


Kinetochores are multiprotein assemblies directing mitotic spindle attachment and chromosome segregation. In apicomplexan parasites, most known kinetochore components and associated regulators are apparently missing, suggesting a minimal structure with limited control over chromosome segregation. In this study, we use interactomics combined with deep homology searches to identify 13 previously unknown components of kinetochores in Apicomplexa. Apicomplexan kinetochores are highly divergent in sequence and composition from animal and fungal models. The nanoscale organization includes at least four discrete compartments, each displaying different biochemical interactions, subkinetochore localizations and evolutionary rates across the phylum. We reveal alignment of kinetochores at the metaphase plate in both Plasmodium berghei and Toxoplasma gondii, suggestive of a conserved “hold signal” that prevents precocious entry into anaphase. Finally, we show unexpected plasticity in kinetochore composition and segregation between apicomplexan lifecycle stages, suggestive of diverse requirements to maintain fidelity of chromosome segregation across parasite modes of division.
Introduction
Eukaryotic chromosome segregation occurs along a spindle formed of microtubules. In order to segregate DNA into daughter cells, the spindle must interact with chromosomes. Most organisms achieve specificity in this interaction by binding to chromosomal sites called centromeres, distinguished by the histone H3 variant centromere protein A (CENP-A; Fukagawa and Earnshaw, 2014). Onto the centromere assembles the kinetochore, a hierarchical assembly and molecular machine that links chromosomes to the spindle (Cheeseman and Desai, 2008). The kinetochore is formed of multiple compartments, each composed of different protein complexes. In animals and fungi, the constitutive centromere-associated network (CCAN) forms the inner kinetochore (Foltz et al., 2006), whilst at the onset of mitosis the outer kinetochore KMN network is recruited, formed of KNL1, MIS12, and NDC80 complexes (Cheeseman et al., 2006). The KMN network serves as both the microtubule-binding component of the kinetochore and a landing pad for SKA and DASH complexes, which strengthen microtubule attachment in animals and fungi, respectively (Helgeson et al., 2018; Lampert et al., 2010), in addition to the spindle assembly checkpoint (SAC), a surveillance system that ensures faithful chromosome segregation (Musacchio and Salmon, 2007). Fidelity requires that sister chromatids are bioriented at metaphase, kinetochores bound by microtubules emanating from opposing spindle poles (Lampson and Cheeseman, 2011). Upon biorientation, the SAC is inactivated through disassembly of the mitotic checkpoint complex (MCC) and liberation of the anaphase-promoting complex/cyclosome (APC/C) culminates in the cleavage of cohesin and segregation of sister chromatids into daughter cells.
The conservation of kinetochore proteins varies greatly across organisms (Meraldi et al., 2006; Tromer et al., 2019; van Hooff et al., 2017). Whilst the KMN network is more widely distributed (D’Archivio and Wickstead, 2017; Salas-Leiva et al., 2021), most of the CCAN is not conserved across eukaryotes. In particular, within the phylum of Apicomplexa, most components of the CCAN and SAC as described in animals and fungi, in addition to the majority of the KMN network, are not clearly detected. This phylum groups a large number of intracellular parasites of considerable medical and veterinary relevance, including the malaria parasite Plasmodium and Toxoplasma, causative agent for toxoplasmosis. In addition to a widespread lack of “canonical” kinetochore components, apicomplexan parasites appear to divide quite differently to most of the cells of their hosts (Francia and Striepen, 2014; Gerald et al., 2011; Striepen et al., 2007). Flexibility in the scale of amplification and modes of division, often adapting to the size and environment of different host cells and tissues, suggests division checkpoints in these parasites may be very different to those described in animals (Alvarez and Suvorova, 2017). Asexual forms of Toxoplasma gondii divide in tissues of the mammalian intermediate hosts by endodyogeny, producing two daughter cells from a single round of fission within the mother cell, which is consumed as the offspring mature. Uncoupling nuclear divisions from the cell cycle allows certain apicomplexan parasites to successively replicate their genome in the absence of cytokinesis. In red blood cells, Plasmodium species divide by schizogony to produce multinucleated coenocytes. Karyokinesis is asynchronous, resulting in nongeometric expansion. Following last division, daughter cells either bud internally or from the surface of the mother plasmalemma (Arnot et al., 2011; Speer and Dubey, 2001). During mosquito transmission, capacitated male gametocytes (microgametocytes) produce a polyploid nucleus that gives rise to eight haploid gametes. Microgametes fertilize female gametes (macrogametes), shortly followed by meiosis and differentiation producing characteristic banana-shaped ookinetes.
Three organelles contain DNA in apicomplexan parasites. In addition to the mitochondrion, the nucleus and a remnant of secondary symbiosis called the apicoplast each have their own distinct segregation cycle. The nucleus undergoes a largely closed mitosis, an intranuclear spindle nucleates from spindle poles to bind kinetochores, layered structures maintained close to the nuclear periphery (Aikawa, 1966; Dubremetz, 1973; Farrell and Gubbels, 2014; Prensier and Slomianny, 1986; Sinden and Hartley, 1985; Zeeshan et al., 2020). Current evidence suggests apicomplexan parasites segregate chromosomes with fidelity (Iwanaga et al., 2010, 2012). However, in light of the apparent absence of most kinetochore and checkpoint proteins, how these organisms maintain faithful chromosome segregation remains unknown.
In this study, we identify 13 previously unknown components of apicomplexan kinetochores. Apicomplexan kinetochores are highly divergent in sequence and composition from animal and fungal models. Despite sequence disparity, apicomplexan kinetochores display modes of chromosome segregation analogous to metaphase-to-anaphase transition. Furthermore, we show plasticity in kinetochore composition and segregation between lifecycle stages, suggestive of diverse requirements and regulation between parasite modes of division. Given their essentiality towards parasite proliferation, we identify the apicomplexan kinetochore as an excellent therapeutic candidate for selective inhibition.
Results
NUF2 and SKA2 complexes assemble at the nuclear periphery in malaria parasites
Whilst the majority of kinetochore components are not clearly detectable in apicomplexan parasites, centromeric proteins (CENPs) A, C, and E, the SKA component SKA2 and the NDC80 complex (NDC80C) are present (Fig. S1 A), and NDC80/HEC1 has recently been described as a bona fide kinetochore marker in the rodent malarial parasite Plasmodium berghei (Zeeshan et al., 2020). To understand whether apicomplexan kinetochore proteins bear similarities to their animal and fungal cousins, we localized P. berghei components related to different animal kinetochore complexes. Immunoblotting confirmed expression of fusion proteins with the expected mobility (Fig. S1, B and C) corresponding to the integration of the coding sequence for mScarlet-I (mSc) or mNeonGreen fused with triple hemagglutinin-epitope tag (mNG-3xHA) at the C-terminus of endogenous NUF2 (PBANKA_0414300) and SKA2 (PBANKA_0405800), respectively. Localizations of candidate proteins were examined throughout asexual blood-stages and during sexual development that occurs upon host-to-mosquito transmission. In agreement with previously localized apicomplexan centromeres (Brooks et al., 2011; Perrin et al., 2021; Verma and Surolia, 2013) and kinetochores (Farrell and Gubbels, 2014; Zeeshan et al., 2020), location and movement of NUF2 and SKA2 fusion proteins were restricted to the nuclear periphery during progression of asexual blood-stage divisions (Fig. 1 A). Protein levels were undetectable in G1 phase in intraerythrocytic ring stages and first seen in the nucleus at the onset of DNA replication during trophozoite development, then accumulating as punctate foci concomitant with chromosome segregation and formation of daughter nuclei. Signal for both NUF2 and SKA2 fusions reduced to below detectability in fully budded schizonts.
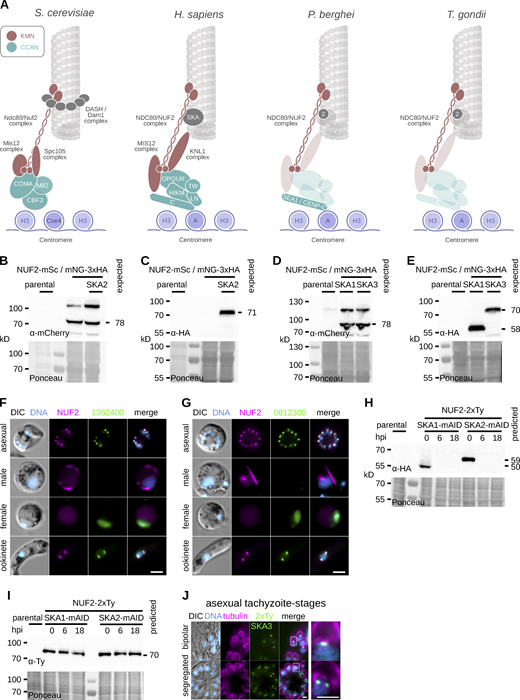
Generation and validation of tagged NUF2 and SKA components in P.berghei and T. gondii. (A) Presence/absence (bold/grey) of known kinetochore proteins in P. berghei and T. gondii. (B–E) Immunoblots of malaria parasites expressing SKA proteins tagged with mNeonGreen-3xHA alongside NUF2-mScarlet-I, probed with either polyclonal α-mCherry or monoclonal α-HA antibodies. Protein loading is shown by Ponceau S stain. (F and G) Micrographs of live native fluorescence in malaria parasites expressing tagged NUF2 and SKA2-interacting proteins during asexual blood stages and sexual mosquito stages of development. Counter-staining of DNA with Hoechst 33342 (cyan) and differential interference contrast images are also shown. Scale bar, 2 μm. (H and I) Immunoblots of T. gondii parasites expressing tagged kinetochore proteins and showing depletion of mAID-3xHA tagged protein upon induction of auxin. Protein loading is shown by Ponceau S stain. (J) Micrographs of fixed immunofluorescence in T. gondii tachyzoites expressing SKA3 tagged with a 2xTy epitope throughout intracellular divisions. Counter-staining tubulin (magenta). DNA staining with DAPI (cyan) and differential interference contrast images are also shown. Scale bar, 5 μm. Source data are available for this figure: SourceData FS1.
