


T cells require rapid proliferation to initiate adaptive immunity to prevent pathogen attacks. The nucleolus, a distinct subnuclear membrane-less compartment for ribosomal biogenesis, is indispensable for cell proliferation. However, specific nucleolar proteins involved in rapid T cell proliferation and their underlying molecular regulatory mechanism remain elusive. Here, we identified an essential nucleolar protein, DCAF13, in T cells and revealed its significant regulation of rapid T cell proliferation. Its depletion drastically impairs T cell proliferation due to severe 18S rRNA maturation failure, consequent abnormal ribosome assembly in nucleoli, and insufficient production of nascent proteins. Mechanistically, we propose that DCAF13 promotes NPM1 phase separation to accelerate pre-RNA enrichment and its endonuclease UTP23 for 18S rRNA maturation during T cell proliferation. Our findings reveal the modulatory effect of nucleolar NPM1/DCAF13 phase separation on ribosomal maturation to ensure rapid T cell proliferation and further pathogen clearance for the first time.
Introduction
T cell immune defense against pathogenic attacks relies on the rapid expansion of antigen-specific T cells to efficiently and thoroughly remove pathogen-infected host cells. Once stimulatory and pathogenic foreign antigens are encountered, naïve T cells, which rest in a “dormant” state with a slow protein synthesis rate (Chapman et al., 2020), are rapidly activated and enter the cell cycle to clonally expand, which further activates the transcription of various genes and accelerates the T cell ribosomal machinery, emergently providing an enormous amount of newly synthesized proteins in a short period. The ribosomal output ramps up more than 13-fold in 24 h compared with that in the naïve stage (Wolf et al., 2020). Thus, vigorous and rapid protein synthesis is essential for supporting T cell clonal expansion and proliferation upon antigen stimulation.
Previous studies on the regulation of T cell proliferation have mainly focused on gene transcriptional and protein posttranslational modifications, while the posttranscriptional facet has rarely been explored. Recently, increasing evidence has indicated that the rapidity and abundance of protein translation directly affect T cell proliferation and functional execution (Araki et al., 2017; Wolf et al., 2020). Integrative analysis of the whole proteome obtained from proliferating and clonally expanding T cells has shown that the most apparently upregulated genes mainly cluster in ribosome biogenesis, structural constituents of ribosomes, and nucleoli (Tan et al., 2017a), indicating the importance of ribosomes and related nucleolar functions in mediating T cell exit from the quiescent state. Specifically, protein translation capacity and cellular ribosome biogenesis are actively upregulated in response to T cell stimulation either by peptides loaded on antigen-presenting cells (APCs) or by the lymphocytic choriomeningitis virus (LCMV) pathologic antigen (Araki et al., 2017; Tan et al., 2017b). The nucleus, as the primary regulatory center for the transcription and processing of ribosomal RNA (rRNA), is essential for ribosome biogenesis and cell proliferation (Boisvert et al., 2007; Iarovaia et al., 2019). Thus, the identification of novel nucleolar proteins and their related molecular functions in regulating T cell proliferation is of great significance.
Moreover, the nucleolus is a well-known membrane-less subnuclear compartment with liquid-like physical properties located inside the nucleus (Brangwynne et al., 2011; Olson and Dundr, 2005). In addition to ribosomal DNA (rDNA) and rRNA, more than 700 species of ribosomal and nonribosomal accessory factors exist inside the nucleolus (Andersen et al., 2002; Jarboui et al., 2011). In recent years, with the revelation of nucleolar importance, increasing attention has been directed toward identifying nucleolar proteins and their corresponding functions in nucleoli. In addition to polymerases that mediate rRNA transcription and a few ribosomal proteins that compose ribosomes, fibrillarin (FIB) and nucleophosmin (NPM1) are essential proteins that have been identified as the main components required to build the dense fibrillar component (DFC) and granular component (GC) regions of nucleoli (Okuwaki, 2008; Shubina et al., 2016). In addition, large amounts of nucleolar proteins exist, and the functions of these proteins await clarification.
The nucleolus is an essential compartment for T cells and expands significantly coupled with T cell proliferation once naïve T cells encounter stimulants. Therefore, uncovering nucleolar regulation of T cell proliferation is an emerging and increasingly attractive field. Jarboui et al. conducted a nucleolar proteomic analysis on Jurkat T cells and identified 872 nucleolar proteins (Jarboui et al., 2011), which was the first study to unbiasedly explore the components of the T cell nucleolus. DDB1 and CUL4 Associated Factor 13 (DCAF13), as one of the identified proteins in the above nucleolar proteomic analysis, is a highly expressed nucleolar protein that shares 43% homology with yeast Sof1, which has been reported to physically associate with U3 snoRNA and Nop1p (a homolog of fibrillarin in yeast) and to be related to the biogenesis of the 40S ribosomal subunits in yeast (Bax et al., 2006). A similar phenomenon has also been observed in mammalian oocytes (Zhang et al., 2019a).
In this study, we found that DCAF13 was preferentially expressed in lymphoid organs, such as lymph nodes and the spleen, and significantly upregulated in response to T cell receptor (TCR) stimulation. DCAF13 depletion specifically in T cells impairs T cell homeostasis, proliferation, and functional execution, demonstrating that DCAF13 serves as an essential nucleolar protein for T cell survival and proliferation. We further revealed that DCAF13 promoted NPM1 phase separation to concentrate pre-RNA and its endonuclease UTP23, further facilitating 18S rRNA maturation for T cell ribosome maturation.
Results
Nucleolar DCAF13 is actively upregulated in response to T cell activation
The activation and proliferation of T cells are accompanied by the transcription and protein synthesis of numerous genes and morphological enlargement of T cell volume and nuclear size (Tan et al., 2017b; Wolf et al., 2020). To validate this hypothesis, we isolated naïve T cells (marked by CD25−CD62Lhi CD44low) from lymph nodes and stimulated them with anti-CD3/CD28 antibodies in vitro. When comparing forward scatter area (FSA) values and L-homopropargylglycine (HPG) signals of naïve T cells and activated T cells, which indicate cell volume and newly synthesized proteins, respectively, we observed that activated T cells showed greater FSA values and more HPG incorporation signals than naïve T cells (Fig. 1, A and B). Meanwhile, we observed that the nucleolar volume of both activated CD4+ and CD8+ T cells was significantly expanded and morphology-related by applying immunofluorescent 3D reconstruction to accurately calculate the nucleolar volume (Fig. 1 C). Moreover, their borders of nucleolar structures were found to be relatively well-defined, and the size of the nucleolus was larger than those in naïve T cells based on electron microscopy (Fig. 1 D).

Nucleolar protein DCAF13 is actively upregulated in response to T cell activation. (A) FSA of naïve and stimulated T cells detected by flow cytometry. (B) HPG signals of naïve and stimulated T cells detected by flow cytometry. (C) Confocal z-stack three-dimensional reconstruction of nucleoli (left) and corresponding quantitation of the nucleolar volume (right) are shown. Scale bar, 2 μm (CD4 T cells, n = 50; CD8 T cells, n = 20). (D) Representative electron microscopy images (upper) and the quantities of areas (down) of the nucleoli of naïve and stimulated CD4+ and CD8+ T cells. The white dotted line outlines the shape of the nucleus, and the white tip denotes obvious DFC regions (n = 15). (E) DCAF13 expression levels in different tissues were detected by Western blotting. (F) DCAF13 protein upregulation in response to anti-CD3/CD28 stimulation detected by Western blotting. (G) Immunofluorescence for fibrillarin and DCAF13 nucleolar colocalization in naïve and stimulated T cells (left). Inserts in the bottom left of the images showing 150% higher magnification of a representative cell (dashed boxed). Quantification of DCAF13 integrated density (IntDens) of both naïve and stimulated T cells (right) in CD4+ (n = 10) and CD8+ (n = 10) T cells are shown. Scale bar, 5 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F1.
Nucleolar protein DCAF13 is actively upregulated in response to T cell activation. (A) FSA of naïve and stimulated T cells detected by flow cytometry. (B) HPG signals of naïve and stimulated T cells detected by flow cytometry. (C) Confocal z-stack three-dimensional reconstruction of nucleoli (left) and corresponding quantitation of the nucleolar volume (right) are shown. Scale bar, 2 μm (CD4 T cells, n = 50; CD8 T cells, n = 20). (D) Representative electron microscopy images (upper) and the quantities of areas (down) of the nucleoli of naïve and stimulated CD4+ and CD8+ T cells. The white dotted line outlines the shape of the nucleus, and the white tip denotes obvious DFC regions (n = 15). (E) DCAF13 expression levels in different tissues were detected by Western blotting. (F) DCAF13 protein upregulation in response to anti-CD3/CD28 stimulation detected by Western blotting. (G) Immunofluorescence for fibrillarin and DCAF13 nucleolar colocalization in naïve and stimulated T cells (left). Inserts in the bottom left of the images showing 150% higher magnification of a representative cell (dashed boxed). Quantification of DCAF13 integrated density (IntDens) of both naïve and stimulated T cells (right) in CD4+ (n = 10) and CD8+ (n = 10) T cells are shown. Scale bar, 5 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F1.
To find vital nucleolar proteins for T cell activation and proliferation, we compared nucleolar protein proteomic profiling of Jurkat T cells (Jarboui et al., 2011) with the abundant proteins profiling of stimulated human T cells (Wolf et al., 2020) and found 58 potential nucleolar proteins increased in activated T cells. Among them, DCAF13 may be a promising candidate as a nucleolar protein not only because it responded to T cell activation but also because it was robustly synthesized under stimulation (Fig. S1, A and B), indicating that DCAF13 is potentially one of the vital nucleolar proteins for T cell activation.

Newly synthesized kinetics ranking of human T cell nucleolar proteins in response to stimulation. (A) Overlapped nucleolar protein proteomic profiling of Jurkat T cells (Jarboui et al., 2011) with the abundant proteins profiling of stimulated human T cells (https://www.immunomics.ch). There are 58 potential nucleolar proteins increased in T cells upon stimulation. Ranking by the translational kinetics (t50, protein renewal half time), the top 10 proteins in those 58 proteins were listed. (B) The translational kinetics downloaded from https://www.immunomics.ch about the 10 proteins listed in A.

Newly synthesized kinetics ranking of human T cell nucleolar proteins in response to stimulation. (A) Overlapped nucleolar protein proteomic profiling of Jurkat T cells (Jarboui et al., 2011) with the abundant proteins profiling of stimulated human T cells (https://www.immunomics.ch). There are 58 potential nucleolar proteins increased in T cells upon stimulation. Ranking by the translational kinetics (t50, protein renewal half time), the top 10 proteins in those 58 proteins were listed. (B) The translational kinetics downloaded from https://www.immunomics.ch about the 10 proteins listed in A.
We next sought to confirm whether DCAF13 existed in primary T cells. Indeed, we found that DCAF13 was abundant in multiple T cell-enriched immune organs, such as the thymus, spleen, and lymph nodes (Fig. 1 E). Immunofluorescence in primary T cells further showed that DCAF13 was co-expressed and colocalized with fibrillarin, a nucleolar marker mainly located in the DFC part, in the T cell nucleolus (Fig. 1 G). Moreover, DCAF13 expression was dramatically induced in both naïve CD4+ T cells and CD8+ T cells upon stimulation with anti-CD3/CD28 antibodies in vitro (Fig. 1, F and G), suggesting that nucleolar DCAF13 is a potential nucleolar protein and potentially indispensable for T cell proliferation.
DCAF13 is essential for immune cell survival
To clarify its functional role in T cells, we conditionally knocked out Dcaf13 in T cells by crossing Dcaf13fl/fl mice (Zhang et al., 2019a) with CD4 Cre+ transgenic mice to construct CD4 Cre+Dcaf13fl/fl mice. The knockout efficiency of DCAF13 was verified by Western blotting of peripheral mature T cells under CD4 Cre+ conditions (denoted as cKO below; Fig. S2 A). Flow cytometry analysis further showed that the proportions and cell numbers of double-negative (DN), double-positive (DP), CD4+ single-positive (CD4SP), and CD8+ single-positive (CD8SP) subpopulations were comparable between DCAF13 cKO mice and wild-type mice (Fig. S2, B and C). In peripheral lymphatic tissues, such as lymph nodes and spleens, the frequencies and amounts of CD4+ and CD8+ T cells were markedly reduced in DCAF13-knockout mice compared with wild-type litters (Fig. 2, A and B). Nonetheless, such a reduction does not disrupt the balance between naïve (CD62LhiCD44low) and effector (CD62LlowCD44hi) subpopulations (Fig. S2 D). Collectively, these results demonstrate that DCAF13 deletion mediated by CD4 Cre+ impairs peripheral T cell survival.
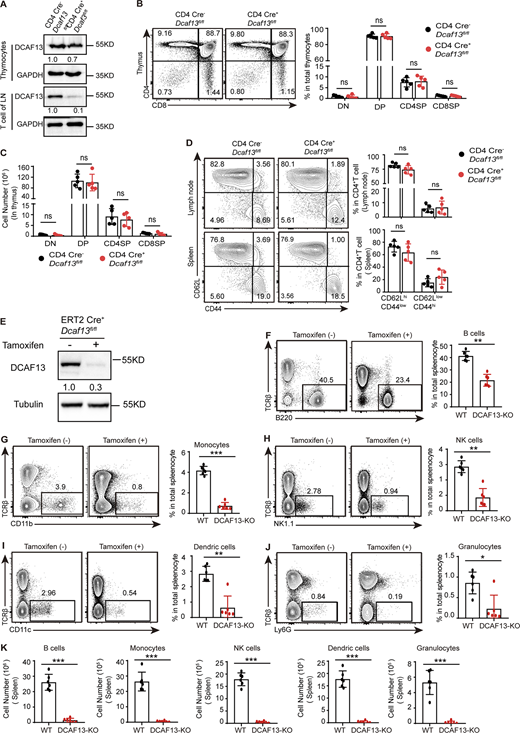
DCAF13 depletion also had a fatal effect on the survival and proliferation of other immune cell. (A) DCAF13 knockout efficiency in thymocytes and peripheral T cells was verified by Western blotting. (B) Representative flow cytometry pictures (left) and corresponding quantification (right) of the DN, DP, CD4SP, and CD8SP thymocyte subpopulations from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice (n = 3). (C) Quantitation of the cell number of DN, DP, CD4SP, and CD8SP subpopulations in the thymus (n = 3). (D) Flow cytometry analysis of the percentage of naïve CD4+ T cells (CD62Lhi CD44low) and effector CD4+ T cells (CD62LlowCD44hi) in lymph nodes and spleens. Representative flow cytometry pictures (left) and corresponding quantification (right) (n = 3). (E) DCAF13 knockdown efficiency in ERT2 Cre+Dcaf13fl/fl T cells with tamoxifen treatment was verified by Western blotting. The gray values of bands were quantified with ImageJ. (F–K) Percentages and the cell number of B cells, monocytes, NK cells, dendric cells, and granulocytes. 4-OH Tam(−) referred to WT, 4-OH Tam(+) referred to DCAF13-KO. n = 6. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001. ****P < 0.0001. Source data are available for this figure: SourceData FS2.

