2 wk, cells were harvested and lysed in modified RIPA buffer containing complete protease inhibitor cocktail and phosphatase inhibitor cocktail. Protein concentration was determined by BCA assay, and protein amount was adjusted to equal concentration with lysis buffer, followed by affinity purification with anti-HA magnetic beads. The eluate of “heavy” and “light” samples was combined and fractionated by SDS-PAGE. Proteins were in-gel digested by trypsin and peptides were desalted by STAGE-tips prior to LC-MS/MS analysis.
Whole proteome analysis
For whole proteome analysis by MS, HeLa wild-type cells and TNIP1 KO cells were cultured in SILAC medium for 2 wk. Cells were harvested and lysed in modified RIPA buffer containing 2% SDS and 0.1% benzonase Nuclease. Protein concentration was determined by BCA assay and equal amounts of proteins in each label were mixed. Proteins were reduced with DTT and alkylated with IAA, followed with SDS-PAGE fractionation and trypsin in-gel digestion. Tryptic peptides were desalted by STAGE-tips prior LC-MS/MS analysis.
TNIP1 phosphorylation analysis
HA-TNIP1 cells were treated with poly(I:C) with or without TBK1 inhibitor MRT67307. Cells were harvested and lysed in modified RIPA buffer containing complete protease inhibitor cocktail and phosphatase inhibitor cocktail, 2% SDS was supplemented to break protein-protein interaction. The samples were diluted to 0.5% SDS with lysis buffer and subjected to affinity purification with anti-HA magnetic beads. After purification, the beads were transferred onto 10 kDa MW cut-off filter with 400 μl 8 M urea and 1 mM DTT. Proteins were digested by elastase or ProAlanase using the FASP protocol (Wiśniewski et al., 2009). Digested peptides were eluted twice with 200 μl 50 mM ammonium bicarbonate into fresh tubes and acidified with TFA to a final concentration of 1%. Peptides were frozen and lyophilized prior to phosphopeptide enrichment. The lyophilized peptides were resuspended in 200 μl of 80% ACN, 0.1% TFA. Phosphopeptides enrichment was performed on Agilent AssayMAP Bravo platform according to manufacturer’s instructions. The Agilent AssayMAP phosphopeptide enrichment v2.0 App was used for automated phosphopeptide enrichment using Fe (III)-NTA cartridges (Basel, Switzerland). Cartridges were primed with 100 μl of 50% ACN, 0.1% TFA using a high flow rate of 300 μl/min and equilibrated using 80% ACN containing 0.1% TFA. Samples were loaded onto the cartridge using a low flow rate of 3 μl/min. The cartridges were washed twice with 200 μl 80% ACN containing 0.1% TFA and eluted with 50 μl of 1% ammonium hydroxide (pH 11) and 50 μl of 1% ammonium hydroxide in 80% ACN to a low-binding PCR tube containing 5 μl FA. The eluted peptides were lyophilized and resuspended in 20 μl of 0.1% FA for LC-MS/MS analysis.
In vitro kinase assay
HA-TNIP1 cells were harvested and lysed in ice-cold modified RIPA buffer containing complete protease inhibitor cocktail. TNIP1 protein was enriched by anti-HA magnetic beads and dephosphorylated by lambda phosphatase with 1 mM MnCl2 for 30 min at 30°C. Beads were washed with 2 × 10 ml of kinase buffer (50 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 150 mM NaCl and 1× PhosSTOP) to remove lambda phosphatase. Kinase buffer was added to a final volume of 800 μl with 1 μg TBK1 protein, for control sample, 10 μM TBK1 inhibitor MRT67307 was added. Kinase assays were performed on a rotor at 37°C for 1 h. Finally, reactions were quenched by addition of 8 M urea and 1 mM DTT. The beads were transferred onto 10 kD MW cut-off filter for the digestion with ProAlanase, followed with phosphopeptide enrichment.