Generation and validation of tagged NUF2 and SKA components in P.berghei and T. gondii. (A) Presence/absence (bold/grey) of known kinetochore proteins in P. berghei and T. gondii. (B–E) Immunoblots of malaria parasites expressing SKA proteins tagged with mNeonGreen-3xHA alongside NUF2-mScarlet-I, probed with either polyclonal α-mCherry or monoclonal α-HA antibodies. Protein loading is shown by Ponceau S stain. (F and G) Micrographs of live native fluorescence in malaria parasites expressing tagged NUF2 and SKA2-interacting proteins during asexual blood stages and sexual mosquito stages of development. Counter-staining of DNA with Hoechst 33342 (cyan) and differential interference contrast images are also shown. Scale bar, 2 μm. (H and I) Immunoblots of T. gondii parasites expressing tagged kinetochore proteins and showing depletion of mAID-3xHA tagged protein upon induction of auxin. Protein loading is shown by Ponceau S stain. (J) Micrographs of fixed immunofluorescence in T. gondii tachyzoites expressing SKA3 tagged with a 2xTy epitope throughout intracellular divisions. Counter-staining tubulin (magenta). DNA staining with DAPI (cyan) and differential interference contrast images are also shown. Scale bar, 5 μm. Source data are available for this figure: SourceData FS1.

NDC80C and SKA proteins localize to spindle poles and kinetochores in the malaria parasite. (A–C) Micrographs of live native fluorescence in malaria parasites expressing tagged kinetochore components NUF2-mScarlet-I (magenta) and SKA2-mNeonGreen-3xHA (green) during blood-stage (A), mosquito-stage (B), and ookinete (C) development. Counter-staining of DNA with Hoechst 33342 (cyan) and differential interference contrast images are also shown. Scale bar, 2 μm. (D) U-ExM resolved SKA2 at spindle poles (γ-tubulin and NHS-ester), the spindle (α/β-tubulin) and kinetochores (NUF2). Scale bar, 1 μm. (E) Relative enrichment of proteins identified by mass spectrometry following immunoprecipitation of NUF2-3xHA (magenta) and SKA2-3xHA (green). NDC80C and SKA2-interacting proteins highlighted. Intensities of proteins are not detected for either immunoprecipitation set to arbitrary minimum value. (F) Live native fluorescence of blood- and mosquito-stage cells expressing SKA2-interacting proteins tagged with mNG-3xHA.
NDC80C and SKA proteins localize to spindle poles and kinetochores in the malaria parasite. (A–C) Micrographs of live native fluorescence in malaria parasites expressing tagged kinetochore components NUF2-mScarlet-I (magenta) and SKA2-mNeonGreen-3xHA (green) during blood-stage (A), mosquito-stage (B), and ookinete (C) development. Counter-staining of DNA with Hoechst 33342 (cyan) and differential interference contrast images are also shown. Scale bar, 2 μm. (D) U-ExM resolved SKA2 at spindle poles (γ-tubulin and NHS-ester), the spindle (α/β-tubulin) and kinetochores (NUF2). Scale bar, 1 μm. (E) Relative enrichment of proteins identified by mass spectrometry following immunoprecipitation of NUF2-3xHA (magenta) and SKA2-3xHA (green). NDC80C and SKA2-interacting proteins highlighted. Intensities of proteins are not detected for either immunoprecipitation set to arbitrary minimum value. (F) Live native fluorescence of blood- and mosquito-stage cells expressing SKA2-interacting proteins tagged with mNG-3xHA.
Unexpectedly, SKA2 and NUF2 fusion proteins showed very different localizations during Plasmodium sexual development (Fig. 1 B). Whilst “rod-like bridges,” as previously described for components of the NDC80C (Zeeshan et al., 2020), were visible for NUF2 during the three rounds of DNA replication and mitosis that occur at microgametogenesis—SKA2 signal was not detectable. A similar dichotomy was seen postactivation of the macrogametocyte, NUF2 spreading sparingly across the cytoplasm compared to SKA2 residing primarily in the nucleus. However, following completion of meiosis both fusion proteins united as four distinct nuclear foci in fully developed banana-shaped ookinetes. Time-course fluorescence microscopy revealed a hierarchical assembly for SKA2 and NUF2 throughout ookinete development (Fig. 1 C). SKA2 accumulated as a single focus at the nuclear periphery between 1 and 2 h after fertilization, which duplicated and migrated to opposing nuclear poles between 2 and 4 h. In contrast, NUF2 foci first appeared later between 4 and 6 h of development, at which point SKA2 signal stretched along a spindle-like structure connecting opposing poles. Signal from both fusion proteins stretched along the central spindle by 8–10 h. Two successive rounds of asynchronous duplication and migration ultimately formed four puncta at the nuclear periphery by 12 h of development.
In contrast to the limitations in defining nanometer-scale structures by conventional fluorescence microscopy, Ultrastructure Expansion Microscopy (U-ExM; Bertiaux et al., 2021; Gambarotto et al., 2019) resolved kinetochores along the ookinete meiotic spindle (Fig. 1 D). The majority of SKA2 signal localized to spindle poles, closely associated to γ-tubulin. Upon formation of a diamond-shaped bipolar spindle, additional foci were obvious both along the spindle and at the spindle equator, colocalizing with NUF2 at kinetochores.
To identify interacting proteins for NUF2 and SKA2 in Plasmodium, we affinity-purified both proteins tagged with 3xHA from synchronized mitotic gametocytes and combined label-free semiquantitative mass spectrometry (Trudgian et al., 2011) to estimate enrichment of interacting proteins (Fig. 1 E). We compared samples by integrated spectral intensities, reasoning that if NDC80 and SKA2 assemble independent kinetochore complexes in Plasmodium, stable interactors would be evident in each pull down relative to one another. NDC80, SPC25, and SPC24 were the most abundant proteins copurifying with NUF2 relative to SKA2. Similarly, the abundance of two hypothetical proteins of unknown function (gene IDs: PBANKA_0812300 and PBANKA_1362400) strongly suggests the presence of a three SKA component complex in the malaria parasite. To validate our biochemical approach, each SKA2 interacting protein was tagged with mNG-3xHA (Fig. S1, D and E). Both tagged PBANKA_0812300 and PBANKA_1362400 proteins showed localization patterns reminiscent of SKA2 in the malaria parasite (Fig. 1 F; and Fig. S1, F and G), colocalizing with NUF2 during asexual blood stage divisions, not detected in microgametocytes and present as four nuclear foci in fully developed ookinetes.
SKA proteins are required for centromere alignment and segregation in T. gondii
The SKA complex (SKAC) has been identified in all 5–6 eukaryotic supergroups (van Hooff et al., 2017). Despite this broad distribution, the complex is not ubiquitously detected in functionally characterized kinetochores, such as in fungi and kinetoplastids. Missing components within SKAC occurs more sporadically, suggesting spurious absences may result from lack of detection due to sequence divergence rather than genuine gene loss. In an attempt to more sensitively identify SKA2-interacting proteins, we employed an iterative hidden Markov model (HMM) profiling strategy. Briefly, HMMs constructed from clear homologs of PBANKA_0812300 and PBANKA_1362400 were used as search queries across an in-house database of HMMs generated from homologous groups of alveolate protein sequences (Table S1) and manually curated HMMs generated from previously classified eukaryotic kinetochore sequences (van Hooff et al., 2017). Significant hits were concatenated iteratively and identified pan-SKA HMMs as highest scoring kinetochore HMMs (Fig. 2, A and B). In particular, we reunited PBANKA_0812300 with SKA1 and PBANKA_1362400 with SKA3 and detected corresponding putative homologs in T. gondii TGME49_264960 and TGME49_289790, respectively.

Toxoplasma SKA proteins accumulate at spindle poles and kinetochores at the onset of mitosis. (A and B) HMMs constructed using alveolate homologs of SKA2-interacting proteins identify HMMs including animal SKA proteins as highest scoring hits. (C) Micrographs of fixed immunofluorescence in dividing intracellular T. gondii tachyzoites expressing kinetochore components SKA1-2xTy and SKA2-2xTy. Counter-staining of DNA (cyan), acetylated α-tubulin (magenta), and differential interference contrast images are also shown. Scale bar, 5 μm. (D) Counter-staining (magenta) for the apicoplast (CPN60), centrosome (Centrin1), and kinetochores (NUF2-2xTy) are also shown. (E and F) U-ExM resolved SKA2 at spindle poles and kinetochores (E; α/β-tubulin and NUF2, respectively), concomitant with alignment of centromeres at metaphase (F; TgChromo1). Scale bar, 2 μm.
Toxoplasma SKA proteins accumulate at spindle poles and kinetochores at the onset of mitosis. (A and B) HMMs constructed using alveolate homologs of SKA2-interacting proteins identify HMMs including animal SKA proteins as highest scoring hits. (C) Micrographs of fixed immunofluorescence in dividing intracellular T. gondii tachyzoites expressing kinetochore components SKA1-2xTy and SKA2-2xTy. Counter-staining of DNA (cyan), acetylated α-tubulin (magenta), and differential interference contrast images are also shown. Scale bar, 5 μm. (D) Counter-staining (magenta) for the apicoplast (CPN60), centrosome (Centrin1), and kinetochores (NUF2-2xTy) are also shown. (E and F) U-ExM resolved SKA2 at spindle poles and kinetochores (E; α/β-tubulin and NUF2, respectively), concomitant with alignment of centromeres at metaphase (F; TgChromo1). Scale bar, 2 μm.