DCAF13 depletion also had a fatal effect on the survival and proliferation of other immune cell. (A) DCAF13 knockout efficiency in thymocytes and peripheral T cells was verified by Western blotting. (B) Representative flow cytometry pictures (left) and corresponding quantification (right) of the DN, DP, CD4SP, and CD8SP thymocyte subpopulations from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice (n = 3). (C) Quantitation of the cell number of DN, DP, CD4SP, and CD8SP subpopulations in the thymus (n = 3). (D) Flow cytometry analysis of the percentage of naïve CD4+ T cells (CD62Lhi CD44low) and effector CD4+ T cells (CD62LlowCD44hi) in lymph nodes and spleens. Representative flow cytometry pictures (left) and corresponding quantification (right) (n = 3). (E) DCAF13 knockdown efficiency in ERT2 Cre+Dcaf13fl/fl T cells with tamoxifen treatment was verified by Western blotting. The gray values of bands were quantified with ImageJ. (F–K) Percentages and the cell number of B cells, monocytes, NK cells, dendric cells, and granulocytes. 4-OH Tam(−) referred to WT, 4-OH Tam(+) referred to DCAF13-KO. n = 6. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001. ****P < 0.0001. Source data are available for this figure: SourceData FS2.

DCAF13 is required for peripheral T cell survival. (A) Flow cytometry analysis of the percentages of CD4+ and CD8+ T cell subsets in lymph nodes and spleens from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice (left) and the corresponding percentage quantitation (right) (n = 5) are shown. (B) Quantitation of the cell numbers of CD4+ and CD8+ T cell subsets in lymph nodes and spleens, as total splenocytes or lymphocytes multiplied by the percentage of the corresponding subset found in the total population (n = 5). (C) Flow cytometry analysis of bone marrow transplantation reconstituted in CD45.1+-irradiated B6 wild-type mice. The bone marrow cells injected into the host mice were composed of CD45.1+ B6 (WT) bone marrow cells mixed with CD45.2+ CD4 Cre−Dcaf13fl/fl (WT) or CD4 Cre+Dcaf13fl/fl (cKO) bone marrow cells. CD45.1+ cells served as internal controls. The data shown are the percentages of CD45.2+ (DCAF13 cKO or WT) subsets gated on CD4+ or CD8+ T cell populations in lymph nodes and spleens from the host mice after 8 wk. Representative flow cytometry pictures (left) and corresponding quantitation (right; WT, n = 4; cKO, n = 6) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DCAF13 is required for peripheral T cell survival. (A) Flow cytometry analysis of the percentages of CD4+ and CD8+ T cell subsets in lymph nodes and spleens from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice (left) and the corresponding percentage quantitation (right) (n = 5) are shown. (B) Quantitation of the cell numbers of CD4+ and CD8+ T cell subsets in lymph nodes and spleens, as total splenocytes or lymphocytes multiplied by the percentage of the corresponding subset found in the total population (n = 5). (C) Flow cytometry analysis of bone marrow transplantation reconstituted in CD45.1+-irradiated B6 wild-type mice. The bone marrow cells injected into the host mice were composed of CD45.1+ B6 (WT) bone marrow cells mixed with CD45.2+ CD4 Cre−Dcaf13fl/fl (WT) or CD4 Cre+Dcaf13fl/fl (cKO) bone marrow cells. CD45.1+ cells served as internal controls. The data shown are the percentages of CD45.2+ (DCAF13 cKO or WT) subsets gated on CD4+ or CD8+ T cell populations in lymph nodes and spleens from the host mice after 8 wk. Representative flow cytometry pictures (left) and corresponding quantitation (right; WT, n = 4; cKO, n = 6) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test whether the defects in peripheral T cell survival in DCAF13 cKO mice are intrinsic to T cells, we established irradiated CD45.1+ bone marrow chimeric mice by injecting wild-type CD45.1+ bone marrow cells mixed with CD45.2+ bone marrow cells from either CD4 Cre+Dcaf13fl/fl or CD4 Cre−Dcaf13fl/fl mice. We found that the proportions of CD4 Cre+Dcaf13fl/fl T cells in both lymph nodes and spleens were markedly lower than those of CD4 Cre−Dcaf13fl/fl cells, which was especially evident for CD8+ T cells (Fig. 2 C), suggesting that DCAF13 supports peripheral T cell maintenance in vivo in a T cell–intrinsic manner.
Next, to demonstrate whether the effects above can be found in other kinds of immune cells, we generated ERT2 Cre+Dcaf13fl/fl mice by crossing Dcaf13fl/fl mice with tamoxifen-inducible Cre (ERT2 Cre) mice to delete DCAF13 in other cells. We then treated ERT2 Cre+Dcaf13fl/fl mice with tamoxifen to delete DCAF13 and analyzed some other cell populations in the spleen, including B cells, monocytes, NK cells, dendric cells, and granulocytes. We found that the decline of both cell numbers and percentages can be found in all these cells (Fig. S2, E–K). These results suggested that DCAF13 is essential for immune cell survival, including T cells.
The absence of DCAF13 leads to severe impairment in T cell proliferation
Since TCR signaling plays a critical role in maintaining peripheral T cells, we proceeded to examine whether this pathway is also affected due to DCAF13 deficiency. Thus, we analyzed OT1-TG CD4 Cre+Dcaf13fl/fl mice that express MHC class I-restricted TCRs and found that a marked reduction still occurred in peripheral CD8+ T cells (Fig. 3 B) with no obvious defect in thymic T cell development (Fig. 3 A). Additionally, DCAF13-deleted T cells showed comparable increases in the phosphorylation levels of TCR signaling-related proteins (e.g., PLC-γ, ERK, and AKT) and the expression levels of T cell activating factors (e.g., CD69 and CD25) upon stimulation in vitro (Fig. 3, C–G). Collectively, the dramatic absence of peripheral T cells resulting from DCAF13 depletion does not cause a TCR signaling defect.

DCAF13-deficient T cells showed normal TCR response and activation. (A) Representative flow cytometry pictures (upper) and corresponding quantification (bottom) of the DN, DP, CD4SP, and CD8SP thymocyte subpopulations from OT1-TG CD4 Cre−Dcaf13fl/fl (WT) and OT1-TG CD4 Cre+Dcaf13fl/fl (cKO; WT, n = 6; cKO, n = 3). (B) Representative flow cytometry pictures (left) of the percentage of CD4+ and CD8+ T cell subsets in lymph nodes and spleen from OT1-TG CD4 Cre−Dcaf13fl/fl (WT) and OT1-TG CD4 Cre+Dcaf13fl/fl (cKO) and corresponding quantitation (right; WT, n = 6; cKO, n = 3). (C) The phosphorylation level of PLC-γ, AKT, and ERK in T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice after stimulation with anti-CD3/CD28. Quantification of phosphorylation level in total targeted proteins with ImageJ. (D and E) Flow cytometry analysis of CD69 (D) and CD25 (E) in naïve CD4+ T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under indicated anti-CD3/CD28 stimulation for 4 h. Representative flow cytometry pictures (left) and corresponding quantitation (right; n = 3) are shown. (F and G) Flow cytometry analysis of CD69 (F) and CD25 (G) in naïve CD8+ T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under indicated anti-CD3/CD28 stimulation for 4 h. Representative flow cytometry pictures (left) and corresponding quantitation (right) (n = 3) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F3.
DCAF13-deficient T cells showed normal TCR response and activation. (A) Representative flow cytometry pictures (upper) and corresponding quantification (bottom) of the DN, DP, CD4SP, and CD8SP thymocyte subpopulations from OT1-TG CD4 Cre−Dcaf13fl/fl (WT) and OT1-TG CD4 Cre+Dcaf13fl/fl (cKO; WT, n = 6; cKO, n = 3). (B) Representative flow cytometry pictures (left) of the percentage of CD4+ and CD8+ T cell subsets in lymph nodes and spleen from OT1-TG CD4 Cre−Dcaf13fl/fl (WT) and OT1-TG CD4 Cre+Dcaf13fl/fl (cKO) and corresponding quantitation (right; WT, n = 6; cKO, n = 3). (C) The phosphorylation level of PLC-γ, AKT, and ERK in T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice after stimulation with anti-CD3/CD28. Quantification of phosphorylation level in total targeted proteins with ImageJ. (D and E) Flow cytometry analysis of CD69 (D) and CD25 (E) in naïve CD4+ T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under indicated anti-CD3/CD28 stimulation for 4 h. Representative flow cytometry pictures (left) and corresponding quantitation (right; n = 3) are shown. (F and G) Flow cytometry analysis of CD69 (F) and CD25 (G) in naïve CD8+ T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under indicated anti-CD3/CD28 stimulation for 4 h. Representative flow cytometry pictures (left) and corresponding quantitation (right) (n = 3) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F3.
Another main factor determining the scale of the peripheral T cell pool, cell apoptosis, was detected by Annexin-V staining prior to cell proliferation. Strikingly, we found no significant differences in the percentages of Annexin-V+ T cells in thymic DN, DP, CD4SP, and CD8SP T cell subpopulations or peripheral T cells from lymph nodes and spleens, regardless of whether DCAF13 was depleted (data not shown).
We next conducted a CellTrace assay to evaluate the proliferation ability of naïve T cells upon stimulation in vitro. DCAF13-deleted T cells specifically presented no CFSE-shifted signaling (Fig. 4 A) or BrdU incorporation (Fig. 4 B), indicating that DCAF13 depletion impairs the proliferation potential and DNA replication of T cells upon stimulation. To further verify this proliferation defect in vivo, we performed T cell homeostatic proliferation by intravenously injecting a T cell mixture (CD45.1+ wild-type naïve T cells mixed with CD45.2+ CD4 Cre−Dcaf13fl/fl or CD4 Cre+Dcaf13fl/fl naïve T cells as the control and experimental groups, respectively) into 6-Gy pre-irradiated CD45.1+ recipient mice. We analyzed the mice 4 d after transfer and found no CFSE-shifted signaling in DCAF13-deleted T cells, indicating that DCAF13-deleted T cells have defects on proliferation (Fig. 4 C), which further confirms in vivo that T cells with DCAF13 deletion failed to proliferate.