LC-MS/MS analyses
Solubilized peptides were injected into a 20 cm fused silica column with an inner diameter of 75 µm and in-house packed with C18 (ReproSil-Pur 120 C18-AQ, 1.9 μm, Dr. Maisch) for reverse phase fractionation by EasyLC 1200 nanoflow-HPLC system (Thermo Fisher Scientific). Peptides were loaded with solvent A (0.1% FA in water) at a max. pressure of 800 Bar and eluted with a step gradient of solvent B (0.1% FA in 80% ACN) from 2 to 25% within 85 min, from 25 to 60% within 5 min, followed by increasing to 100% in 2 min at a flow rate of 250 nl/min. MS/MS analysis was performed on a nano-electrospray ion source equipped QExactive HF-X mass spectrometer (Thermo Fisher Scientific). The spray voltage was set to 2.3 kV with a capillary temperature of 250°C. Mass spectrometer was operated in positive polarity mode and MS data were acquired in the data-dependent mode. The automatic gain control (AGC) target was set to 3 × 106, the resolution was set to 120,000, and the ion injection time was set to 15 ms for the full scan at a mass range of m/z = 370–1750. 12 precursors were fragmented using normalized collisional energy (NCE) of 28 by higher-energy collisional dissociation (HCD). MS/MS scans were acquired with a resolution of 30,000, AGC target of 100,000 maximum IT of 54 ms, isolation window of 1.6 m/z, and dynamic exclusion window of 30 s. MS raw files were analyzed using MaxQuant software version 1.6.2.10 (Cox and Mann, 2008), data were searched against UniProt full-length homo sapiens database (21.033 entries, released March, 2016). Cysteine carbamidomethylation was set as a fixed modification, protein aminoterminal acetylation and methionine oxidation were set as variable modifications. Ubiquitination of lysine was set as a variable modification and digestion was specific to trypsin/P for Ubisite experiment. Serine-, threonine- and tyrosine- phosphorylation were set as a variable modification and digestion was set to unspecific or ProAlanase for TNIP1 phosphorylation experiments. The analysis was carried out with “match-between-run” with a time window of 0.7 min. MaxQuant results were analyzed using Perseus (Tyanova et al., 2016).
Total RNA extraction and library construction
Four replicates of HeLa WT cells and TNIP1 KO cells (clone1 and clone2) were cultured in 10 cm dishes. The total RNA was extracted using RNeasy Mini Kit (Qiagen) according to the instructions of the manufacturer. Cells were lysed with 350 μl RLT buffer and homogenized by passing 5 times through a blunt 20-gauge needle. One volume of 70% ethanol was supplemented, the lysates were transferred to RNeasy spin column and centrifuged at 8,000 × g to collect RNA. The RNA was washed once with 700 μl RW1 buffer and twice with 500 μl RPE buffer. After completely removing the RPE buffer, the RNA was eluted with 50 μl RNAse-free water. The quality of RNA samples was analyzed by RNA screen tape (Agilent). The complementary DNA (cDNA) libraries were barcoded using Illumina primers and sequenced on one lane of an Illumina Novaseq 6000 instrument with 2 × 50 bp paired-end sequencing cycles. The sequence data were deposited at the European Nucleotide Archive (accession number: PRJEB45902). The output reads from multiple lanes were combined into single files and quality control was performed with FastQC v0.11.7 and cleaned with fastp v0.19.5 to removed polyG trails and keep only full-length reads (Chen et al., 2018). The human genome GRCh38.p13 (ENSEMBL) was used to remap the reads using STAR v2.5.3a (Dobin et al., 2013). The differential expression between WT and KO was analyzed by R software (R Core Team, 2014 http://www.R-project.org/) using the DESeq2 package (Love et al., 2014).
qRT-PCR
The total RNA were purified as above, 1 μg RNA was subjected to reverse transcription by Quantitect RT kit (Qiagen). Quantitative PCR was performed using the KAPA SYBR Fast (Universal) qPCR kit (Merck Millipore) according to the manufacturer’s recommendations. The qRTPCR reaction was performed with a Rotor-GeneQ (Qiagen) and the same thermal profile conditions were set as 95°C for 10 min; then 40 cycles were performed of 10 s at 95°C, 20 s 60°C and 20 s 7°C. The following primer pairs were used: TNIP1 forward 5′-CTAGTGTGACGGCAGGTAAGG-3′, TNIP1 reverse 5′-GCTGCTTCATGGACCGGAA-3′; Actin forward 5′-GGACTTCGAGCAAGAGATGG-3′, Actin reverse 5′-AGCACTGTGTTGGCGTACAG-3′; HPRT1 forward 5′-TGACACTGGCAAAACAATGCA-3′, HPRT1 reverse 5′-GGTCCTTTTCACCAGCAAGCT-3′; 18s rRNA forward 5′-CGGCGACGACCCATTCGAAC-3′, 18s rRNA reverse 5′-GAATCGAACCCTGATTCCCCGTC-3′.