To validate our bioinformatic approach, SKA1—3 were tagged in T. gondii by fusion at the endogenous locus with a mini auxin-induced degron (mAID) including triple hemagglutinin (3xHA)-epitope tag, to localize and deplete protein upon addition of auxin (IAA; Fig. S1, H and I). Costaining DNA and the mitotic spindle revealed TgSKA components were undetectable in G1 phase, but accumulated centrally upon a bipolar spindle during mitosis (Fig. 2 C and Fig. S1 J). Foci segregated at anaphase into budding daughter cells, returning to below detectability at cytokinesis. Furthermore, coexpression of NUF2 fused to a double Ty epitope tag (NUF2-2xTy) or costaining of various intracellular organelles revealed that SKA proteins substantially displaced distal to the apicoplast and centrosome compared to kinetochores (Fig. 2 D).
U-ExM further resolved the tachyzoite mitotic spindle (Fig. 2 E). In tachyzoites, the spindle nucleates from a region closely associated to a centriolar microtubule organizing center. Following MTOC duplication, a short prometaphase spindle elongates to form a bipolar opposing diamond at metaphase, prior to spindle collapse at anaphase. Whilst TgNUF2 localized to a region separating newly duplicated spindle poles, TgSKA2 was not detected until diamond-spindle formation. Costaining pericentromeric chromatin (TgChromo1; Gissot et al., 2012; Fig. 2 F) identified diamond-spindle formation accompanied alignment of centromeres at a striking metaphase plate before partitioning into budding daughter cells. As in the malaria parasite, the majority of Toxoplasma SKA signal localized to spindle poles and the midzone.
Depletion of TgSKA proteins (Fig. S2 A) led to a severe reduction in lytic plaque formation (Fig. S2 B). Compared to a previous depletion of NUF2 by tetracyclin regulated promoter (Farrell and Gubbels, 2014), the effects of auxin-induced depletion of NUF2, in addition to SKA1 and SKA2, were evident within the first cell cycle 6 h after induction (Fig. S2 C). By 18 h, the majority of vacuoles had abnormal cell numbers and accumulations of DNA with no associated cell body (Fig. S2, D and E).

Toxoplasma SKA proteins are required for intracellular growth and lytic plaque formation. (A) Strategy for auxin-induced depletion of T. gondii kinetochore components. (B) Tachyzoites depleted for SKA1–3 failed to form lysis plaques 7 d after inoculation compared to parental controls. (C) Intracellular growth is severely reduced in T. gondii depleted of SKA1 and 2 and NUF2 (**, P < 0.01; ***, P < 0.001; χ2 test). (D and E) Intracellular vacuoles containing accumulations of DNA (arrow) and no associated cell body were present after depletion of SKA1–2 tagged with mAID-3xHA. Scale bar, 5 µm. (F and G) Insertion of mScarlet-I at the C-terminus of MORN1 is detrimental to parasite growth.
Toxoplasma SKA proteins are required for intracellular growth and lytic plaque formation. (A) Strategy for auxin-induced depletion of T. gondii kinetochore components. (B) Tachyzoites depleted for SKA1–3 failed to form lysis plaques 7 d after inoculation compared to parental controls. (C) Intracellular growth is severely reduced in T. gondii depleted of SKA1 and 2 and NUF2 (**, P < 0.01; ***, P < 0.001; χ2 test). (D and E) Intracellular vacuoles containing accumulations of DNA (arrow) and no associated cell body were present after depletion of SKA1–2 tagged with mAID-3xHA. Scale bar, 5 µm. (F and G) Insertion of mScarlet-I at the C-terminus of MORN1 is detrimental to parasite growth.
To explore the requirement of TgSKA proteins towards nuclear division, we monitored the Toxoplasma centrosome and centromeres in the presence or absence of SKA1 or SKA2. In contrast to tagging with YFP (Gubbels et al., 2006), introduction of the coding sequence for mScarlet-I at the C-terminus of the centrosome component MORN1 was detrimental to parasite proliferation (Fig. S2, F and G). Instead, costaining of the spindle with markers of the centrosome (TgCentrin1) or pericentromeric chromatin revealed depletion of either SKA1 or SKA2 led in a strong increase in mitotic index (Fig. 3, A and B). This defect was apparent after duplication of the centrosome, with an accumulation of cells with two centrin foci or a single discernible TgChromo1 focus along monopolar or bipolar mitotic spindles. U-ExM revealed cells with elongated mitotic spindles, misaligned centromeres at the metaphase plate, and lagging centromeres and kinetochores unable to properly partition into daughter cells (Fig. 3, C and D).

Toxoplasma SKA proteins are essential for centromere and kinetochore segregation. (A and B) Morphological analyses of cells depleted for SKA1 or SKA2 tagged with mAID-3xHA reveal a buildup of cells with duplicated Centrin1 staining (A) or a single TgChromo1 focus (B) along bipolar or monopolar spindles (**, P < 0.01; ***, P < 0.001; χ2 test). Representative micrographs are shown below. Counter-staining of DNA (cyan), acetylated α-tubulin (magenta), and centrin1 or TgChromo1 (green). Scale bar, 2.5 μm. (C and D) U-ExM revealed tachyzoites with elongated mitotic spindles and unaligned and lagging kinetochores (C) or centromeres (D). Scale bar, 1 µm. (E and F) Levels of NUF2 (E) and SKA1 or SKA2 (F) at kinetochores in cells depleted for either protein. Representative micrographs are shown below. DNA (cyan), centrin1 (tomato), and NUF2, SKA1, or SKA2 (green). Scale bar, 4 μm.
Toxoplasma SKA proteins are essential for centromere and kinetochore segregation. (A and B) Morphological analyses of cells depleted for SKA1 or SKA2 tagged with mAID-3xHA reveal a buildup of cells with duplicated Centrin1 staining (A) or a single TgChromo1 focus (B) along bipolar or monopolar spindles (**, P < 0.01; ***, P < 0.001; χ2 test). Representative micrographs are shown below. Counter-staining of DNA (cyan), acetylated α-tubulin (magenta), and centrin1 or TgChromo1 (green). Scale bar, 2.5 μm. (C and D) U-ExM revealed tachyzoites with elongated mitotic spindles and unaligned and lagging kinetochores (C) or centromeres (D). Scale bar, 1 µm. (E and F) Levels of NUF2 (E) and SKA1 or SKA2 (F) at kinetochores in cells depleted for either protein. Representative micrographs are shown below. DNA (cyan), centrin1 (tomato), and NUF2, SKA1, or SKA2 (green). Scale bar, 4 μm.
To investigate the hierarchy of Toxoplasma kinetochore components, we looked at the recruitment and codependency of NUF2 relative to SKA1 or SKA2, in cells depleted for either protein (Fig. 3, E and F). Whilst neither SKA component was required for recruitment of NUF2, kinetochore assembly of SKA1 and SKA2 was abolished in mitotic tachyzoites depleted for NUF2.
Taken together, these observations indicate that SKA components localize to both the spindle poles and kinetochores in apicomplexan parasites. Maintenance of the SKA at Toxoplasma kinetochores is dependent on, and possibly downstream to, assembly of the NDC80C, together required proper chromosome segregation at mitosis.
Apicomplexan kinetochore protein 1 marks a new kinetochore compartment in P.berghei
The apparent absence of CCAN components outside of CENP-C, in addition to MIS12 and KNL1 complexes (MIS12C and KNL1C) of the KMN network, questions what bridges the apicomplexan outer kinetochore to the centromere? NDC80C and SKAC appear to be biochemically distinct sets and affinity purifications of NUF2 and SKA2 did not reveal additional candidates. To investigate the possibility of less stable kinetochore interactions, we employed a proximity-based approach of affinity purification under conditions of formaldehyde cross-linking and compared spectral intensities to controls without cross-linking (Fig. 4 A). In addition to components of the minichromosome maintenance (MCM) complex; MCM3 and MCM6, two proteins were most enriched upon cross-linking (green). One of these proteins is a homolog of STU2 (PBANKA_1337500; Data S1), a protein required for the correction of kinetochore-spindle attachment errors and bipolar spindle stability during chromosome biorientation (Miller et al., 2016, 2019). In contrast, a hypothetical protein of unknown function (PBANKA_0621300) bore no obvious sequence similarity to known animal or fungal proteins, whilst orthologs were identified across apicomplexan genomes (Data S2). In agreement with biochemical interaction, the tagged protein colocalized with NUF2 throughout asexual blood-stage divisions (Fig. 4 B), in addition to across spindle bundles during microgametogenesis (Fig. 4 C; and Fig. S3 A). Furthermore, the identified protein accumulated as foci in the nuclei of female gametes prior to fertilization, suggesting recruitment to centromeres before SKAC and NDC80C. Given biochemical affinities, kinetochore localization and conservation, we named the protein apicomplexan kinetochore protein 1 (AKiT1).