DCAF13 deficiency impaired T cell proliferation. (A) CFSE signals were detected by flow cytometry to evaluate the proliferative capacity of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under anti-CD3/CD28 stimulation. (B) BrdU incorporation was detected by flow cytometry to evaluate DNA replication of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under anti-CD3/CD28 stimulation. (C) Flow cytometry analysis of T cell homeostatic proliferation in vivo reconstituted in CD45.1+mice. Naïve T cells isolated from CD45.2+ CD4 Cre−Dcaf13fl/fl or CD4 Cre+Dcaf13fl/fl were labeled with CellTrace Violet and then transferred into CD45.1+ mice as the control and experimental groups, respectively. 4 d after transfer, CellTrace dilution in donor cells from the recipients’ lymph nodes was analyzed by flow cytometry. Representative flow cytometry pictures (left) and corresponding quantitation of the percentage of Celltracelo cells (right; n = 6) are shown. (D) CFSE signals were detected by flow cytometry to evaluate the proliferative capacity of naïve T cells from ERT2 Cre+Dcaf13fl/fl cells under anti-CD3/CD28 stimulation. Tamoxifen-treated T cells were regarded as DCAF13 cKO cells, and those without tamoxifen were regarded as WT controls. The percentage of individual peaks was quantified by FlowJo V10 software. (E) BrdU incorporation was detected by flow cytometry to evaluate DNA replication of naïve T cells from ERT2 Cre+Dcaf13fl/fl mice under anti-CD3/CD28 stimulation. Tamoxifen-treated T cells were regarded as DCAF13 cKO cells, and those without tamoxifen were regarded as WT controls (n = 6). (F) Cell cycle analysis of CD4+ T and CD8+ T cells from ERT2 Cre+Dcaf13fl/fl mice based on the DAPI content. The percentage of individual phase was quantified by FlowJo V10 software. Representative flow cytometry pictures (left) and corresponding quantitation (right; n = 6) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DCAF13 deficiency impaired T cell proliferation. (A) CFSE signals were detected by flow cytometry to evaluate the proliferative capacity of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under anti-CD3/CD28 stimulation. (B) BrdU incorporation was detected by flow cytometry to evaluate DNA replication of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice under anti-CD3/CD28 stimulation. (C) Flow cytometry analysis of T cell homeostatic proliferation in vivo reconstituted in CD45.1+mice. Naïve T cells isolated from CD45.2+ CD4 Cre−Dcaf13fl/fl or CD4 Cre+Dcaf13fl/fl were labeled with CellTrace Violet and then transferred into CD45.1+ mice as the control and experimental groups, respectively. 4 d after transfer, CellTrace dilution in donor cells from the recipients’ lymph nodes was analyzed by flow cytometry. Representative flow cytometry pictures (left) and corresponding quantitation of the percentage of Celltracelo cells (right; n = 6) are shown. (D) CFSE signals were detected by flow cytometry to evaluate the proliferative capacity of naïve T cells from ERT2 Cre+Dcaf13fl/fl cells under anti-CD3/CD28 stimulation. Tamoxifen-treated T cells were regarded as DCAF13 cKO cells, and those without tamoxifen were regarded as WT controls. The percentage of individual peaks was quantified by FlowJo V10 software. (E) BrdU incorporation was detected by flow cytometry to evaluate DNA replication of naïve T cells from ERT2 Cre+Dcaf13fl/fl mice under anti-CD3/CD28 stimulation. Tamoxifen-treated T cells were regarded as DCAF13 cKO cells, and those without tamoxifen were regarded as WT controls (n = 6). (F) Cell cycle analysis of CD4+ T and CD8+ T cells from ERT2 Cre+Dcaf13fl/fl mice based on the DAPI content. The percentage of individual phase was quantified by FlowJo V10 software. Representative flow cytometry pictures (left) and corresponding quantitation (right; n = 6) are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To further prove this investigation, we treated ERT2 Cre+Dcaf13fl/fl mice with tamoxifen to delete DCAF13 at a designated time. Compared with ERT2 Cre+Dcaf13fl/fl T cells without tamoxifen treatment, the CFSE left-shift signal and BrdU-positive ratio in the tamoxifen-treated T cells were significantly reduced (Fig. 4, D and E), further supporting the extremely essential role of DCAF13 in T cell proliferation. Moreover, DCAF13 deficiency led T cells to accumulate in the G0 phase and to decrease in the S phase and G2/M phase (Fig. 4 F).
DCAF13 deficiency impairs 18S rRNA maturation to affect the ribosomal biogenesis of activated T cells
Remarkably, we observed a compacted nucleolar morphology in DCAF13-deleted T cells by comparing with those in wild-type T cells, whose area was significantly increased upon stimulation in vitro by electron microscopy (Fig. S3 A). Then, we calculated their nucleolar volume by reconstructing the 3D version and found that stimulated DCAF13-deleted T cells presented a much smaller volume than stimulated wild-type T cells, although no difference was noted when at naïve status. The nucleolar morphology of activated DCAF13-deleted T cells was similar to that of naïve T cells, unlike the fused morphology of stimulated wild-type T cells, suggesting that the nucleoli of DCAF13-deleted T cells failed to respond to the stimuli (Fig. 5 A). Moreover, unlike the multiple nucleoli observed in wild-type naïve T cells, only one nucleolus was often observed in DCAF13-deleted T cells (Fig. 5 B), indicating that DCAF13 may affect nucleolus formation.
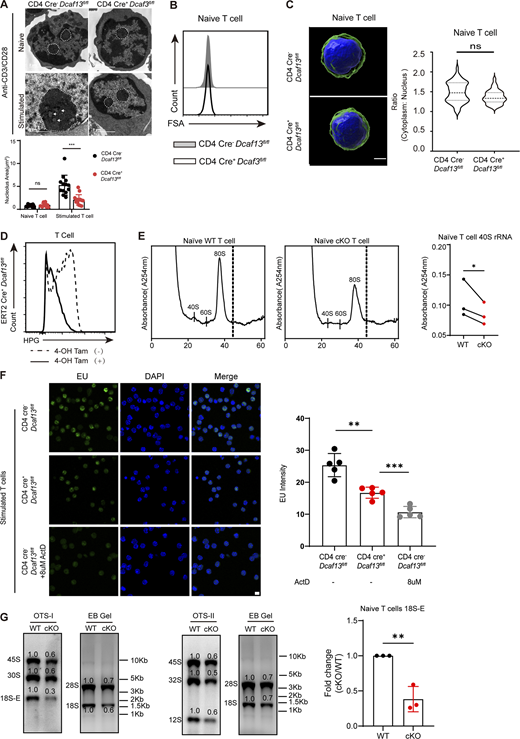
DCAF13 depletion exerted slighter effects on rRNA transcription rather than protein synthesis. (A) Nucleolar morphology of naïve or stimulated CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells detected by electron microscopy. Representative images (upper) and the quantities of areas (right) are shown. The white dotted line outlined the shape of the nucleus and the white tip marked obvious DFC regions (n = 13). (B) FSA of naïve CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells. (C) Confocal z-stack three-dimensional reconstruction of naïve CD4 T cells was isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) to compare their ratios of cytoplasm and nucleus. Representative images (left) and corresponding quantitation from three independent experiments (n = 36) are shown. DAPI (blue) and CD4 (green). Scale bar, 2 μm. (D) Flow cytometry analysis of HPG signals in ERT2 Cre+Dcaf13fl/fl CD4+ and CD8+ T cells treated with tamoxifen (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). T cells were stimulated with anti-CD3/CD28 for 24 h. (E) Polysome profiles analysis of the equal amounts of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO). Representative pictures of A254 absorbance profile in three independent experiments(left) and quantification of the 40S peaks (right, n = 3) are shown. (F) IF of EU signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated by anti-CD3/CD28 for 24 h. Actinomycin D (ActD) was added as an EU inhibitor. The gray value of the EU signal was measured by ImageJ (n = 5). (G) Ribosomal RNA processing products in naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice were detected by northern blotting with OTSI and OTSII oligos. Loaded RNA was purified from equal amounts of cells. The gray values of bands were quantified with ImageJ. WT cells were normalized to 1.0. The fold change of 18S-E was summarized from three independent experiments (n = 3). Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001. Source data are available for this figure: SourceData FS3.

DCAF13 depletion exerted slighter effects on rRNA transcription rather than protein synthesis. (A) Nucleolar morphology of naïve or stimulated CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells detected by electron microscopy. Representative images (upper) and the quantities of areas (right) are shown. The white dotted line outlined the shape of the nucleus and the white tip marked obvious DFC regions (n = 13). (B) FSA of naïve CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells. (C) Confocal z-stack three-dimensional reconstruction of naïve CD4 T cells was isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) to compare their ratios of cytoplasm and nucleus. Representative images (left) and corresponding quantitation from three independent experiments (n = 36) are shown. DAPI (blue) and CD4 (green). Scale bar, 2 μm. (D) Flow cytometry analysis of HPG signals in ERT2 Cre+Dcaf13fl/fl CD4+ and CD8+ T cells treated with tamoxifen (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). T cells were stimulated with anti-CD3/CD28 for 24 h. (E) Polysome profiles analysis of the equal amounts of naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO). Representative pictures of A254 absorbance profile in three independent experiments(left) and quantification of the 40S peaks (right, n = 3) are shown. (F) IF of EU signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated by anti-CD3/CD28 for 24 h. Actinomycin D (ActD) was added as an EU inhibitor. The gray value of the EU signal was measured by ImageJ (n = 5). (G) Ribosomal RNA processing products in naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice were detected by northern blotting with OTSI and OTSII oligos. Loaded RNA was purified from equal amounts of cells. The gray values of bands were quantified with ImageJ. WT cells were normalized to 1.0. The fold change of 18S-E was summarized from three independent experiments (n = 3). Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001. Source data are available for this figure: SourceData FS3.

DCAF13-deficient T cells showed impaired new protein synthesis caused by abnormal ribosomal maturation. (A) Confocal z-stack three-dimensional reconstruction of nucleoli from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells to compare nucleolar sizes. Representative images (left) and corresponding quantitation of the nucleolar volume (right) from three independent experiments (naïve T cells, n = 60; stimulated T cells, n = 50). DAPI (blue) and NPM1 (cyan) are shown. Scale bar, 2 μm. (B) Quantitation of the nucleolar number in naïve or stimulated CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells from three independent experiments (naïve T cells, n = 60; stimulated T cells, n = 50). (C) FSAs of CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. (D) Confocal z-stack three-dimensional reconstruction of stimulated T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) cells to compare their ratios of cytoplasm and nuclei. Representative images (left) and corresponding quantitation from three independent experiments (n = 45) are shown. DAPI (blue) and CD4 (green). Scale bar, 2 μm. (E) Flow cytometry analysis of HPG signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. (F) Immunofluorescence staining of HPG signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. Representative pictures (left) and quantification of the HPG intensity (right; WT, n = 6; cKO, n = 3) are shown. Scale bar, 10 μm. (G) Polysome profile analysis of stimulated T cells with anti-CD3/CD28 treatment for 72 h. These cells were isolated from ERT2 Cre+Dcaf13fl/fl T cells and treated with tamoxifen (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). Cells were loaded in equal amounts. Representative pictures of the A254 absorbance profile in three independent experiments (left) and quantification of the 40S peaks (right, n = 3) are shown. (H) Ribosomal RNA processing products in stimulated T cells were detected by northern blotting with OTSI and OTSII oligos. Loaded RNA was purified from the same number of cells. The gray values of bands were quantified with ImageJ. WT cells were normalized to 1.0. The fold changes of 45S, 30S, and 18S-E rRNA were summarized from three independent experiments (n = 3). Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F5.
DCAF13-deficient T cells showed impaired new protein synthesis caused by abnormal ribosomal maturation. (A) Confocal z-stack three-dimensional reconstruction of nucleoli from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells to compare nucleolar sizes. Representative images (left) and corresponding quantitation of the nucleolar volume (right) from three independent experiments (naïve T cells, n = 60; stimulated T cells, n = 50). DAPI (blue) and NPM1 (cyan) are shown. Scale bar, 2 μm. (B) Quantitation of the nucleolar number in naïve or stimulated CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells from three independent experiments (naïve T cells, n = 60; stimulated T cells, n = 50). (C) FSAs of CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. (D) Confocal z-stack three-dimensional reconstruction of stimulated T cells isolated from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) cells to compare their ratios of cytoplasm and nuclei. Representative images (left) and corresponding quantitation from three independent experiments (n = 45) are shown. DAPI (blue) and CD4 (green). Scale bar, 2 μm. (E) Flow cytometry analysis of HPG signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. (F) Immunofluorescence staining of HPG signals in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells stimulated with anti-CD3/CD28 for 24 h. Representative pictures (left) and quantification of the HPG intensity (right; WT, n = 6; cKO, n = 3) are shown. Scale bar, 10 μm. (G) Polysome profile analysis of stimulated T cells with anti-CD3/CD28 treatment for 72 h. These cells were isolated from ERT2 Cre+Dcaf13fl/fl T cells and treated with tamoxifen (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). Cells were loaded in equal amounts. Representative pictures of the A254 absorbance profile in three independent experiments (left) and quantification of the 40S peaks (right, n = 3) are shown. (H) Ribosomal RNA processing products in stimulated T cells were detected by northern blotting with OTSI and OTSII oligos. Loaded RNA was purified from the same number of cells. The gray values of bands were quantified with ImageJ. WT cells were normalized to 1.0. The fold changes of 45S, 30S, and 18S-E rRNA were summarized from three independent experiments (n = 3). Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F5.
Given the importance of nucleoli for protein synthesis and the high demand for protein synthesis for T cell growth and proliferation (Haneke et al., 2020; Polymenis and Aramayo, 2015), smaller FSA values and cytoplasm–nucleus ratios in stimulated DCAF13-deleted T cells than in stimulated wild-type T cells were observed, as expected, while no difference was observed between naïve wild-type and DCAF13-deleted T cells (Fig. 5, C and D; and Fig. S3, B and C). Through the HPG incorporation assay, we hardly observed an HPG peak in DCAF13-deleted T cells compared with the obvious HPG incorporation in CD4 Cre−Dcaf13fl/fl T cells (Fig. 5 E). This disappearance of HPG signaling in DCAF13-deleted T cells directly demonstrated their inability to produce proteins, which was further validated by fluorescence imaging (Fig. 5 F). Additionally, a decrease in HPG was also observed in induced DCAF13-deleted T cells isolated from ERT2 Cre+Dcaf13fl/fl mice (Fig. S3 D). The results indicated that DCAF13 deficiency might affect activated T cell proliferation by impairing the protein supply.
Recent studies have reported that ribosome biogenesis is essential to sustain T cell proliferation upon stimulation (Araki et al., 2017; Tan et al., 2017b). Therefore, we next decided to perform polysome profiling analysis in ERT2 Cre+Dcaf13fl/fl T cells with or without tamoxifen treatment to explore the role of DCAF13 in stimulated T cell ribosome status, including ribosome constitution and polysome formation during the translation process. We found a greater decrease in the peak of 40S (Fig. 5 G), which is the small subunit of the ribosome (RPS) and is composed of 18S rRNA and various ribosomal proteins (Klinge and Woolford, 2019; Pelletier et al., 2018). Inconsistent with stimulated T cells, similar 40S decreases were shown between the naïve DCAF13-deleted T cells and wild-type T cells, with obvious 80S peaks and weak 40S and 60S peaks (Fig. S3 E), suggesting that naïve T cells contain idle ribosomes that may deal with the short-term protein demand following rapidly activation, which is consistent with previous findings (Wolf et al., 2020).
Considering that the nucleolus plays an essential role in ribosome biogenesis (Hernandez-Verdun et al., 2010), we postulated that DCAF13 deficiency might impair ribosome biogenesis in nucleoli and sequentially result in protein synthesis collapse. Northern blotting of active T cells showed aberrant accumulation of 30S pre-rRNA and a significant reduction in its sequential 18S-E pre-rRNA (Fig. 5 H). This low efficiency or even inability to perform 18S-E pre-rRNA processing results in 40S ribosomal subunit reduction, which was consistent with those reported previously in early embryonic development (Zhang et al., 2018). We also applied northern blotting to assess naïve T cells from CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) mice and observed similar RNA processing patterns along with reduced rRNA transcription (Fig. S3 G), Although a slight decrease in nucleolar RNA transcription was evident in DCAF13-deleted T cells based on 45S signals in northern blotting and EU signals (Fig. 5 H; and Fig. S3, F and G), which is an analog of uridine that incorporates into nascent RNA, the total loss of nascent proteins and severe reduction in 40S subunit suggest that DCAF13 depletion mainly contributes to the abnormality of 18S rRNA maturation rather than transcription. Together, the results proposed that nucleolar DCAF13 is required for T cell ribosome biogenesis by affecting 18S rRNA maturation to meet the T cell proliferative demand of newly synthesized proteins.
DCAF13 interacts with NPM1 through its C-terminal Sof domain
To clarify the underlying molecular mechanism by which DCAF13 affects activated T cell ribosome biogenesis, we decided to systematically outline potential DCAF13 interacting partners in EL4 cells (a typical murine T lymphocyte cell line) through coimmunoprecipitation (Co-IP) coupled with mass spectrometry. We aimed to reveal the underlying molecular mechanism by which DCAF13 affects activated T cell ribosome biogenesis. With Gene Ontology (GO) analysis, we clarified that DCAF13 interactomes were enriched as RNA-binding proteins and ribosomal proteins, which mainly contribute to the structural constituents of ribosomes and ribosome biogenesis (Fig. 6 A). The biased enrichment of ribosomal proteins in the small subunit (RPS; Fig. S4 A) implies the potential specificity of DCAF13 for the 40S small subunit of ribosomes, which is consistent with the above northern blotting results (Fig. 5 H). Remarkably, NPM1 was found to be one of the most specifically enriched nucleolar proteins by DCAF13 (Fig. S4 A). The interaction between DCAF13 and NPM1 was further confirmed by reciprocal co-IP (Fig. 6, C and D), and their colocalization in the nucleolar region was observed by immunofluorescence (Fig. 6 B). Since NPM1 is mainly located in the GC region, the immunofluorescence confirmed that DCAF13 is not only located in the DFC region (shown in Fig 1 G) but is also located in the GC region. Moreover, we ruled out the possibility of RNA-mediated DCAF13/NPM1 binding by co-IP combined with RNaseA treatment (Fig. 6 E).