Gene set enrichment analyses
Gene set enrichment analyses (GSEA) was performed using default setting weighted enrichment statics and signal2noise metric for ranking genes (Subramanian et al., 2005). Significantly regulated genes in transcriptome data and proteins in whole proteome data were listed in Table S3.
Quantification and statistical analyses
Significantly regulated ubiquitination sites were determined by two samples paired t test, FDR < 0.05, S0 = 0.1. In all other cases unpaired, two-sided t tests were used unless stated otherwise. In general, parametric test were used, and data distribution was assumed to be normal, but this was not formally tested. The interactome network was generated by the String database. The statistical analyses including standard deviations, error bars, and P values were performed using Excel (Microsoft), all the details were indicated in figure legends.
Online supplemental material
Fig. S1 shows TNIP1 gets ubiquitinated and degraded in the lysosome. Fig. S2 shows TNIP1 localizes to autophagosomes. Fig. S3 shows endogenous TNIP1 interacts with TAX1BP1 and p62/SQSTM1 under basal conditions. Fig. S4 shows regulation of TNIP1 protein abundance. Table S1 shows UbiSite-proteomics approach to identify ubiquitination sites on proteins being potentially involved in autophagy. Table S2 shows HA-TNIP1 interactome. Table S3 shows RNA-Proteome correlation comparing WT and TNIP1 KO HeLa clones. Table S4 shows chemical reagents used in this study. Table S5 shows antibodies used in this study. Table S6 lists plasmids used in this study.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium through the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD027163.
The RNA sequence data were deposited at the European Nucleotide Archive (accession no. PRJEB45902).
Acknowledgments
The technical assistance of Aud Øvervatn is greatly appreciated. We thank Laurent Falquet for RNAseq data analysis and Michael Stumpe and Dieter Kressler for proteomics support. We are grateful to Richard J. Youle for the generous gift of the pentaKO cells. We thank the bioimaging core facility, Department of Medical Biology, UiT—The Arctic University of Norway for expert assistance.
This work was funded by grants from: FRIBIOMED (grant 214448) and TOPPFORSK (grant 249884) programs of the Research Council of Norway to T. Johansen; the Danish National Research Foundation (grant no. 141 to ATLAS) and the Independent Research Fund Denmark (8022-00051) to Blagoy Blagoev; the Swiss National Science Foundation to J. Dengjel (310030_184781) and F. Reggiori (CRSII5_189952), the Canton and University of Fribourg, and the Novartis Foundation for Medical-Biological Research to J. Dengjel. This work was part of the SKINTEGRITY.CH collaborative research project.
Author contributions: J. Dengjel, T. Johansen, N.L. Rasmussen, and J. Zhou designed the experiments. J. Zou and N.L. Rasmussen performed most of the experiments. G. Evjen performed GST pulldown experiments and analyzed data. D.S. Sankar, P. Verlhac, N. van de Beck, F. Reggiori, and Y.P. Abudu generated cell lines. S. Kaeser-Pebernard and C. Roubaty performed expression analyses. V. Akimov and B. Blagoev performed and analyzed Ubi-Site experiments. Z. Hu performed phosphoproteomics. H.L. Olsvik assisted in supervision performed in vitro mutagenesis and analyzed data. T. Lamark analyzed data and assisted in supervision. T. Johansen and J. Dengjel supervised and coordinated the overall research, analyzed the data, and approved the final manuscript. J. Zou, N.L. Rasmussen, H.L. Olsvik, T. Johansen, and J. Dengjel wrote the manuscript with input from all authors.