AKiT1 is a component of the Plasmodium kinetochore. (A) Relative enrichment of proteins (shaded threshold) identified following immunoprecipitation of NUF2 upon cross-linking compared to non–cross-linked cells. (B and C) Micrographs of blood-stage (B) and mosquito-stage (C) cells expressing NUF2-mScarlet-I (magenta) and AKiT1-mNG-3xHA (green). Scale bar, 2 μm. (D and E) U-ExM identified alignment of tagged AKiT1 and NUF2 kinetochores during blood-stage divisions (D), whilst dispersed along the mitotic spindle during microgametogenesis (E). Scale bar, 1 μm. (F and G) Centroid measurements for head-to-head AKiT1 foci along the microgametocyte spindle (F), and relative to NUF2 and centrin in segregated clusters (G). Total number of foci analyzed, SEM error bars, and representative micrograph shown. Centrin1 (cyan), NUF2 (magenta), and AKiT1 (green). Scale bar, 2 μm.
AKiT1 is a component of the Plasmodium kinetochore. (A) Relative enrichment of proteins (shaded threshold) identified following immunoprecipitation of NUF2 upon cross-linking compared to non–cross-linked cells. (B and C) Micrographs of blood-stage (B) and mosquito-stage (C) cells expressing NUF2-mScarlet-I (magenta) and AKiT1-mNG-3xHA (green). Scale bar, 2 μm. (D and E) U-ExM identified alignment of tagged AKiT1 and NUF2 kinetochores during blood-stage divisions (D), whilst dispersed along the mitotic spindle during microgametogenesis (E). Scale bar, 1 μm. (F and G) Centroid measurements for head-to-head AKiT1 foci along the microgametocyte spindle (F), and relative to NUF2 and centrin in segregated clusters (G). Total number of foci analyzed, SEM error bars, and representative micrograph shown. Centrin1 (cyan), NUF2 (magenta), and AKiT1 (green). Scale bar, 2 μm.

AKiT1 is a component of the Plasmodium kinetochore. (A and B) Widefield images of native fluorescence (A) and U-ExM (B) in malaria parasites expressing NUF2-mScarlet-I (magenta) and AKiT1-mNeonGreen-3xHA (green) during mosquito stages of development. U-ExM revealed AKiT1 along the spindle (identified by α/β-tubulin counter-stain) and at spindle poles (centrin). DNA (cyan) is also shown. Scale bar, 2 μm.
AKiT1 is a component of the Plasmodium kinetochore. (A and B) Widefield images of native fluorescence (A) and U-ExM (B) in malaria parasites expressing NUF2-mScarlet-I (magenta) and AKiT1-mNeonGreen-3xHA (green) during mosquito stages of development. U-ExM revealed AKiT1 along the spindle (identified by α/β-tubulin counter-stain) and at spindle poles (centrin). DNA (cyan) is also shown. Scale bar, 2 μm.
U-ExM during intraerythrocytic development revealed centrosome migration and mitotic spindle formation were accompanied by the alignment of PbAKiT1 foci at the metaphase plate (Fig. 4 D). Interestingly, spindle length changed drastically between asexual and sexual stage cells (Fig. 4 D and E; and Fig. S3 B). During the first round of microgametocyte mitosis, PbAKiT1 tagged at the C-terminus localized as pairs of foci along the spindle (Fig. 4 F; mean distance apart 139 ± 4 nm; determined as distance divided by expansion factor). Kinetochores then fired asynchronously to spindle poles (). Two additional rounds of mitosis ultimately produced kinetochore foci arranged into six to eight cone-shaped clusters at the nuclear periphery. Crucially, additional staining of NUF2 identified subkinetochore localizations and a clear bipartite architecture in segregated kinetochore clusters, with the distinction of the PbAKiT1-mNG-3xHA at the inner kinetochore relative to PbNUF2-mSc at the outer kinetochore and centrin at the centrosome (Fig. 4 G; mean distances of 103 ± 3 nm and 425 ± 6 nm, respectively).
AKiT1 is an essential component of the Toxoplasma kinetochore
Previous genome-wide screenings have indicated that AKiT1 is essential for blood-stage proliferation in both human and rodent malaria parasites (Bushell et al., 2017; Zhang et al., 2018). Furthermore, we were unable to interrogate AKiT1 function using conditional approaches available in P. berghei, including no clear evidence for protein depletion upon introduction of an auxin-inducible degradation motif and no recovery of parasites following attempts to integrate blood-stage specific promoters (Table S2). We therefore interrogated the AKiT1 ortholog in T. gondii by fusion with an auxin-inducible degron, speculating that kinetochore architectures with respect to AKiT1 may bear similarities across apicomplexan organisms. Tagged TgAKiT1 showed characteristic kinetochore localizations (Fig. 5 A), with clearer association of foci with respect to TgNUF2 at kinetochores compared to the centrosome or apicoplast (Fig. S4 A). In contrast to SKA proteins, TgAKiT1 foci formed a cone at the nuclear periphery during centrosome duplication (Fig. 5 B). Little overlap was seen with TgNUF2 at the spindle and TgAKiT1 foci aligned at the metaphase pate prior to segregation at anaphase.

AKiT1 is an essential component of the Toxoplasma kinetochore. (A) Micrographs of T. gondii tachyzoites expressing AKiT1-2xTy. DNA (cyan), tubulin (magenta), and differential interference contrast images shown. Scale bar, 5 μm. (B) U-ExM identified alignment of AKiT1 foci at metaphase. Scale bar, 2 μm. (C and D) Depletion of AKiT1-mAID-3xHA delayed mitotic progression, with a buildup of cells with duplicated centrin foci (C) unable to properly partition TgChromo1 (D; **, P < 0.01; ***, P < 0.001; χ2 test). (E and F) U-ExM revealed tachyzoites displaying elongated mitotic spindles, with misaligned and lagging kinetochores (E) and centromeres (F). Scale bar, 1 µm. Levels of NUF2 and AKiT1 in cells depleted for either component. Representative images are also shown.
AKiT1 is an essential component of the Toxoplasma kinetochore. (A) Micrographs of T. gondii tachyzoites expressing AKiT1-2xTy. DNA (cyan), tubulin (magenta), and differential interference contrast images shown. Scale bar, 5 μm. (B) U-ExM identified alignment of AKiT1 foci at metaphase. Scale bar, 2 μm. (C and D) Depletion of AKiT1-mAID-3xHA delayed mitotic progression, with a buildup of cells with duplicated centrin foci (C) unable to properly partition TgChromo1 (D; **, P < 0.01; ***, P < 0.001; χ2 test). (E and F) U-ExM revealed tachyzoites displaying elongated mitotic spindles, with misaligned and lagging kinetochores (E) and centromeres (F). Scale bar, 1 µm. Levels of NUF2 and AKiT1 in cells depleted for either component. Representative images are also shown.
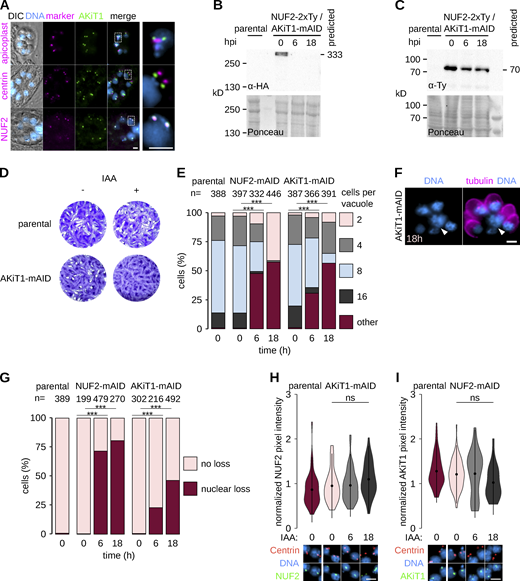
AKiT1 is required for kinetochore segregation in Toxoplasma.(A) Micrographs of fixed immunofluorescence in T. gondii expressing AKiT1-mAID-3xHA throughout intracellular divisions. Counter-staining with antibodies raised against organelle markers (magenta) for the apicoplast (CPN60), centrosome (Centrin1), and kinetochores (NUF2-2xTy). DNA staining with DAPI (cyan) and differential interference contrast images are also shown. Scale bar, 5 μm. (B and C) Immunoblots of T. gondii parasites expressing tagged kinetochore proteins and showing depletion of AKiT1 protein upon induction of auxin. Protein loading is shown by Ponceau S stain. (D) Depletion of AKiT1-mAID-3xHA prevented proper formation of lysis plaques 7 d postinoculation compared to parental controls. (E) Intracellular growth is severely reduced in T. gondii depleted of AKiT1 and NUF2 (***, P < 0.001; χ2 test). (F and G) Intracellular vacuoles containing accumulations of DNA (arrow) and no associated cell body were present after depletion of SKA1–2 tagged with mAID-3xHA. Scale bar, 5 µm. (H and I) Levels and localization of NUF2 and AKiT1 in cells depleted for either component. Representative images are shown below. Scale bar, 4 μm. Source data are available for this figure: SourceData FS4.
AKiT1 is required for kinetochore segregation in Toxoplasma.(A) Micrographs of fixed immunofluorescence in T. gondii expressing AKiT1-mAID-3xHA throughout intracellular divisions. Counter-staining with antibodies raised against organelle markers (magenta) for the apicoplast (CPN60), centrosome (Centrin1), and kinetochores (NUF2-2xTy). DNA staining with DAPI (cyan) and differential interference contrast images are also shown. Scale bar, 5 μm. (B and C) Immunoblots of T. gondii parasites expressing tagged kinetochore proteins and showing depletion of AKiT1 protein upon induction of auxin. Protein loading is shown by Ponceau S stain. (D) Depletion of AKiT1-mAID-3xHA prevented proper formation of lysis plaques 7 d postinoculation compared to parental controls. (E) Intracellular growth is severely reduced in T. gondii depleted of AKiT1 and NUF2 (***, P < 0.001; χ2 test). (F and G) Intracellular vacuoles containing accumulations of DNA (arrow) and no associated cell body were present after depletion of SKA1–2 tagged with mAID-3xHA. Scale bar, 5 µm. (H and I) Levels and localization of NUF2 and AKiT1 in cells depleted for either component. Representative images are shown below. Scale bar, 4 μm. Source data are available for this figure: SourceData FS4.