Mass spectrometry identified DCAF13-associated proteins. (A) GO clusters of DCAF13 interacting proteins analyzed in the Database for Annotation, Visualization and Integrated Discovery (DAVID: https://david.ncifcrf.gov/). (B) Immunofluorescence for NPM1 and DCAF13 nucleolar colocalization in naïve and stimulated T cells. Inserts in the bottom left of the images showing 150% higher magnification of a representative cell (dashed boxed). Scale bar, 5 μm. (C) Co-IP of Flag-DCAF13 to confirm its interaction with myc-NPM1 in 293T cells. (D) Co-IP of Flag-NPM1 to confirm its interaction with DCAF13 in 293T cells. (E) Co-IP of Flag-DCAF13 with RNaseA treatment to confirm its interaction with NPM1 in an RNase A-insensitive manner. (F) The strategy of DCAF13 mutations and Co-IP of these mutations is to map the interacting site with NPM1. (G) The rescue of proliferation defects in CD8 T cells caused by DCAF13 deletion by ectopically expressing DCAF13-FL and DCAF13(1–417) mutations in tamoxifen-treated ERT2 Cre+Dcaf13fl/fl T cells (regarded as DCAF13 KO). The percentage of individual peaks was quantified by FlowJo V10 software. Source data are available for this figure: SourceData F6.
Mass spectrometry identified DCAF13-associated proteins. (A) GO clusters of DCAF13 interacting proteins analyzed in the Database for Annotation, Visualization and Integrated Discovery (DAVID: https://david.ncifcrf.gov/). (B) Immunofluorescence for NPM1 and DCAF13 nucleolar colocalization in naïve and stimulated T cells. Inserts in the bottom left of the images showing 150% higher magnification of a representative cell (dashed boxed). Scale bar, 5 μm. (C) Co-IP of Flag-DCAF13 to confirm its interaction with myc-NPM1 in 293T cells. (D) Co-IP of Flag-NPM1 to confirm its interaction with DCAF13 in 293T cells. (E) Co-IP of Flag-DCAF13 with RNaseA treatment to confirm its interaction with NPM1 in an RNase A-insensitive manner. (F) The strategy of DCAF13 mutations and Co-IP of these mutations is to map the interacting site with NPM1. (G) The rescue of proliferation defects in CD8 T cells caused by DCAF13 deletion by ectopically expressing DCAF13-FL and DCAF13(1–417) mutations in tamoxifen-treated ERT2 Cre+Dcaf13fl/fl T cells (regarded as DCAF13 KO). The percentage of individual peaks was quantified by FlowJo V10 software. Source data are available for this figure: SourceData F6.
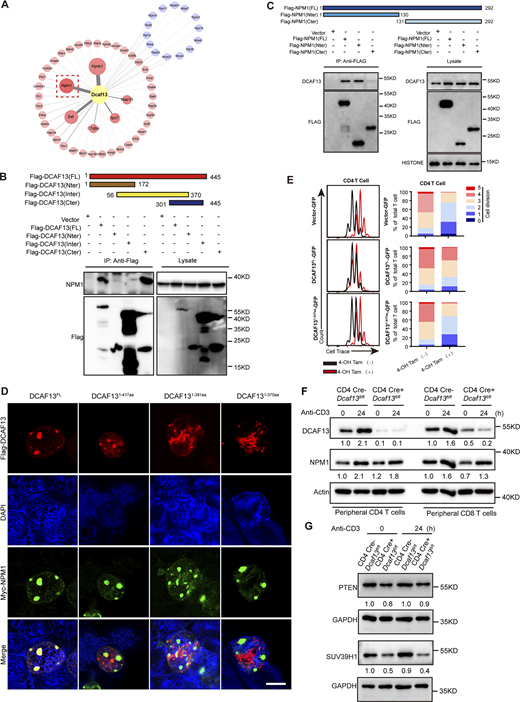
The Sof domain in the DCAF13 C-terminal was essential for its interaction with NPM1. (A) Network of DCAF13-interacting proteins in EL-4 T cells identified by LC–MS/MS. Subgrouping into ribosomal proteins (blue) and other proteins (red). The sizes of bubbles correspond to fold enrichment. A red dashed box is used to highlight the location of NPM1. (B) Co-IP of Flag-DCAF13-FL, Flag-DCAF13-Nter, Flag-DCAF13-Inter, and Flag-DCAF13-Cter to roughly map out its C-terminal charging the interaction with NPM1. (C and D) Co-IP of Flag-NPM1-FL, Flag-NPM1-Nter, and Flag-NPM1-Cter to roughly map out its N-terminal charging the interaction with DCAF13 (down; D). IF for cellular colocalization between Flag-DCAF13-FL, Flag-DCAF13-1-417aa, Flag-DCAF13-1-391aa, and Flag-DCAF13-1-370aa with NPM1. Scale bar, 10 μm. (E) The rescue of proliferation defect in CD4 T cells caused by DCAF13 deletion by ectopic expressing DCAF13-FL and DCAF13(1–417) mutations in tamoxifen-treated ERT2 Cre+Dcaf13fl/fl T cells (regarded as DCAF13 KO). The percentages of individual peaks were quantified by FlowJo V10 software. (F) NPM1 protein expression levels in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) CD4+or CD8+ T cells. The gray values of bands were quantified with ImageJ. (G) PTEN and SUV39H1 protein expression levels in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells. The gray values of bands were quantified with ImageJ. Source data are available for this figure: SourceData FS4.

The Sof domain in the DCAF13 C-terminal was essential for its interaction with NPM1. (A) Network of DCAF13-interacting proteins in EL-4 T cells identified by LC–MS/MS. Subgrouping into ribosomal proteins (blue) and other proteins (red). The sizes of bubbles correspond to fold enrichment. A red dashed box is used to highlight the location of NPM1. (B) Co-IP of Flag-DCAF13-FL, Flag-DCAF13-Nter, Flag-DCAF13-Inter, and Flag-DCAF13-Cter to roughly map out its C-terminal charging the interaction with NPM1. (C and D) Co-IP of Flag-NPM1-FL, Flag-NPM1-Nter, and Flag-NPM1-Cter to roughly map out its N-terminal charging the interaction with DCAF13 (down; D). IF for cellular colocalization between Flag-DCAF13-FL, Flag-DCAF13-1-417aa, Flag-DCAF13-1-391aa, and Flag-DCAF13-1-370aa with NPM1. Scale bar, 10 μm. (E) The rescue of proliferation defect in CD4 T cells caused by DCAF13 deletion by ectopic expressing DCAF13-FL and DCAF13(1–417) mutations in tamoxifen-treated ERT2 Cre+Dcaf13fl/fl T cells (regarded as DCAF13 KO). The percentages of individual peaks were quantified by FlowJo V10 software. (F) NPM1 protein expression levels in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) CD4+or CD8+ T cells. The gray values of bands were quantified with ImageJ. (G) PTEN and SUV39H1 protein expression levels in CD4 Cre−Dcaf13fl/fl (WT) and CD4 Cre+Dcaf13fl/fl (cKO) T cells. The gray values of bands were quantified with ImageJ. Source data are available for this figure: SourceData FS4.
To characterize the region of DCAF13 mediating DCAF13/NPM1 binding, we constructed Sof domain (C-terminal, Cter), WD40 repeat (internal region, Inter), and other regions (N-terminal region, Nter) truncations to pull down NPM1. Specifically, we found that only the Sof domain could pull down NPM1 (Fig. S4 B) and the N-terminal region of NPM1 was charged by the interaction with DCAF13 (Fig. S4 C). We further verified with co-IP that the last 30-amino acid region of the Sof domain was critical for NPM1 binding (Fig. 6 F). Immunofluorescence imaging of the DCAF13 (1–417 aa) mutant with the last 30-amino acid region truncation showed proper nuclear localization (Fig. S4 D), excluding the contribution of nuclear localization failure to the inability of its interaction with NPM1. Consistently, we found that the DCAF13 (1–417 aa) mutant failed to rescue DCAF13-depleted T cells from proliferative disability, while full-length DCAF13 could rescue these cells (Fig. 6 G and Fig. S4 E). Our data demonstrated that full-length DCAF13 is essential for T cell proliferation, possibly through its Sof-domain-mediated interaction with NPM1.
DCAF13 promotes the NPM1 phase separation process
Due to the importance of NPM1 in ribosome biogenesis, we proposed that DCAF13 might regulate ribosome biogenesis by interacting with NPM1 via its C-terminal Sof domain. As DCAF13 has been reported to be a subtractor adaptor for DDB1/CUL4 complex-mediated ubiquitination to degrade targeting proteins (Zhang et al., 2019a, 2019b), logically, we measured NPM1 protein levels in DCAF13 cKO T cells together with PTEN and SUV39H1, which have been reported to be targeting substrates of DCAF13 in oocyte development and early embryonic development, respectively (Zhang et al., 2019b, 2018). No expected abnormal accumulation of NPM1 or PTEN and SUV39H1 was observed in DCAF13 cKO T cells (Fig. S4, F and G), indicating that the role of DCAF13 in NPM1 relies on mechanisms other than the polyubiquitination-dependent degradation of NPM1.
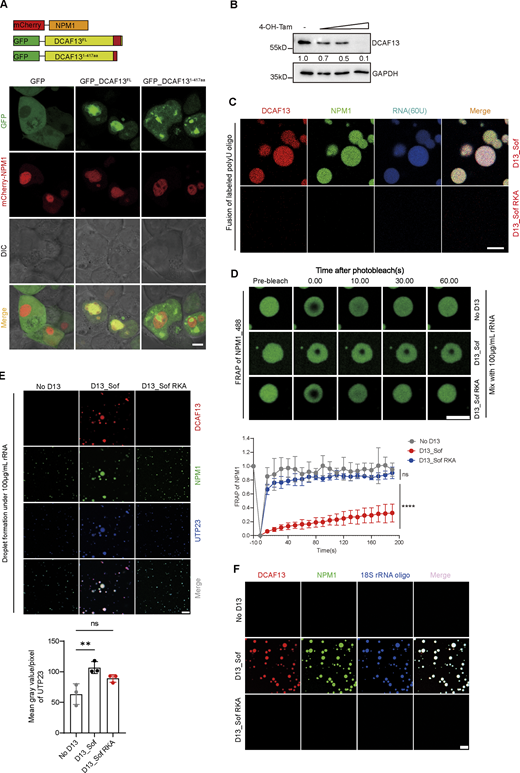
As NPM1 has been reported to drive nucleolar dynamic multilayered construction through the phase separation process (Iarovaia et al., 2019; Lafontaine et al., 2020; Shin and Brangwynne, 2017), we next evaluated whether DCAF13 could modulate NPM1 to form phase-separated liquid compartments. We observed that overexpression of full-length DCAF13 N-terminally fused with GFP (GFP-DCAF13-FL) decreased the recovery of NPM1 in 293T cells but not overexpression of GFP-DCAF13 (1–417) or the GFP-only construct (Fig. 7 A and Fig. S5 A), indicating DCFA13 could affect NPM1 kinetic properties, suggesting that DCAF13 might participate in NPM1 phase separation through its C-terminal Sof domain. Interestingly, we noticed that the Sof domain of DCAF13 contained multiple positively charged arginine-rich motifs (termed R-motifs; Fig. 7 B), which is an important electrostatic characteristic of the chaperone factors that can drive NPM1 phase separation (Mitrea et al., 2016). We also constructed a DCAF13-Sof mutant domain called DCAF13_RKA mutant, which mutated arginine into the uncharged amino acid alanine, thus providing a neutral protein as the control (Fig. 7 B). Therefore, we examined whether NPM1 and DCAF13-Sof possessed phase separation potential in vitro. In the presence of rRNA, we found that the DCAF13 Sof domain dramatically lowered NPM1 saturation concentrations to form biomolecular condensates and increased NPM1 phase boundary, whereas the DCAF13-Nter and DCAF13 RKA mutants could not (Fig. 7, C and D). When further determining if this DCAF13-promoting effect on nucleolar NPM1 phase boundary was also concentration-dependent in primary T cells, we measured the change of nucleolar volume along with stimulation by z-stack to reconstitute the intact nucleoli and found a positive relationship between nucleolar NPM1 volume and DCAF13 intensity, nucleolar volume increased accompanied with DCAF13 upregulation responding stimulation (Fig. 7 E), which was also oppositely verified by a gradient decrease in the nucleolar NPM1 volume along with a sequential DCAF13 reduction induced by different concentrations of tamoxifen (Fig. 7 F and Fig. S5 B), suggesting that cellular concentration-dependent promotion of DCAF13 occurred in NPM1 phase separation. Together, these results demonstrate that DCAF13 can promote NPM1 phase separation through its Sof domain electrostatic motif.

DCAF13 promoted NPM1 phase separation. (A) Representative pictures of FRAP and the corresponding curve of NPM1 in 293T cells (GFP-Vector, n = 10; GFP-DCAF13FL and GFP-DCAF13 1-417aa, n = 13). Dashed circles represent the bleach areas. Scale bar, 5 μm. (B) The strategy of DCAF13-Sof, DCAF13-Sof RKA mutant, and DCAF13-Nter for purification. (C) Turbidity of NPM1 with DCAF13-Sof, DCAF13-Sof RKA mutant, or DCAF13-Nter in the presence of 100 μg/ml of rRNA. (D) Representative pictures of droplet (left) and quantification of the droplet size (right, n = 48 for No D13; n = 39 for D13_Sof; n = 29 for D13_Sof RKA; n = 17 for D13_Nter) of NPM1 with DCAF13-Sof, DCAF13-of RKA mutant or DCAF13-Nter in the presence of 100 μg/ml of rRNA. Scale bar, 10 μm. (E) Confocal z-stack three-dimensional reconstruction of wild-type T cells stimulated with anti-CD3/CD28 for 24 h to analyze the relationship between DCAF13 and nucleolar volume. Representative pictures (left) and the positive correlation curve (right; n = 120) are shown. Scale bar, 2 μm. (F) Confocal z-stack three-dimensional reconstruction of T cells with DCAF13 knock-down by treating with different concentrations of tamoxifen. Representative pictures (left) and the positive correlation curve (right; n = 60, respectively). Scale bar, 2 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DCAF13 promoted NPM1 phase separation. (A) Representative pictures of FRAP and the corresponding curve of NPM1 in 293T cells (GFP-Vector, n = 10; GFP-DCAF13FL and GFP-DCAF13 1-417aa, n = 13). Dashed circles represent the bleach areas. Scale bar, 5 μm. (B) The strategy of DCAF13-Sof, DCAF13-Sof RKA mutant, and DCAF13-Nter for purification. (C) Turbidity of NPM1 with DCAF13-Sof, DCAF13-Sof RKA mutant, or DCAF13-Nter in the presence of 100 μg/ml of rRNA. (D) Representative pictures of droplet (left) and quantification of the droplet size (right, n = 48 for No D13; n = 39 for D13_Sof; n = 29 for D13_Sof RKA; n = 17 for D13_Nter) of NPM1 with DCAF13-Sof, DCAF13-of RKA mutant or DCAF13-Nter in the presence of 100 μg/ml of rRNA. Scale bar, 10 μm. (E) Confocal z-stack three-dimensional reconstruction of wild-type T cells stimulated with anti-CD3/CD28 for 24 h to analyze the relationship between DCAF13 and nucleolar volume. Representative pictures (left) and the positive correlation curve (right; n = 120) are shown. Scale bar, 2 μm. (F) Confocal z-stack three-dimensional reconstruction of T cells with DCAF13 knock-down by treating with different concentrations of tamoxifen. Representative pictures (left) and the positive correlation curve (right; n = 60, respectively). Scale bar, 2 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DCAF13 promoted NPM1 phase separation. (A) The strategy of mCherry-NPM1 and GFP-DCAF13 plasmids construction and the microscopic pictures of 293T cells transfected plasmids as designed. Scale bar, 5 μm. (B) Western blotting gradient knockdown efficiency of DCAF13 by T cells isolated from ERT2 Cre+Dcaf13fl/fl with different concentrations of tamoxifen treatment to knock down DCAF13 in different levels (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). Those cells were cultured with anti-CD3/CD28 for 48 h. The gray values of bands were quantified with ImageJ. (C) Labeled polyU enriched in NPM1 condensates with DCAF13-Sof domain other than with DCAF13-Sof RKA mutant. Scale bar, 5 μm. (D) Representative pictures of FRAP and the corresponding curve (n = 6) of droplet including purified NPM1 protein in the presence of 100 μg/ml rRNA with DCAF13-Sof, DCAF13-Sof RKA mutant protein, or NPM1 protein only. All three groups were with the presence of 100 μg/ml rRNA. Scale bar, 2 μm. (E) Representative pictures of endonuclease UTP23 enrichment in NPM1 and DCAF13-Sof domain droplets with the presence of 100 μg/ml rRNA. The mean gray value/pixel of UTP23 was measured by ImageJ (n = 3). Scale bar, 5 μm. (F) Condensates formation of NPM1 with DCAF13_Sof, DCAF13_Sof RKA mutant in the presence of HEX-labeled 18S rRNA oligo. Scale bar, 5 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData FS5.

DCAF13 promoted NPM1 phase separation. (A) The strategy of mCherry-NPM1 and GFP-DCAF13 plasmids construction and the microscopic pictures of 293T cells transfected plasmids as designed. Scale bar, 5 μm. (B) Western blotting gradient knockdown efficiency of DCAF13 by T cells isolated from ERT2 Cre+Dcaf13fl/fl with different concentrations of tamoxifen treatment to knock down DCAF13 in different levels (regarded as DCAF13 KO) or without tamoxifen (regarded as WT). Those cells were cultured with anti-CD3/CD28 for 48 h. The gray values of bands were quantified with ImageJ. (C) Labeled polyU enriched in NPM1 condensates with DCAF13-Sof domain other than with DCAF13-Sof RKA mutant. Scale bar, 5 μm. (D) Representative pictures of FRAP and the corresponding curve (n = 6) of droplet including purified NPM1 protein in the presence of 100 μg/ml rRNA with DCAF13-Sof, DCAF13-Sof RKA mutant protein, or NPM1 protein only. All three groups were with the presence of 100 μg/ml rRNA. Scale bar, 2 μm. (E) Representative pictures of endonuclease UTP23 enrichment in NPM1 and DCAF13-Sof domain droplets with the presence of 100 μg/ml rRNA. The mean gray value/pixel of UTP23 was measured by ImageJ (n = 3). Scale bar, 5 μm. (F) Condensates formation of NPM1 with DCAF13_Sof, DCAF13_Sof RKA mutant in the presence of HEX-labeled 18S rRNA oligo. Scale bar, 5 μm. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData FS5.
DCAF13/NPM1 condensates enrich preRNA and its endonuclease UTP23
As DCAF13 deficiency impaired the 18S rRNA maturation of activated T cells, we further tested whether DCAF13-promoted NPM1 phase separation facilitated the enrichment of pre-RNA. Indeed, we observed that RNA gradually increased within the DCAF13/NPM1 condensates after adding 10 μM HEX-labeled polyU (Fig. 8 A and Video 1). However, in the condition with the DCAF13-RKA mutant mixed with NPM1, we did not find the formation of biomolecular condensates. Therefore, no RNA was enriched with condensates (Fig. S5 C). Moreover, the DCAF13-Sof domain significantly decreased the fluorescence recovery of NPM1 in droplets after photobleaching (FRAP) experiments (Fig. S5 D), which is consistent with our above observations in 293T cells (Fig. 7 A).