Depletion of TgAKiT1 (Fig. S4, B and C) severely reduced parasite survival (Fig. S4 D), with most vacuoles displaying abnormal cell numbers (Fig. S4 E) and accumulations of DNA, 18 h after depletion (Fig. S4, F and G). These cells were primarily stalled at anaphase, with an increase in cells with duplicated centrin foci or a single TgChromo1 focus (Fig. 5, C and D), presenting elongated spindles, misaligned centromeres, and lagging kinetochores (Fig. 5, E and F).
AKiTs bridge the outer kinetochore to the centromere
In animals and fungi, kinetochores are composed of hierarchical assemblies totaling ∼50–100 proteins. Whilst exciting, the addition of AKiT1 does not increase our repertoire of apicomplexan kinetochore past mere order of magnitude less than in animal or fungal systems. Streamlining of redundant kinetochore composition is not uncommon in eukaryotes (Przewloka et al., 2007) and it is quite possible few apicomplexan kinetochore proteins manage the roles of many in other organisms. However, neither the centromeric histone variant CENH3 nor SEA1 (a previously reported homolog of CENP-C; Perrin et al., 2021; Verma and Surolia, 2014) were identified in immunoprecipitates of NUF2 or SKA2, suggesting additional components may bridge the outer kinetochore to the centromere. To test whether PbAKiT1 interacts with centromeric proteins, we employed a similar proximity cross-linking-based approach and affinity-purified protein, however with an additional limited cross-linking condition (Fig. S5, A and B; D’Archivio and Wickstead, 2017). Given that at least in animals the SPC24:SPC25 heterodimer forms a direct interaction with the CCAN, we additionally immunopurified PbSPC24 under the same conditions, to compare interactors (Fig. 6 A). A clear reciprocal enrichment was seen for each component of NDC80C across all immunoprecipitates of SPC24. Additionally, PbAKiT1 was enriched—albeit marginally—upon cross-linking, reinforcing a less stable interaction between PbSPC24 and PbAKiT1 compared to PbSPC24 and remaining NDC80C components. Two additional proteins of unknown function (gene IDs: PBANKA_1243900, PBANKA_0522000) were identified at comparable levels. Proteins belonging to the prereplicative complex MCM6, ORC1, and CDC6 were enriched upon higher cross-linking conditions and this set also contained components of the SKAC and STU2, along with six additional hypothetical proteins of unknown function. SEA1 was identified upon high cross-linking alone.
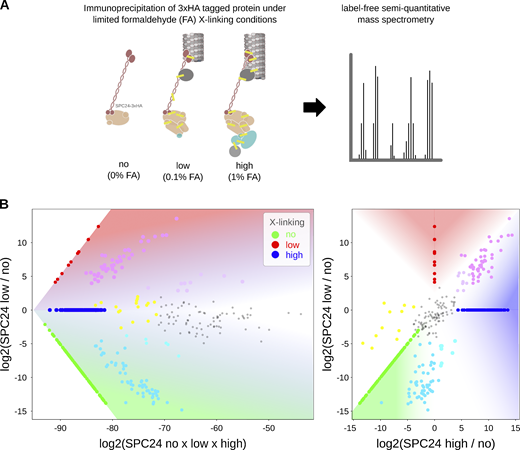
Relative protein abundance following immunoprecipitation under limited cross-linking and mass spectrometry. (A) General workflow for immunoprecipitation under limited cross-linking and mass spectrometry. (B) Representative plot demonstrating positions of relative protein abundances in main plot (Fig. 6 A). The values in Table S3, as identified under specific formaldehyde (FA) cross-linking conditions no (0%), low (0.1%), and high (1%), are multiplied by (×) or divided by (/) one another, according to the axes, and log2 transformed. For display, intensities not detected under a specific condition are set to the minimum value identified across all experiments (in this instance 2.87 × 10−10). Plotted intensities identified under specific cross-linking conditions are colored according to the key and those identified under combinations of conditions are mix colored, e.g., relative abundances enriched upon cross-linking compared to noncross-linking are displayed in magenta/pink, whereas proteins identified as equally abundant across all conditions are in grey.
Relative protein abundance following immunoprecipitation under limited cross-linking and mass spectrometry. (A) General workflow for immunoprecipitation under limited cross-linking and mass spectrometry. (B) Representative plot demonstrating positions of relative protein abundances in main plot (Fig. 6 A). The values in Table S3, as identified under specific formaldehyde (FA) cross-linking conditions no (0%), low (0.1%), and high (1%), are multiplied by (×) or divided by (/) one another, according to the axes, and log2 transformed. For display, intensities not detected under a specific condition are set to the minimum value identified across all experiments (in this instance 2.87 × 10−10). Plotted intensities identified under specific cross-linking conditions are colored according to the key and those identified under combinations of conditions are mix colored, e.g., relative abundances enriched upon cross-linking compared to noncross-linking are displayed in magenta/pink, whereas proteins identified as equally abundant across all conditions are in grey.
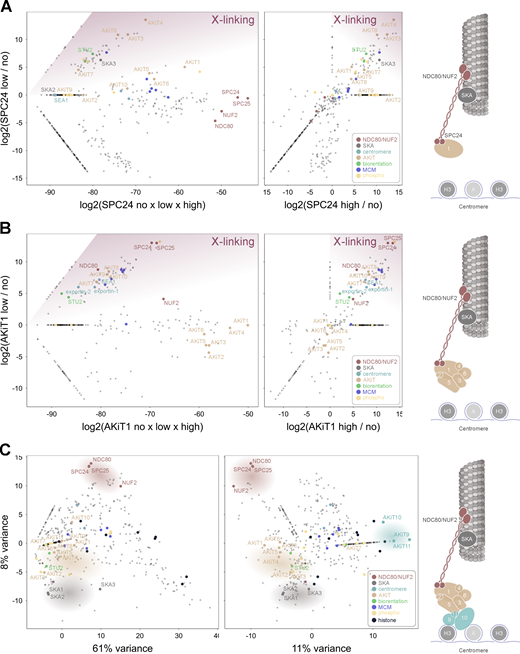
AKiT1 interacts with additional AKiT components and proteins at the Plasmodium centromere. (A and B) Relative enrichment of proteins immunopurified with SPC24-3xHA (A) and AKiT1-3xHA (B) under conditions of low compared to high and no cross-linking conditions (left), or low compared to high cross-linking (right). (C) Principal components 1–3 of integrated spectral intensities identified following immunopurifications of NUF2, SPC24, AKiT1, AKiT8, AKiT9, SKA2, PBANKA_1343200, NEK1, and KIN8X. Intensities for all 780 proteins detected across experiments are presented in Table S3.
AKiT1 interacts with additional AKiT components and proteins at the Plasmodium centromere. (A and B) Relative enrichment of proteins immunopurified with SPC24-3xHA (A) and AKiT1-3xHA (B) under conditions of low compared to high and no cross-linking conditions (left), or low compared to high cross-linking (right). (C) Principal components 1–3 of integrated spectral intensities identified following immunopurifications of NUF2, SPC24, AKiT1, AKiT8, AKiT9, SKA2, PBANKA_1343200, NEK1, and KIN8X. Intensities for all 780 proteins detected across experiments are presented in Table S3.
Along with PbAKiT1, five proteins of unknown function were most abundant across PbAKiT1 immunoprecipitates (Fig. 6 B), four of which (gene IDs: PBANKA_0612200, PBANKA_1310500, PBANKA_1243900, and PBANKA_0522000) were also enriched in purifications of SPC24 upon cross-linking. Each tagged-hypothetical protein (Fig. S6 A) showed PbAKiT1-like localization patterns (Fig. 7 A), colocalizing with NUF2 during blood-stage and microgametocyte mitosis, also present as foci in activated female macrogametes and accumulating as four puncta in fully developed ookinetes. We therefore named these proteins AKiT2–6. In addition to components of the prereplicative complex, MCM2–7, and STU2, proteins previously reported to interact with apicomplexan centromeres SMC1, Exportin 1, and Exportin 7 (Francia et al., 2020), in addition to SEA1 were enriched upon low cross-linking conditions, and this threshold showed reciprocal enrichment for all components of the NDC80C.
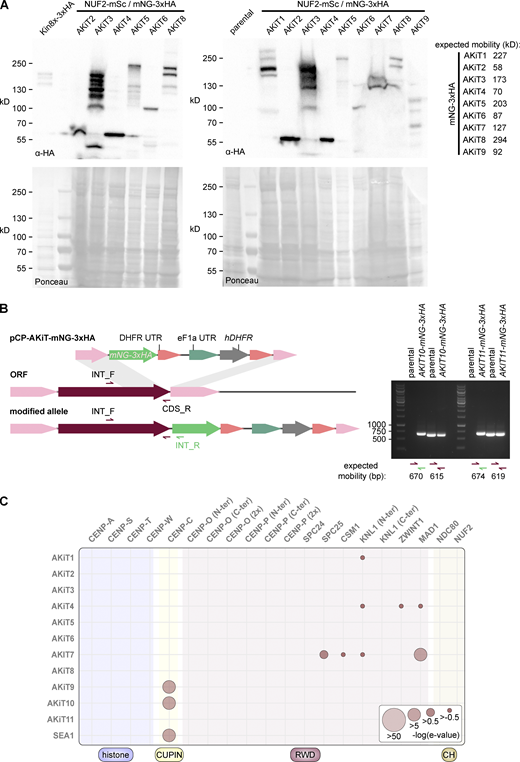
Generation of tagged AKiT components in P. berghei. (A) Immunoblots of malaria parasites expressing tagged AKiT proteins, probed with a monoclonal α-HA antibody. Protein loading is shown by Ponceau S stain. (B) PCR on genomic DNA of malaria parasites expressing tagged AKiT10 and 11 alongside parental controls. (C) HMM profile-profile comparisons using domain-only kinetochore HMMs against AKiT orthologs. Source data are available for this figure: SourceData FS6.
Generation of tagged AKiT components in P. berghei. (A) Immunoblots of malaria parasites expressing tagged AKiT proteins, probed with a monoclonal α-HA antibody. Protein loading is shown by Ponceau S stain. (B) PCR on genomic DNA of malaria parasites expressing tagged AKiT10 and 11 alongside parental controls. (C) HMM profile-profile comparisons using domain-only kinetochore HMMs against AKiT orthologs. Source data are available for this figure: SourceData FS6.

AKiTs accumulate alongside NUF2 at the nuclear periphery in malaria parasites. (A–C) Micrographs of P. berghei expressing NUF2-mScarlet-I (magenta) and AKiT1–6 (A), AKiT7–9 (B) and AKiT10–11 (C) tagged with mNG-3xHA (green). Scale bar, 2 μm.
AKiTs accumulate alongside NUF2 at the nuclear periphery in malaria parasites. (A–C) Micrographs of P. berghei expressing NUF2-mScarlet-I (magenta) and AKiT1–6 (A), AKiT7–9 (B) and AKiT10–11 (C) tagged with mNG-3xHA (green). Scale bar, 2 μm.
Interestingly, three proteins of unknown function (gene IDs: PBANKA_0612300, PBANKA_0213200, and PBANKA_1307000) formed a distinct enrichment profile compared to AKiT1–6 across immunoprecipitations. Each tagged protein showed characteristic kinetochore localizations (Fig. 7 B), with that encoded by PBANKA_0612300 present in the absence of NUF2-mSc signal and reminiscent of the nuclear membrane. We named these proteins AKiT7–9, respectively. To test the extent and composition of additional kinetochore complexes, the relative abundances of copurifying proteins identified in each kinetochore protein immunoprecipitation (without cross-linking) were assessed by principal component analysis (Brusini et al., 2021; Fig. 6 C), including NUF2, SPC24, SKA2, AKiT1, AKiT8, and AKiT9, alongside controls previously shown to localize in the vicinity of the Plasmodium spindle, KIN8X (Zeeshan et al., 2019), and NEK1 (Dorin-Semblat et al., 2011). Principal components 1 and 3 encompass 69% of the total variance in the data and show distinct clustering of NDC80C and SKA components relative to all AKiTs. Principle components 2 and 3 further resolved AKiT clustering, with the distinction of AKiT1–6 relative to AKiT9, itself clustering with histones H3 and H4, component of the histone chaperone SPT16 and two additional proteins of unknown function (gene IDs: PBANKA_0406000 and PBANKA_0803900) that we named AKiT10 and AKiT11, respectively. Both tagged proteins (Fig. S6 B) showed kinetochore-like localizations (Fig. 7 C), with AKiT11-mNG-3xHA positive cells, however, lost from parasite populations following transfection, suggesting a detrimental modification towards parasite growth.
Apicomplexan kinetochores are highly divergent in sequence and composition from animal and fungal models
AKiT1–11 have not previously been annotated with protein function or domains. HMMs spanning kinetochore protein domains were unable to classify most AKiT sequences (Fig. S6 C), excepting an RWD domain of the spindle assembly checkpoint protein MAD1 that is conserved within AKiT7 (e−21). Similarly, cupin domains found in AKiT9 (e−28), AKiT10 (e−8), and SEA1 (e−15) suggest that these genes are homologous to the CCAN component CENP-C. To further explore similarities between AKiTs and known conventional kinetochore proteins (cKiTs), we queried AKiT HMMs against our database of full-length kinetochore protein HMMs (Fig. 8 A). Putative homologs were further scrutinized for structural similarity (DALI Z-scores) using AlphaFold2 (AF2)-predicted structures (Fig. 8, B and C). Full-length HMM profiles revealed a higher degree of similarity between AKiT1 and KNL1 (Fig. 8 A; e−2), with the strongest signal coming from a coiled-coil region plus the N-terminal half of the first RWD domain. AF2 confirmed a coiled-coil–RWD topology in AKiT1 orthologs similar to cKiTs (Fig. 8 B). Although Plasmodium AKiT1’s RWD domain could not be confidently folded (50 > pLDDT > 7), we found greater similarity between AKiT1 of the apicomplexan Nephromyces (Fig. 8 B) and the double RWD domain of KNL1.
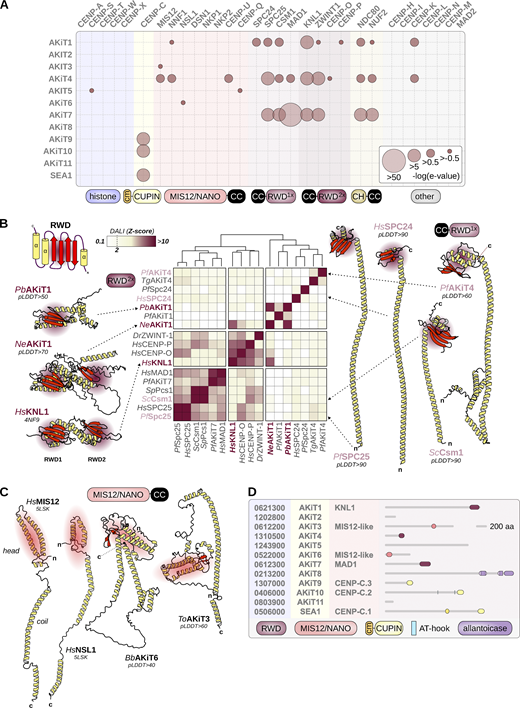
Apicomplexan kinetochores include distant relatives of eukaryotic kinetochore and spindle assembly checkpoint proteins. (A) HMM profile-profile comparisons using full-length conventional kinetochore HMMs (Tromer et al., 2019) and alveolate homologs of AKiT proteins. E-values are binned to indicate the confidence of detection. (B) Structural comparisons of the RWD domains of cKiTs and AKiTs (AF2-predicted structures) using DALI. Z-scores are clustered using Ward’s method of minimum variance clustering. For previously resolved structures, a PDB code is shown and for AF2 the average pLDDT confidence score shown. Two letters indicate species (Table S1). (C) Resolved structures for HsMIS12 and HsNSL1 and AF2-predicted structures for ToAKiT3 and BbAKiT6, the latter two chosen due to their shorter length than Plasmodium orthologs. (D) Predicted protein domain architectures for AKiT1–11.
Apicomplexan kinetochores include distant relatives of eukaryotic kinetochore and spindle assembly checkpoint proteins. (A) HMM profile-profile comparisons using full-length conventional kinetochore HMMs (Tromer et al., 2019) and alveolate homologs of AKiT proteins. E-values are binned to indicate the confidence of detection. (B) Structural comparisons of the RWD domains of cKiTs and AKiTs (AF2-predicted structures) using DALI. Z-scores are clustered using Ward’s method of minimum variance clustering. For previously resolved structures, a PDB code is shown and for AF2 the average pLDDT confidence score shown. Two letters indicate species (Table S1). (C) Resolved structures for HsMIS12 and HsNSL1 and AF2-predicted structures for ToAKiT3 and BbAKiT6, the latter two chosen due to their shorter length than Plasmodium orthologs. (D) Predicted protein domain architectures for AKiT1–11.
A similar coiled-coil–RWD toplogy was detected at the C-terminus of AKiT4, with RWD domains of SPC25 and KNL1 as most similar. Comparison of AF2 structures revealed a single RWD domain at the C-terminus most similar to SPC24, SPC25, Csm1/monopolin, and MAD1. Clustering of the DALI Z-scores suggests greater similarity for AKiT4 with SPC24, however both appear as highly divergent RWD domains (low DALI Z-scores) and therefore a clear cKiT ortholog for AKiT4 is elusive.
Strikingly, we did find very weak similarity between AKiT3 and 6 and the largely coil-containing members of the MIS12/NANO complexes (Fig. 8 C). AF2 structures of AKiT3 and 6 are similar in topology, with a two α helix “head” domain and a long coil that drives complex formation. However, the similarity between MIS12C components and AKiT3 and 6 could not be properly captured upon structural alignment and pLDDT prediction confidence scores are low (<50). Profile-vs-profile searches against Pfam did reveal similarity between AKiT3 and 6 and the head domains of MIS12 (Fig. S7 A), and further HMM alignment revealed strong similarity for the head domains of MIS12 and AKiT3 and 6 (Fig. S7, B and C). Since all MIS12/NANO components share a similar structural topology and sequence similarity (Tromer et al., 2019) and the sequence similarities between AKiT3 and 6 and MIS12 are borderline, a clear 1-to-1 MIS12 component for AKiT3 and 6 remains unclear.

AKiT3 and AKiT6 bear similarity of the head domain of MIS12. (A and B) HMM profile-profile alignment of AKiT3 and 6 with MIS12 by (A) HHpred, and against (B) the Pfam/ECOD and PDB database. (C) Alignment of head domains predicted by AF2 and profile-profile comparisons (see A and B) of AKiT3 and 6 with MIS12.