PreRNA and its endonuclease UTP23 were enriched in DCAF13/NPM1 condensates. (A) Serial pictures of HEX-labeled polyU oligos enriched in NPM1 and DCAF13-Sof domain condensates. Scale bar, 5 μm. Refer to Video 1. (B) Representative pictures of endonuclease UTP23 and labeled polyU enrichment in NPM1 and DCAF13-Sof domain condensates. Scale bar, 5 μm. The mean gray values of UTP23 were calculated by ImageJ (NO D13, n = 10; D13_Sof, n = 11; D13_Sof RKA, n = 6). (C) RIP with anti-NPM1 in T cells from ERT2 Cre+Dcaf13fl/fl T cells with tamoxifen (regarded as DCAF13 KO) and without tamoxifen (regarded as WT). IgG served as the negative control. Western blotting (left) and quantitative PCR (right) with 18Spre primer are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F8.
PreRNA and its endonuclease UTP23 were enriched in DCAF13/NPM1 condensates. (A) Serial pictures of HEX-labeled polyU oligos enriched in NPM1 and DCAF13-Sof domain condensates. Scale bar, 5 μm. Refer to Video 1. (B) Representative pictures of endonuclease UTP23 and labeled polyU enrichment in NPM1 and DCAF13-Sof domain condensates. Scale bar, 5 μm. The mean gray values of UTP23 were calculated by ImageJ (NO D13, n = 10; D13_Sof, n = 11; D13_Sof RKA, n = 6). (C) RIP with anti-NPM1 in T cells from ERT2 Cre+Dcaf13fl/fl T cells with tamoxifen (regarded as DCAF13 KO) and without tamoxifen (regarded as WT). IgG served as the negative control. Western blotting (left) and quantitative PCR (right) with 18Spre primer are shown. Data are presented as the mean ± SD. A two-tailed Student’s t test was used. ns is not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F8.
Related toFig. 8 A . RNA was recruited into NPM1/DCAF13 droplets. 10 μM HEX-labeled polyU RNA oligo was added from the edge of the NPM1/DCAF13 mixture. Droplets formed with 40 µM DCAF13-Sof and 10 µM NPM1. DCAF13-Sof was labeled with Alexa Fluor 405 and NPM1 with Alexa Fluor 488. Images were collected at 20 s/frame. Time reported as min:s, Scale bar, 10 μm.
Related toFig. 8 A . RNA was recruited into NPM1/DCAF13 droplets. 10 μM HEX-labeled polyU RNA oligo was added from the edge of the NPM1/DCAF13 mixture. Droplets formed with 40 µM DCAF13-Sof and 10 µM NPM1. DCAF13-Sof was labeled with Alexa Fluor 405 and NPM1 with Alexa Fluor 488. Images were collected at 20 s/frame. Time reported as min:s, Scale bar, 10 μm.
The knockdown of UTP23, an endonuclease essential for 18S rRNA maturation through cleavage at sites A0, A1/1, and A2/2a of 47S pre-RNA (Wang et al., 2014; Wells et al., 2017), was reported to result in the processing failure of 18S-E pre-rRNA (Wells et al., 2017), which was coincidentally found in DCAF13 cKO T cells (Fig. 5 H). Thus, we hypothesized that DCAF13-promoted NPM1 condensates might also provoke UTP23 to process pre-RNA efficiently. Interestingly, we observed that more UTP23 and RNA were concentrated into the condensates of NPM1 with the DCAF13-Sof domain compared to that with DCAF13-RKA or without DCAF13 (Fig. 8 B and Fig. S5 E), indicating that DCAF13-promoted NPM1 phase separation potentially favors UTP23 or UTP23-like endonuclease recruitment.
To further clarify whether DCAF13 deletion also disrupted the binding of NPM1 with pre-rRNA in T cells, RNA immunoprecipitation (RIP) by anti-NPM1 was performed in primary DCAF13-deleted or wild-type T cells. The quantitative PCR results clearly showed the failure of NPM1 to enrich pre-RNA in the absence of DCAF13 (Fig. 8 C). We also conducted phase separation experiments by using 18S rRNA oligo to confirm the results (Fig. S5 F). All these results suggest that DCAF13 may promote NPM1 phase separation to concentrate pre-RNA and UTP23 endonuclease to facilitate 18S rRNA maturation and ribosomal biogenesis in activated T cells.
Discussion
Using T cell–specific DCAF-13-knockout mice and biochemistry approaches, we explored the physiological significance of nucleoli in T cells. Here, we unveiled one nucleolar protein, DCAF-13, in T cells and its essential role in promoting rapid T cell proliferation and revealed that this promotion by DCAF13 occurs through enhancement of NPM1 condensates to enrich the endonuclease UTP23 and pre-RNA, thereby facilitating 18S rRNA maturation and ribosomal biogenesis in activated T cells. Our findings add to the evidence of the importance of nucleoli in T cell proliferation and adaptive immunity.
We demonstrate that DCAF13 is indispensable in nascent protein synthesis and ribosomal 40S maturation, supporting its critical role in T cell proliferation. Polysome profiling and pre-18S processing analysis suggest that DCAF13 serves as a key factor in facilitating ribosome biogenesis to meet the massive biomolecular demand during T cell proliferation. Also, consistent with findings in mammalian oocyte development, DCAF13 plays a role in rRNA transcription, especially in naïve T cells. To date, an increasing number of studies have reported that ribosome biogenesis and its mediated translation determine T cells’ rapid and timed expansion (Chapman et al., 2020; Ricciardi et al., 2018; Tan et al., 2017b; Wolf et al., 2020). Nonetheless, the underlying mechanism of T cell activation-related ribosome biogenesis and the key factors involved in determining ribosome quantity and/or quality are still elusive. Our study first revealed that DCAF13 facilitates T cell ribosome maturation.
Notably, DCAF13 regulation promotes NPM1 phase separation. NPM1 is a dominant component of the GC region (Itahana et al., 2003; Lindström, 2011), where phase separation process not only regulates quality control of protein synthesis to prevent potentially toxic aggregate formation (Frottin et al., 2019) but also contributes to nucleolar structural maintenance to facilitate ribosome biogenesis (Feric et al., 2016; Mitrea et al., 2018; Riback et al., 2020). NPM1 can undergo heterotypic phase separation with other proteins, including arginine-rich motifs (R-motifs; Ferrolino et al., 2018; Mitrea et al., 2016, 2018). DCAF13 also harbors an arginine-rich domain (Sof domain). Here, we demonstrate that it binds to NPM1 and drives NPM1 to form biomolecular condensates, which have never been reported in T cell nucleoli and are related to T cell activation and proliferation.
Moreover, our findings also demonstrate that DCAF13/NPM1 condensates benefits recruiting the 18S pre-RNA precursor and its endonuclease UTP23 into the NPM1 phase, thereby facilitating RNA precursor maturation for further ribosome assembly. Accordingly, the existing model suggests that nucleolar phase separation is driven by ribosome biogenesis-acting factors and that the corresponding RNA substrate forms stable nucleolar condensates to ensure a steady-state flux of components in and out (Berry et al., 2015; Feric et al., 2016; Lafontaine, 2019; Yao et al., 2019). Here, DCAF13/NPM1 condensates work similarly to a biochemical reaction to efficiently induce T cell ribosome maturation and biogenesis by enriching the pre-RNA precursor as the substrate and its related endonuclease UTP23. Here, we found DCAF13 appears to colocalize with both the DFC and GC, suggesting that the effects of DCAF13 on nucleolar phase behavior might exist more comprehensively, extending beyond its interactions with NPM1.
To summarize, our study uncovers the essential factor DCAF13 in regulating ribosomal maturation to fulfill the massive demand for protein synthesis for rapid T cell proliferation for the first time. DCAF13 is regulated mainly through phase separation promotion of NPM1 to facilitate recruitment of rRNA and related endonuclease to support rRNA processing. This regulatory mechanism broadens our knowledge of the posttranscriptional mechanism modulating T cell proliferation capacity, which may be exploited to overcome the T cell expansion limitation in T cell adoptive therapy.
Materials and methods
Experimental animals
All mice used in this study, except for those specifically described, were C57BL/6 (B6) mice. The Dcaf13fl/fl mouse strain has been described previously (Zhang et al., 2019a). Briefly, 34-bp-long nucleotides at Dcaf13 exon 2 were deleted by Cre-recombinase, leading to an open reading frame shift in Dcaf13 mRNA and consequent depletion of the DCAF13 protein. CD4 Cre and ERT2 Cre transgenic mice were kindly provided by Dr. Rémy Bosselut (National Institutes of Health, Bethesda, MD, USA) and Y.W. He (Duke University Medical Center, Durham, NC, USA), respectively. OTI TCR transgenic mice, CD45.1 mice, and Rag1−/− mice were purchased from the Jackson Laboratory. All experiments were performed using 6- to 10-wk-old mice. All mice were maintained in the Animal Facility of Zhejiang University, and all animal experimental procedures were approved by the Animal Review Committee at Zhejiang University School.
Primary T cell sorting and culture
Naïve CD4+ T cells (CD4+CD25−CD62LhiCD44lo) and naïve CD8+ T cells (CD8+CD25−CD62LhiCD44lo) were sorted by the FACS Aria II flow cytometer, and total T cells were enriched by the MagniSort Mouse T cell Enrichment Kit (no. 8804-6820-74; Invitrogen). Sorted T cells were seeded in a 96-well or 48-well plate precoated with 2 μg/ml anti-CD3 (no. 14-0031-82; Invitrogen) and cultured in a T cell medium containing 3 μg/ml anti-CD28 (no. 16-0281-85; Invitrogen) with 30 Gy-dose irradiated antigen-presenting (APC) cells from the spleen at a ratio of 1:3. For cell cytokine detection, T cells were stimulated for 4 h at 37°C in fresh T cell medium with 50 ng/ml phorbol 12-myristate 13-acetate (PMA; no. P819; Sigma-Aldrich), 1 mg/ml ionomycin (no. I390; Sigma-Aldrich), and brefeldin A (no. 00-4506-5; eBioscience). The T cell medium was RPMI-1640 (no. 21870-076; Gibco) culture medium supplemented with 10% fetal bovine serum (no. 16000-044; Gibco), 1% penicillin and streptomycin solution (no. 516106; Sigma-Aldrich), 55 mΜ 2-mercaptoethanol (no.ES-007-E; Millipore), and 1 mM sodium pyruvate (no. 113-24-6; Sigma-Aldrich).
Flow cytometry and related reagents
Single-cell suspensions were prepared and stained according to the manufacturer’s protocol. For surface marker staining, cell samples were incubated for 30 min at 2–8°C in PBS buffer containing the required antibody. The antibodies used are listed in Table 1. All stained cell samples were subjected to a BD LSR Fortessa Flow Cytometer, and the collected data were analyzed by FlowJo V10 software.
Antibodies used to perform Western blotting, immunofluorescence, flow cytometry, and other related experiments
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-DCAF13 antibody [EPR18723] | Abcam | AB_2889164 |
| Anti-nucleophosmin antibody [FC82291] | Abcam | AB_297271 |
| Anti-fibrillarin antibody [38F3] | Abcam | AB_4566 |
| Anti-GAPDH antibody | Abcam | AB_9485 |
| Anti-KMT1A/SUV39H1 antibody [EPR24200-179] | Abcam | AB_309471 |
| Histone H4 (D2X4V) rabbit mAb | Cell Signaling Technology | AB_2798345 |
| β-Tubulin (9F3) rabbit mAb | Cell Signaling Technology | AB_823664 |
| Phospho-PLCγ1 (Tyr783) (D6M9S) rabbit mAb | Cell Signaling Technology | AB_2728690 |
| PLCγ1 (D9H10) rabbit mAb | Cell Signaling Technology | AB_10691383 |
| Phospho-Akt (Ser473) (D9E) rabbit mAb | Cell Signaling Technology | AB_2315049 |
| Akt (pan) (C67E7) rabbit mAb | Cell Signaling Technology | AB_915783 |
| Phospho- Erk1/2 (Thr202/Tyr204) (E10) mouse mAb | Cell Signaling Technology | AB_331768 |
| p44/42 MAPK (Erk1/2) antibody rabbit | Cell Signaling Technology | AB_330744 |
| DYKDDDDK Tag (D6W5B) rabbit mAb | Cell Signaling Technology | AB_2572291 |
| Myc-Tag (9B11) mouse mAb | Cell Signaling Technology | AB_331783 |
| Normal mouse IgG pAb | Millipore | AB_145840 |
| CD25 monoclonal antibody (PC61 5.3), Pacific blue | eBioscience | AB_2556574 |
| CD62L (L-Selectin) monoclonal antibody (MEL-14), APC | eBioscience | AB_469410 |
| TCR beta monoclonal antibody (H57-597), PE-Cyanine7, | eBioscience | AB_2573506 |
| TCR V alpha 2 monoclonal antibody (B20.1), FITC | eBioscience | AB_465259 |
| IL-2 monoclonal antibody (JES6-5H4) PE | eBioscience | AB_466150 |
| TNF alpha monoclonal antibody (MP6-XT22), PE | eBioscience | AB_466199 |
| BrdU monoclonal antibody (BU20A), APC | eBioscience | AB_11040534 |
| CD11c Monoclonal antibody (N418), FITC | eBioscience | AB_11011485 |
| CD11b Monoclonal antibody (M1/70), PE | eBioscience | AB_12011283 |
| NK1.1 monoclonal antibody (PK136), PE | eBioscience | AB_12594183 |
| PE anti-mouse CD8a antibody (53–6.7) | BioLegend | AB_312747 |
| APC anti-mouse CD8a antibody (53–6.7) | BioLegend | AB_312751 |
| Brilliant violet 650 anti-mouse CD8a antibody (53–6.7) | BioLegend | AB_2563056 |
| PE/Cyanine7 anti-mouse CD4 antibody (GK1.5) | BioLegend | AB_312707 |
| Pacific blue anti-mouse CD4 antibody (GK1.5) | BioLegend | AB_493647 |
| APC/Cyanine7 anti-mouse CD4 Antibody (GK1.5) | BioLegend | AB_312699 |
| FITC anti-mouse/human CD44 antibody (IM7) | BioLegend | AB_312957 |
| APC/Cyanine7 anti-mouse/human CD44 antibody (IM7) | BioLegend | AB_830785 |
| PE anti-mouse CD45.1 antibody (A20) | BioLegend | AB_313497 |
| APC anti-mouse CD45.2 Antibody (104) | BioLegend | AB_389211 |
| PE anti-mouse IFN-γ antibody (XMG1.2) | BioLegend | AB_315402 |
| FITC anti-mouse/human CD45R/B220 antibody (RA3-6B2) | BioLegend | AB_103205 |
| APC anti-mouse F4/80 antibody (BM8) | BioLegend | AB_123116 |
| APC anti-mouse Ly-6G antibody (1A8) | BioLegend | AB_127613 |
| Zombie Violet fixable viability kit | BioLegend | 423113 |
| Zombie NIR fixable viability kit | BioLegend | 423105 |
| Peroxidase-AffiniPure goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch Laboratories | AB_2534069 |
| Peroxidase-AffiniPure goat anti-mouse IgG (H+L) | Jackson ImmunoResearch Laboratories | AB_10015289 |