AKiT3 and AKiT6 bear similarity of the head domain of MIS12. (A and B) HMM profile-profile alignment of AKiT3 and 6 with MIS12 by (A) HHpred, and against (B) the Pfam/ECOD and PDB database. (C) Alignment of head domains predicted by AF2 and profile-profile comparisons (see A and B) of AKiT3 and 6 with MIS12.
Overall, our data indicates that apicomplexan kinetochores include AKiT1/KNL1, AKiT7/MAD1, and AKiT9 and 10 and SEA-1 as parologs of CENP-C. It also suggests the presence of highly divergent MIS12/NANO-like homologs in AKiT3 and 6. AKiT4 is an RWD-domain containing protein the identity for which remains unclear, and we did not find clear similarity between AKiTs 2, 5, 8, or 11 with known kinetochore proteins (Fig. 8 D).
AKiT1 requires CENP-C for kinetochore localization in Toxoplasma
CENP-C binds directly to CENP-A nucleosomes at the centromere where it serves as a platform for the recruitment of essential kinetochore proteins. While all known kinetochore systems harbor one CENP-C ortholog, Plasmodium operates three paralogs. In contrast, we identified two paralogs in Toxoplasma (along with a gene annotation error in TGME49_209880, with the C-terminus encoding TgAKiT9; Fig. S8). To reconstruct the evolution of CENP-C, we modeled aligned cupin domains from CENP-C homologs, with a focus on apicomplexan and dinoflagellate sequences (Fig. 9 A and Data S3). Surprisingly, we found AKiT9 and CENP-C in Apicomplexa and Dinoflagellata, indicating these genes duplicated before the last common ancestor of Myzozoa. In contrast, AKiT10 and SEA1 cluster within an apicomplexan CENP-C group. The cupin domain of AKiT10 follows the species tree, while SEA1 appears highly divergent (longer branch length) and oddly positioned between a group of coccidian, hematozoan, and gregarine-related CENP-Cs. Strict reconciliation suggests a scenario where SEA1 and AKIT10 duplicated in the ancestor of coccidians and hematozoans, followed by subsequent loss in piroplasmids and coccidians. However, the long branch and “peculiar” position of SEA1 allows a Plasmodium-specific duplication as equally likely. Furthermore, whilst AKiT9 and 10 sit functionally within the Plasmodium CCAN, they lack the conserved CENP-C motif required for CENP-A binding, but which is present in SEA-1 and Toxoplasma CENP-C (Fig. 9 A).

The T. gondii gene TGME49_209880 is incorrectly predicted and harbors a bona fide AKiT9 at its C-terminus. Multiple sequence alignment showing TGME49_209880 where only the C-terminus aligns with coccidian AKiT9 orthologs, which are much shorter in length.
The T. gondii gene TGME49_209880 is incorrectly predicted and harbors a bona fide AKiT9 at its C-terminus. Multiple sequence alignment showing TGME49_209880 where only the C-terminus aligns with coccidian AKiT9 orthologs, which are much shorter in length.
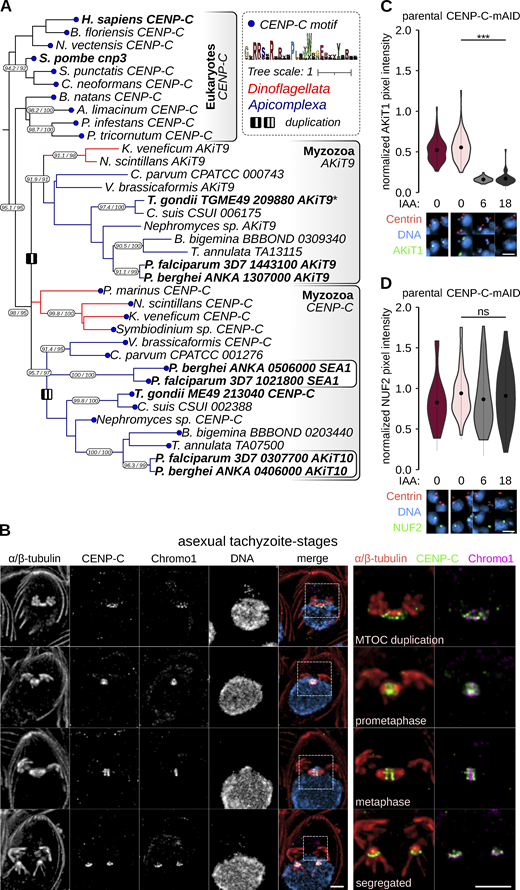
AKiT1 localization to kinetochores is dependent upon CENP-C in Toxoplasma. (A) Pruned tree of a maximum likelihood inference based on an alignment of cupin domains retrieved following iterative HMM searches for myzozoa and eukaryotic homologs of CENP-C. Duplications within the myzozoan CENP-C clade are indicated by I (AKiT9-CENP-C) and II (AKiT10-SEA1). Terminal blue nodes indicate sequences harboring a known CENP-A binding motif. Numbers on branches indicate rapid bootstrap and Shimodaira–Hasegawa-aLRT support (1,000 replicates). Full tree can be found in Data S3. (B) U-ExM of T. gondii tachyzoites expressing TgCENP-C fused to mAID-3xHA (green) throughout intracellular divisions. Counter-staining of DNA (cyan), TgChromo1 (magenta). Scale bar, 1 μm. (C and D) Levels and localization of NUF2 and AKiT1 postdepletion of CENP-C. Representative images are shown below. DNA (cyan), Centrin1 (tomato), and AKiT1 or NUF2 (green). Scale bar, 4 μm.
AKiT1 localization to kinetochores is dependent upon CENP-C in Toxoplasma. (A) Pruned tree of a maximum likelihood inference based on an alignment of cupin domains retrieved following iterative HMM searches for myzozoa and eukaryotic homologs of CENP-C. Duplications within the myzozoan CENP-C clade are indicated by I (AKiT9-CENP-C) and II (AKiT10-SEA1). Terminal blue nodes indicate sequences harboring a known CENP-A binding motif. Numbers on branches indicate rapid bootstrap and Shimodaira–Hasegawa-aLRT support (1,000 replicates). Full tree can be found in Data S3. (B) U-ExM of T. gondii tachyzoites expressing TgCENP-C fused to mAID-3xHA (green) throughout intracellular divisions. Counter-staining of DNA (cyan), TgChromo1 (magenta). Scale bar, 1 μm. (C and D) Levels and localization of NUF2 and AKiT1 postdepletion of CENP-C. Representative images are shown below. DNA (cyan), Centrin1 (tomato), and AKiT1 or NUF2 (green). Scale bar, 4 μm.
To investigate whether kinetochores in Toxoplasma rely upon similar design principles as in animals and fungi, we assessed the dependency of essential components TgNUF2 and TgAKiT1 on TgCENP-C (TGME49_213040) for recruitment to kinetochores, by similarly tagging protein with an auxin-inducible degron (Fig. S9 A). Consistent with a conserved localization to eukaryotic kinetochores, the majority of tagged TgCENP-C localized alongside TgChromo1 at centromeres and TgAKiT1 at kinetochores throughout tachyzoite divisions (Fig. 9 B and Fig. S9 B). Supporting the notion CENP-C forms a platform for kinetochore assembly in Toxoplasma, levels of TgAKiT1 at kinetochores were reduced upon depletion of TgCENP-C (Fig. 9 C). Surprisingly, however, levels of TgNUF2 were not clearly affected upon TgCENP-C depletion (Fig. 9 D), despite the fatal effect on chromosome segregation postdepletion (Fig. S9, C–H).
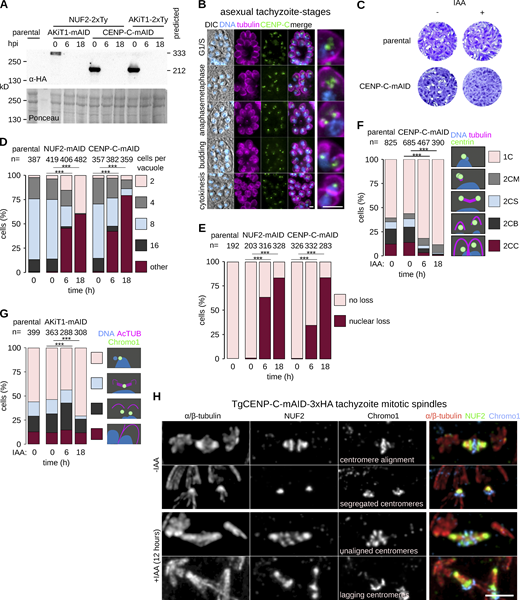
CENP-C is required for T. gondii proliferation. (A) Immunoblots of T. gondii parasites expressing tagged AKiT1 and CENP-C. (B) Micrographs of fixed immunofluorescence in T. gondii tachyzoites throughout intracellular divisions. Tubulin (magenta), DNA (cyan), and differential interference contrast images are also shown. Scale bar, 5 μm. (C) Tachyzoites depleted for CENP-C failed to form lysis plaques 7 d after inoculation compared to parental controls. (D) Intracellular growth is severely reduced in T. gondii depleted of CENP-C and NUF2 (***, P < 0.001; χ2 test). (E) Intracellular vacuoles containing accumulations of DNA and no associated cell body were present after depletion of CENP-C. (F and G) Morphological analyses to assess the effect of CENP-C depletion on partitioning of the centrosome (F) or centromeres (G; ***, P < 0.001; χ2 test). (H) U-ExM revealed cells with misaligned and lagging centromeres along elongated mitotic spindles postdepletion of CENP-C. Source data are available for this figure: SourceData FS9.