| Donkey anti-rabbit IgG (H+L) Secondary antibody, Alexa fluor 594 | Invitrogen | AB_141637 |
| Goat anti-mouse IgG (H+L) Secondary antibody, Alexa fluor 488 | Invitrogen | AB_2534069 |
| DAPI solution | Dojindo | D523 |
Nascent protein synthesis detection by HPG incorporation
The Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kit (no. C10428; Invitrogen) was used to measure nascent proteins based on the principle of azide-alkyne cycloaddition between Alexa Fluor 488 azide and L-homopropargylglycine (HPG), an amino acid analog of methionine containing an alkyne moiety, which would be integrated into newly translated proteins in the absence of methionine. HPG (50 μM) with or without 100 μg/ml cycloheximide (CHX; no. C7698; Sigma-Aldrich), a eukaryote protein synthesis inhibitor introduced as a negative control, was diluted with methionine-free T cell medium (no. A1451701; Invitrogen) and fed to cultured T cells for 30 min of incubation at 37°C. Cells were then fixed with 4% paraformaldehyde in PBS for 15 min at room temperature followed by a permeablization step using PBS plus 0.5% Triton X-100 for 20 min at room temperature. Click reaction was performed by adding 100 μl reaction cocktail (86 μl 1× Click-iT HPG reaction buffer, 4 ul Component D, 0.25 Alexa Fluor azide, 10 μl Click-iT HPG buffer additive) and incubated for 30 min at room temperature. Nuclei were stained with DAPI. The analysis was processed by confocal laser scanning microscopy (IX83-FV3000; OLYMPUS) using a 100× oil immersion (NA = 1.4) objective. HPG was detected by using the 488-laser, and DAPI was detected by the 405-laser. Images were collected by FV31-SW software and analyzed by ImageJ.
Electron microscopy
Experimental T cell pellets were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer overnight at 4°C and then washed with 0.1 M cold PB gently to avoid destroying the lumps of fixed cells. Then, the pellets were postfixed with 1% osmium tetroxide in 0.1 M PB for 1 h at 4°C followed by several gentle rinses with deionized water at room temperature and stained with 2% aqueous uranyl acetate for ∼30 min in the dark. Next, the stained pellets were dehydrated through sequential treatment from 50%, 70%, 90%, and 100% ethanol to 100% propylene oxide. After dehydration, the cell pellets were treated with resin embedding as follows: 1:1 mix of propylene oxide:resin for 2 h at room temperature, 1:3 mix of propylene oxide:resin for another 2 h at room temperature, and 100% resin overnight at 37°C with appropriate orientation. Finally, ultrathin sections (with a thickness of 100 nm) were prepared with an ultramicrotome (EM UC7; Leica), and images were collected by transmission electron microscopy (FEI Tecnai G2 Spirit 120 Kv). Image analysis and quantification were performed using ImageJ software.
Newly transcribed nucleolar RNA detection by EU incorporation
The Click-iT RNA Alexa Fluor 488 Imaging Kit (no. C10329; Invitrogen) was applied to measure newly transcribed RNA based on the principle of azide-alkyne cycloaddition between Alexa Fluor 488 azide and 5-ethynyl uridine (EU), an alkyne-modified nucleoside, which would be utilized for newly synthesized RNA. T cells intended for detection were treated with or without a final concentration of 8 µM actinomycin D (ActD, no. SBR00013; Sigma-Aldrich), a transcription inhibitor, diluted in pre-warm fresh T cell medium and incubated at 37°C for 30 min. After being washed with PBS, the cells were labeled with 1 mM final EU diluted in fresh T cell medium for 1 h. Cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then permeabilized with PBS plus 0.5% Triton X-100 for 15 min at room temperature. Click reaction was performed by adding 500 μl reaction cocktail (428 μl Component C, 20 μl Component D, 1.8 μl Alexia Fluor azide, 50 μl Click-iT reaction buffer additive) and incubated for 30 min at room temperature. Nuclei were stained with DAPI. The images were collected by confocal laser scanning microscopy (IX83-FV3000; OLYMPUS) using a 100× oil immersion (NA = 1.4) objective. EU was detected by using the 488-laser and DAPI was detected by the 405-laser. Images were collected by FV31-SW software and analyzed by ImageJ.
Knockdown DCAF13 in vivo by tamoxifen inducement
ERT2 Cre+Dcaf13fl/fl mice were used to detect the effects of DCAF13 depletion on immune cell populations. Tamoxifen (TAM) was prepared as a 10 mg/ml solution by dissolving TAM powder (T5678; Sigma-Aldrich) in sunflower seed oil (S5007; Sigma-Aldrich). To induce the expression of Cre/ER fusion protein, mice were treated for three consecutive days with 100 μl TAM by intraperitoneal injection (i.p.). Mice treated only with sunflower seed oil were referred to WT controls. 8 d after the first treatment, cells from the spleen were analyzed for the percentages and number of B cells, monocytes, NK cells, dendric cells, and granulocytes.
Cell trace assay
Sorted naïve T cells were suspended in PBS at a density of 1 × 106 cells/ml and supplemented with 5 μM CFSE (no. C34554; Invitrogen). The cell supernatant was incubated at 37°C for 3 min to allow CFSE dye to incorporate into the cells, followed by the addition of five volumes of ice-cold RPMI medium supplemented with 10% FBS to terminate labeling. Excessive CFSE was removed by pelleting the cells and removing the supernatant for three successive washes in the medium. CFSE-labeled T cells were cultured as experimentally required, and CFSE signaling dilution was assessed by flow cytometry as a marker of T cells for multiple generations. The percentage of individual peaks was quantified by FlowJo V10 software.
BrdU labeling and detection
Cultured T cells were transferred to fresh T cell medium containing 60 μM BrdU (no. 19–160; Sigma-Aldrich) and incubated for 1 h such that BrdU could incorporate into synthesizing DNA in place of thymidine. BrdU can be detected in conjunction with an anti-BrdU antibody for subsequent flow cytometric analysis to evaluate the cell cycle. BrdU staining was performed by utilizing a BrdU flow kit (no. 559619; BD) according to the manufacturer’s instructions. The percentage of the individual phase was quantified by FlowJo V10 software.
Immunofluorescence (IF) staining
Coverslips were treated with a 0.01% (vol/vol) poly-L-lysine solution (no. P4707; Sigma-Aldrich) for 5 min, drained, and dried at room temperature for 15 min. Naïve or activated T cells suspended in RPMI-1640 (1 × 106/ml) were plated on coverslips and then incubated on ice for 15 min. Staining was performed as described in a previous study (Zhou et al., 2017). In brief, the cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.2% Triton X-100 in PBS for 10 min. For nucleolar protein detection, the cells were fixed with 4% paraformaldehyde in PBS for 1 min at room temperature coupled with −20°C pre-cold methanol for <1 min at room temperature. Next, the fixed cells were blocked with Image iT FX signal enhancer (no. I3693; Invitrogen) for 30 min at room temperature followed by blocking with 1% BSA in PBS. The cells were then incubated with primary antibodies diluted with 1% BSA in PBS at an optimized concentration at 4°C overnight. All antibodies used are listed in Table 1. After extensive washing with PBS, the slides were incubated with goat anti-rabbit Alexa Fluor 594 (1:250) and goat anti-mouse Alexa Fluor 488 (1:250) secondary antibodies followed by nuclear staining with DAPI. The washed slides were mounted with an anti-fade mounting solution (no. P36934; Invitrogen). Images were acquired at room temperature using confocal laser scanning microscopy (Zeiss LSM 880 Fast Airyscan Confocal) fitted with a 100×/NA0.55 oil objective. DAPI was excited with a 405 nm laser, Alexa Fluor 488 fluorescence was excited with a 488 nm laser, and Alexa Fluor 594 fluorescence was excited with a 561 nm laser. Images were collected by Zen 2.3 software and analyzed by ImageJ.
3D immune-confocal image reconstruction
Regarding nucleolar volume, we applied 3D immune-confocal image reconstruction as previously described (Markov et al., 2021). We stained the T cells with NPM1 as a nucleolar marker and DAPI for the nucleus and then scanned the cells using an Olympus IX3 FV3000 confocal microscope equipped with a UPLSAPO 100×/1.4 oil objective. The microscope was operated with FV31S-DT software. DAPI fluorescence was excited using a 405 nm laser, Alexa Fluor 488 fluorescence was excited using a 488 nm laser, and Alexa Fluor 594 fluorescence was excited using a 561 nm laser. Z-stack of sections (recorded in OIR format) were captured throughout the entire depth of the individual nucleus or whole cells, resulting in a Z-step of 0.43 μm/slides. Image stacks of nuclei or whole cells were reconstructed into 3D isosurfaces using Imaris9.5. Briefly, the OIR format files were directly imported into Imaris9.5 and the surpass view was selected. Background subtraction thresholding was determined by increasing the minimum value on the display adjustment panel (the same minimum value was used in the same panel), followed by automatic tracing and surface rendering. Finally, the resulting 3D isosurfaces, including all-channel surfaces, were exported by capturing a snapshot. The individual intensities of selected channels were automatically calculated by Imaris9.5.
Bone marrow transplantation
To determine whether the phenotype embodied in DCAF13-deficient T cells was T cell intrinsic, bone marrow transplantation was performed. For cotransfer settings, CD45.1+ WT bone marrow cells mixed at a ratio of 1:1 with CD45.2+ CD4 Cre+Dcaf13fl/fl bone marrow cells were regarded as the experimental group, and those mixed with CD45.2+ CD4 Cre−Dcaf13fl/fl bone marrow cells were regarded as the control group. A total of 106 of the above cell mixture was intravenously injected into CD45.1+ host mice. Prior to transplantation, host mice were fed ABX solution including 0.2 g vancomycin hydrochloride, 0.2 g ampicillin sodium salt, 0.2 g streptomycin sulfate, and 0.1 g metronidazole in 200 ml sterilized water for 1 wk to eliminate intestinal flora and treated with 8-Gy irradiation in advance of transplantation. Thymic T cell development and peripheral homeostasis were analyzed by flow cytometry as described above.
T cell homeostatic proliferation in vivo
CD4 Cre+Dcaf13fl/fl (CD45.2+) T cells and CD4 Cre−Dcaf13fl/fl (CD45.2+) T cells were purified from lymph nodes using a MagniSort Mouse T cell enrichment kit (no. 8804-6820-74; Invitrogen) and mixed with CD45.1+ T cells from wild-type mice at a ratio of 1:1. The above cells were then labeled by Celltrace Violet (C34557; Invitrogen) at 37°C for 15 min and intravenously injected into 6-Gy preirradiated CD45.1+ mice as hosts, and the Celltrace dilution in donor T cells was analyzed at 4 d by flow cytometry as described above.
Polysome fractionation
Polysome profiles were analyzed as described previously (Gandin et al., 2014). Briefly, to examine the influence of DCAF13 on the polysome profile, total T cells were sorted by a MagniSort Mouse T cell Enrichment Kit (no. 8804-6820-74; Invitrogen) from lymph nodes derived from ERT Cre+Dcaf13fl/fl mice and cultured in T cell culture conditions as described above for 72 h with 2 nm 4-hydroxytamoxifen (tamoxifen; no. H7904; Sigma-Aldrich) to induce Dcaf13 knockout or without tamoxifen as the wild-type control. Experimental cells were incubated with CHX at a final concentration of 100 µg/ml in fresh growth media for 5 min at 37°C before collection by centrifugation at 200 g for 5 min at 4°C. CHX (100 µg/ml) was added to all buffers used during this process to retain mono- and polysome complexes. Cells were washed twice with 5 ml of ice-cold 1× PBS containing 100 µg/ml CHX and then resuspended in 450 μl of polysome lysis buffer (10 mM HEPES, 15 mM MgCl2, 100 mM KCl, pH = 7.4) containing EDTA-free protease inhibitor cocktail (no. 4693132001; Roche), 100 µg/ml CHX, 100 U/ml RNAase inhibitor (no. R8060; Solabio), 1 mM DTT, and 1% (vol/vol) Triton X-100. The cells were then vortexed for 5 s and kept on ice for 20 min. Lysates were centrifuged at 16,000 g for 7 min at 4°C and the supernatant (∼500 μl) was transferred to a new prechilled tube during sucrose gradient (10–50%) formation through Gradient Master (GM-108; Biocomp). Next, the cell lysate was loaded onto sucrose gradients followed by ultracentrifugation at 222,228 g (36,000 rpm) for 2 h at 4°C using the SW41Ti rotor (Beckman, Optima XPN-100). After ultracentrifugation, the sucrose gradient was fractionated from the top of the tube by a Piston Gradient Fractionator (PGF152; Biocomp). During fractionation, the UV absorbance was detected at 254 nm and a smooth line scatter plot of the absorbances was created to evaluate ribosome constitution and translation.
Northern blotting
Northern blots were performed using a DIG northern starter kit (no. 12039672910; Roche) according to the manufacturer’s protocol with minor modifications. RNA was isolated using TRIzol reagent (no. 15596026; Life Technologies), and 5 μg of RNA was electrophorized in a 1% agarose gel dissolved in MOPS buffer (20 mM MOPS, 5 mM NaAc, 2 mM EDTA, pH = 7.5) supplemented with 2% formaldehyde. Next, the gel was imaged in a gel imaging system (Gel Doc XR; Bio–Rad) to determine the relative positions of DNA markers and to record 18S and 28S as loading controls. The gel was blotted by capillary transfer to nylon membranes overnight with 20× SSC (3.0 M NaCl, 0.3 M sodium citrate, pH = 7.0). RNA was fixed on the membrane at a dose of 2,400 kJ by UV crosslinking (CL1000 Ultraviolet Crosslinker, UVP) and baked at 80°C for 30 min, followed by hybridization with 40 ng/ml digoxygenin (DIG)-labeled DNA probe diluted in prewarmed DIG Easy Hyb and incubation at 50°C overnight. The membrane was then incubated by anti-digoxigenin-AP diluted in blocking solution for 1 h and immunological detection in 200 μl CDP-Star ready-to-use solution prepared in the kit. The gray values of bands were quantified with ImageJ. The DIG-labeled oligos for northern blotting are as follows:
OTSI-5′-GATCATTAAACGGGAGACTGTGGAGGAGC-3′;
OTSII-5′-CGATCAATCGCGTCACCCGCTGCGGTGGGT-3′.
Retrovirus production and transduction
Retroviruses were assembled in Plat-E cells as a kind gift from Dr. Xiaolong Liu (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences). pMX-IRES-GFP plasmids containing the indicated genes were transfected into Plat-E cells with Lipofectamine 3000 Transfection Reagent (no. L3000015; Invitrogen) according to the manufacturer’s instructions. 72 h before the new medium was replaced, the supernatant containing retrovirus was obtained and applied to infect EL-4 cells or primary T cells. To express foreign proteins in primary T cells, sorted naïve CD4+ T cells were simulated over 20 h prior to virus transfection. Experimental cells were resuspended in 1 ml retrovirus supernatant in the presence of 10 μg/ml polybrene (Millipore) and 10 mM HEPES buffer. Infection was performed by centrifugation for 2 h at 1,500 g at 32°C. As the PMX plasmid carries the GFP gene, GFP positivity indicated that cells were successfully infected with the virus. Stable EL-4 cell lines expressing the indicated proteins were obtained by flow cytometry sorting and cultured for LM-MS analysis. Combined with CFSE assay and flow cytometry analysis, the rescue effect on cell proliferative deficiency exerted by different DCAF13 truncates was evaluated by CFSE signaling on gated GFP+ T cells.
Co-IP/mass spectrometry
The indicated cells were collected and washed with PBS. Cells were suspended in 800 μl WE buffer (25 mM HEPES, 1.5 mM MgCl2, 150 mM NaCl, 0.1% NP-40, 10% glycerol plus proteinase inhibitor cocktail, pH = 7.9) and incubated on ice for 30 min followed by brief sonication (power 25, 3 s on, 5 s off, 3–5 min) for DNA fragmentation. Next, the cells were centrifuged at 12,000 rpm for 15 min at 4°C and the supernatant was saved and incubated with 15 μl prepared M2 beads (Sigma-Aldrich) overnight at 4°C. The protein-binding beads were washed sequentially with buffer IP-I (10 mM HEPES, 150 mM NaCl, pH = 7.9) three times and buffer IP-II (10 mM HEPES, 200 mM NaCl, pH = 7.9) once. For specific protein detection, 30 μl loading buffer was added and immunoblotting was performed. To identify DCAF13-interacting proteins by LC–MS/MS, the EL-4 cell line stably expressing Flag-DCAF13 was used to perform Co-IP as described above. Flag-DCAF13-bound M2 beads were treated with 100 μg/ml flag peptide solution to completely elute Flag-DCAF13 and its specific binding proteins. The eluent was sent for LC–MS/MS analysis (PTM Biolabs). The interaction network was generated by Cytoscape. The interaction was further confirmed by Co-IP coupled with Western blotting. The gray values of bands were quantified with ImageJ, with normalization of each band by the loading control followed by normalization to WT as 1.0.
Protein expression and purification
The expression of recombinant wild-type NPM1 and DCAF13 was induced with 0.5 mM isopropyl β-D-thiogalactoside (IPTG, no. A600168; Sangon Biotech) in Escherichia coli BL21 (DE3) cells (no. C504-03; Vazyme) at 220 rpm and 37°C for 4 h. BL21(DE3) cells were lysed by an ultrasonic cell crusher with Ni2+ binding buffer (20 mM HEPES, 1 M NaCl, 1 M urea, pH = 7.4) supplemented with 1X protease inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride (PMSF). For DCAF13_Nter, DCAF13_Sof, and its mutant, an extra 95°C boiling treatment of the lysis solution for 30 min was performed for primary purification. The recombinant protein was enriched with Ni Sepharose and eluted with Ni2+ elution buffer (20 mM HEPES, 1 M NaCl, 1 M urea, 500 mM imidazole, pH = 7.4). The eluted recombinant protein was further purified by ion-exchange chromatography on a HiTrap Q HP or HiTrap SP HP column (GE). The final purified recombinant protein was obtained through a Superdex 75 10/300 Gl column (GE) with gel filtration buffer (20 mM HEPES, 500 mM NaCl, pH = 7.4).
Turbidity assay
To construct the phase diagram for NPM1-DCAF13 solutions, NPM1 and DCAF13-Sof/Sof-RKA/Nter were diluted with storage buffer (20 mM HEPES, 500 mM NaCl, 1 mM DTT, pH = 7.5) as required and aliquoted on ice. Phase separation of NPM1 and NPM1-DCAF13 was performed in the presence or absence of 100 μg/ml high purity total ribosomal RNA (rRNA, no. 11020001; bioWORLD) in PS buffer (10 mM Tris, 150 mM NaCl, 1 mM DTT, pH = 7.5). Samples were incubated for 10 min at room temperature and vortexed prior to measuring the absorbance at 340 nm using a NanoDrop 2000c (Thermo Fisher Scientific). Measurements were performed in triplicate. Solution turbidity was recorded as positive for phase separation when A340 ≥ 0.1.
Protein labeling
Protein labeling was performed as previously reported (Feric et al., 2016; Ferrolino et al., 2018). NPM1 protein was labeled with Alexa Fluor 488 NHS Ester (no. A20000; Invitrogen), DCAF13-Sof/Sof-RKA/Nter proteins were labeled with Alexa Fluor 405 NHS Ester (no. A30000; Invitrogen), and UTP23 was labeled with Alexa Fluor 647 NHS Ester (no. A37573; Invitrogen) according to the manufacturer’s protocol. Briefly, labeling dye was introduced into a 50 μM protein solution prepared by dilution with storage buffer (20 mM HEPES, 500 mM NaCl, 1 mM DTT, pH = 7.5) at a concentration ratio of 1:1 and the solution was incubated for 1 h in the dark. Excessive dye was eluted by Amicon Ultra-3K centrifugal filter devices (no. UFC500396; Millipore). In addition, fluorescently labeled NPM1-A488 was mixed with unlabeled NPM1 at a 1:9 ratio in 6 M guanidine-HCl solution and dialyzed with Slide-A-Lyzer Dialysis Cassettes (3.5 K MWCO, no. 66330; Thermo Fisher Scientific) in storage buffer (20 mM HEPES, 500 mM NaCl, 1 mM DTT, pH = 7.5), followed by concentration to 300 μM by Ultra-3K centrifugal filter devices.
Imaging of labeling droplet formation
To image the biomolecular condensates of NPM1/DCAF13 and NPM1/DCAF13/UTP23, samples were prepared in PS buffer (10 mM Tris, 150 mM NaCl, 1 mM DTT, pH = 7.5) in the presence or absence of 100 μg/ml rRNA and incubated for 10 min at room temperature before loading to an eight-well chambered coverglass (no. C8-1.5H-N; Cellvis). Droplets formed and settled onto the surface due to gravity. For RNA-incorporated phase separation, HEX-labeled polyU RNA oligos with lengths of 60 nt, which were kindly provided by Dr. Lou (Key Laboratory of RNA Biology, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences), were added to the NPM1/DCAF13 condensates. HEX-labeled 18S rRNA oligo was synthesized by Shanghai Generay Biotech Co., Ltd. The sequence of 18S rRNA oligo is as follows: 5′-CGAACGUCUGCCCUAUCAACUUUCGAUGGUAGUCGCCGUGCCUACCAUGGUGACCAC-3′. All reported images were recorded with an Olympus FV3000 fluorescence microscope fitted with a UPLSAPO 100×/1.4 oil objective. Alexa Fluor 405 NHS Ester was excited with 405 nm laser, Alexa Fluor 488 NHS Ester was excited with 488 nm laser, Alexa Fluor 647 NHS Ester was excited with 640 nm laser, and HEX was excited with 561 nm laser. Images were collected by FV31-SW software. The droplet size and mean gray vaules were measured by ImageJ.
Fluorescence redistribution after photobleaching (FRAP)
For the FRAP experiments in vitro, a circular region of interest (ROI) located at the center of the droplets was photobleached to ∼10% of the original intensity by illuminating the ROI with the laser, and then the NPM1-A488 signal was successively recorded once every 30 s for a total of 20 min. For the FRAP experiments in vivo, HEK293T cells were seeded onto an eight-well-chamber coverglass (no. C8-1.5H-N; Cellvis) and cotransfected with mCherry-NPM1 and GFP-DCAF13 or GFP-DCAF13 (1–417 aa) mutant. HEK293T cells cotransfected with mCherry-NPM1 and GFP-Vector were regarded as controls. After 24 h of incubation, mCherry and GFP double-positive cells were selected to measure the mCherry-NPM1 signal intensity before and after photobleaching. All images were acquired at room temperature using an OLYMPUS IX83-FV3000 fitted with a UPLSAPO 100×/1.4 oil objective. Alexa Fluor 488 NHS Ester and EGFP were excited with a 488-nm laser and mCherry was excited with a 561-nm laser. Images were collected by FV31-SW software. The collected data were analyzed by FRAP Profiler plug-in from ImageJ.
RNA immunoprecipitation (RIP)-qPCR
IP was performed as described previously with minor modifications (Keene et al., 2006). Briefly, total T cells sorted from ERT Cre+Dcaf13fl/fl cells were cultured in T cell culture conditions as described above for 72 h with or without 2 nm tamoxifen (no. H7904; Sigma-Aldrich) as the DCAF13 KO and WT samples, respectively. Then, ∼107 T cells were collected for subsequent RIP. RNA was isolated by TRIzol reagent (Life Technologies). The primers for qPCR are as follows:
18S_F: 5′-TTGGTGACTCTAGATAACC-3′;
18S_R:5′-GTGGTCACCATGGTAGGCAC-3′.
Database analysis
The proteomic profiling of human T cell nucleoli was provided by Gautier’s group (Jarboui et al., 2011). The proteomic abundance profiling cut-off of a twofold increase in stimulated naïve human T cells and protein translation kinetics after naïve T cell activation were both from the Immunomics database (https://www.immunomics.ch) provided by Geiger’s group (Wolf et al., 2020). These three files overlapped for 56 targeting proteins, which represented the protein renewal half-time (t50).
Statistical analysis
All statistical analyses and graphs were done using GraphPad Prism version 9.0.0. Statistical significance was calculated using a two-tailed Student’s t test for independence. Data distributions were assumed to be normal, but this was not formally tested.
Online supplemental material
Fig. S1 shows that DCAF13 is placed second in the newly synthesized kinetics ranking of human T cell nucleolar proteins in response to stimulation, indicating DCAF13 may be a promising candidate as a nucleolar protein in T cells robustly synthesized in response to T cells activation. It refers to Fig. 1. Fig. S2 shows that the DCAF13 depletion by CD4 Cre+ had no influence on the T cells’ development in the thymus and the homeostasis of naïve and effector T cells in the lymph node and spleen. Meanwhile, Fig. S2 shows that DCAF13 depletion also has a fatal effect on the survival and proliferation of other cells, including B cells, monocytes, NK cells, dendric cells, and granulocytes, indicating that a requirement for DCAF13 is not unique to T cells. It refers to Fig. 2. Fig. S3 shows that DCAF13 depletion has no comparable effect on cell size and cytoplasm–nucleus ratios between naïve wild-type and DCAF13-deleted T cells, resulting from the “rest” status of naïve T cells on protein synthesis. Additionally, Fig. S3 shows DCAF13 depletion exerted slighter effects on rRNA transcription rather than protein synthesis, indicating DCAF13 depletion mainly contributes to the abnormality of 18S rRNA maturation rather than transcription. The figure refers to Fig. 5. Fig. S4 shows that the interaction of DCAF13 with NPM1 mainly occurs between the C-terminal of DCAF13 and the N-terminal of NPM1, by which DCAF13 has no effect on NPM1 degradation as a subtractor adaptor for DDB1/CUL4 complex previously reported and refers to Fig. 6. Fig. S5 shows that DCAF13 facilitates NPM1 to form biomolecular condensates, not only promoting the concentration of UTP23 and RNA into the condensates but also significantly decreasing the fluorescence recovery of NPM1 in droplets by photobleaching (FRAP) in vitro; refers to Fig. 7. Table 1 shows the antibodies used to perform Western blotting, immunofluorescence, flow cytometry, and other related experiments. Video 1 is related to Fig. 8 A.
Data availability
The data are available from the corresponding author ([email protected]) upon reasonable request.
Acknowledgments
We thank Dr. Liu (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China) for his generous gifts of cell lines; and Dr. Leng (Institute Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China) for providing L.monocytogenes-OVA-N4 (LM-N4). We thank Y. Huang, Y. Li, J. Wang, S. Liu, Z. Lin, and L. Xuan from the Core Facilities, Zhejiang University School of Medicine for their technical support; B. Wang and C. Yang from the Center of Cryo-Electron Microscopy, Zhejiang University; and the staff from Animal facilities, Zhejiang University for mice maintenance.
This work was supported by grants from the National Natural Science Foundation of China (32030035, 91442101, 31800734, 81830006, 32100693, 32000623, 31971237, 12002307, and 92269101), the Zhejiang Provincial Natural Science Foundation of China (nos. LZ21C080001 and LY23A020002), Science and Technology Innovation 2030-Major Project (2021ZD0200405), Key project of Experimental Technology Program of Zhejiang University (No. SZD202203), and Zhejiang provincial Key Project of Research and Development (2019C03043).
Author contributions: L. Zhou, S. Wang, and W. Hu contributed equally to this work. L. Wang, W. Chen, L. Zhou, and W. Hu designed the research. L. Zhou, W. Hu, X. Liu, S. Wang, L. Xu, B. Tong, T. Zhang, and Y. Guo performed the experiments and data analysis. L. Zhou, S. Wang, and W. Hu wrote the manuscript. L. Lu, W. Qian, and J. Chen provided expertise and advice. H. Fan and J. Zhao respectively provided Dcaf13fl/fl mice and reagents. L. Wang supervised the project.