CENP-C is required for T. gondii proliferation. (A) Immunoblots of T. gondii parasites expressing tagged AKiT1 and CENP-C. (B) Micrographs of fixed immunofluorescence in T. gondii tachyzoites throughout intracellular divisions. Tubulin (magenta), DNA (cyan), and differential interference contrast images are also shown. Scale bar, 5 μm. (C) Tachyzoites depleted for CENP-C failed to form lysis plaques 7 d after inoculation compared to parental controls. (D) Intracellular growth is severely reduced in T. gondii depleted of CENP-C and NUF2 (***, P < 0.001; χ2 test). (E) Intracellular vacuoles containing accumulations of DNA and no associated cell body were present after depletion of CENP-C. (F and G) Morphological analyses to assess the effect of CENP-C depletion on partitioning of the centrosome (F) or centromeres (G; ***, P < 0.001; χ2 test). (H) U-ExM revealed cells with misaligned and lagging centromeres along elongated mitotic spindles postdepletion of CENP-C. Source data are available for this figure: SourceData FS9.
To assess the position of kinetochore components relative to the centromere, we used U-ExM and manually assigned foci of Toxoplasma kinetochore proteins tagged at the C-terminus relative to TgChromo1 along the spindle axis. In the absence of true prometaphase or anaphase markers, we performed this analysis using cells in metaphase, with TgChromo1 aligned at the metaphase plate (Fig. 10 A). For each kinetochore component, we took the closest centroid relative to TgChromo1, however, taking the most distal tubulin centroid to derive the edges of the spindle, resulting in measurements for 226, 84, 80, 99, 164, and 204 foci (spindle pole, SKA1, NUF2, AKiT1, CENP-C, and TgChromo1, respectively; Fig. 10 B). Toxoplasma kinetochores are composed of at least four discernible compartments. In agreement with the notion that CENP-C forms part of the inner kinetochore, the distribution of centroids representing the C-terminus of CENP-C relative to TgChromo1 was significantly smaller than for AKiT1 (81 ± 6 nm versus 129 ± 7 nm), with NUF2 and SKA1 belonging to the outer most kinetochore compartment identified (183 ± 7 nm and 190 ± 8 nm, respectively).

AKiTs localize to discrete sub-kinetochore compartments along the metaphase spindle. (A) U-ExM of T. gondii tachyzoite cells expressing either of tagged SKA1, NUF2, AKiT1, and CENP-C (green) alongside staining for TgChromo1 (magenta). (B) Centroid measurements along the metaphase spindle axis. In brackets are the number of centroids measured. Dotted lines show the mean relative positions and error bars show the SEM (***, P < 0.001). (C) Models for T. gondii and P. berghei metaphase kinetochores (Note SKA is not detected during microgametogenesis in P. berghei).
AKiTs localize to discrete sub-kinetochore compartments along the metaphase spindle. (A) U-ExM of T. gondii tachyzoite cells expressing either of tagged SKA1, NUF2, AKiT1, and CENP-C (green) alongside staining for TgChromo1 (magenta). (B) Centroid measurements along the metaphase spindle axis. In brackets are the number of centroids measured. Dotted lines show the mean relative positions and error bars show the SEM (***, P < 0.001). (C) Models for T. gondii and P. berghei metaphase kinetochores (Note SKA is not detected during microgametogenesis in P. berghei).
Discussion
Kinetochores of apicomplexan parasites are highly divergent in both sequence and composition from those in animals and fungi. Most components outside the NDC80C have changed beyond previously established recognition (Tromer et al., 2019; van Hooff et al., 2017) and lineage-specific duplications have generated new kinetochore proteins with likely associated functions. Within Apicomplexa, we have detected three paralogs of CENP-C in malaria parasites compared to two in Toxoplasma. AKiT9 and 10 appear to interact with one another and encode cupin domains required for dimerization of CENP-C in conventional eukaryotic models, including human cells and baker’s yeast (Cohen et al., 2008; Sugimoto et al., 1997)—it is tempting to speculate a similar interaction was maintained following duplication to form the heterodimer. However, neither protein harbors the motif critical for binding to CENP-A containing nucleosomes (Carroll et al., 2010; Milks et al., 2009; Trazzi et al., 2002) and we did not detect CENP-A following affinity-purification of AKiT9 in the malaria parasite. In contrast, SEA1 does encode a canonical CENP-A binding motif, suggesting additional components at apicomplexan centromeres that we have not yet localized. AKiT1–6 may be more susceptible to change compared to other apicomplexan kinetochore components, which would go some way to explain the previously hidden signal detected between this set and known eukaryotic kinetochore proteins. Greatest similarity was detected using full-length profiles including most conserved regions across eukaryotic kinetochore homologs compared to those stripped to annotated domains only. One clear drawback of using full-length profiles is the spurious hits of coiled-coil region between different kinetochore proteins (e.g., the similarity between those of SPC25, NDC80-NUF2, MAD1, and AKiT1, 4, and 7). However, recognition of AKiT1 as a homolog of KNL1 is strongly dependent on signal from shared coiled-coils and it will be interesting in the future to explore if these regions have function at kinetochores. A ground-breaking technique now at our disposal is the de novo structure prediction algorithm AlphaFold2 (AF2). AF2 revealed the presence of a single RWD domain in Plasmodium AKiT1, whereas in its Nephromyces ortholog the conventional KNL1 double-RWD domain was detected, suggesting either loss of the second RWD domain or the inability of AF2 to properly fold this part of the structure in Plasmodium. Similar data was obtained for AKiT4; however a clear phylogenetic affiliation with a conventional kinetochore protein remains less clear. As remaining kinetochore RWD proteins (SPC24, SPC25, MAD1, and KNL1) have now been detected in Apicomplexa, AKiT4 is most likely a distant relative of either Csm1/monopolin or ZWINT-1. In budding yeast, Csm1/monopolin crosslinks sister kinetochores in meiosis I, promoting attachment to microtubules extending from the same spindle pole (Monje-Casas et al., 2007). Apicomplexan centromeres are clustered at the nuclear periphery during interphase and it will be interesting to assess whether AKiT4 is required to maintain these characteristic clusters. Weakest similarity detected was between MIS12-related structures and AKiT3 and 6. AF2-predicted structures had similar topology, however, since the structural complexity was low, we could not confidently assign AKiT3 and 6 to any MIS12C members. Furthermore, we did not find clear similarity between AKiTs 2, 5, 8, or 11 with known proteins, suggesting divergence beyond detectability or emergence of lineage-specific kinetochore components. Surprisingly, AKiT8 encodes an allantoicase domain, typically required for purine degradation and not previously described for known kinetochore proteins, the role of which, if any, requires further exploration.
By combining proteomics with U-ExM, we have provided the first model composition of protein complexes at an apicomplexan kinetochore displayed in Fig. 10 B. How does our model compare to those of other kinetochores? Monocentric eukaryotic kinetochores are partite, hierarchical assemblies deposited onto specific chromatin environments, themselves often demarked by centromeric nucleosomes (Akiyoshi and Gull, 2014; Cheeseman, 2014; Cortes-Silva et al., 2020; D’Archivio and Wickstead, 2017; Kozgunova et al., 2019). In animals, CENP-C binds directly to CENP-A nucleosomes (Carroll et al., 2010) and interacts with the four-subunit MIS12C (Przewloka et al., 2011; Screpanti et al., 2011), itself interacting with KNL1C and NDC80C at the spindle (Petrovic et al., 2010). In human cells, an alternative pathway utilizes CENP-TWSX that bridges DNA to NDC80C (Gascoigne et al., 2011). The above design principle is largely conserved in Apicomplexa, whose kinetochores we show are also partite hierarchical assemblies. AKiT9–11 join two identifiable homologs of CENP-C in forming a centromere proximal compartment and likely part of the apicomplexan CCAN. The C-terminus of AKiT1 localizes to a midpoint between CENP-C and NDC80C at the spindle and is dependent upon CENP-C for kinetochore assembly in T. gondii, reminiscent of CENP-C binding to MIS12C/KNL1C. However, neither depletion of CENP-C nor AKiT1 in T. gondii reduced levels of NUF2 at the spindle and similarly endogenous NUF2 levels were not required for kinetochore recruitment of AKiT1. Recently, a highly elongated Plasmodium SPC24 component of the NDC80C was identified and suggested to bridge the >100 nm distance separating the outer kinetochore from the centromere (Zeeshan et al., 2020). However, this “long form” is poorly conserved across Apicomplexa. Whilst present in coccidians, it is lacking in Theileria and Babesia, meaning any direct centromere binding potential would have converged or else been lost by most-closely related hematozoan SPC24 proteins. How NDC80C is maintained at the spindle in Plasmodium and Toxoplasma, remains unknown; however, it is quite possible an analogous pathway to that requiring CENP-T in humans exists or that NDC80C binds microtubules independently of kinetochores in these cells.
Flexibility in modes of division and an apparent inability to delay cell cycle progression in response to microtubule destabilizing agents suggests division checkpoints in apicomplexan parasites may be very different to those described in animal cells (Arnot and Gull, 1998; Morrissette and Sibley, 2002). Here, we reveal alignment of kinetochores during mitosis and meiosis, reminiscent of chromosome biorientation at the metaphase plate and suggestive of a “hold signal” that prevents precocious entry into anaphase (Lampson and Cheeseman, 2011). Furthermore, identification of STU2 at Plasmodium kinetochores hints towards an intrinsic tension-sensing and error-correction mechanism required for establishing bioriented attachments (Miller et al., 2016, 2019). In animal cells, Aurora kinase and the SAC collaborate to ensure biorientation (Musacchio and Salmon, 2007). SAC components (outside MAD1) have not been detected in Apicomplexa (Kops et al., 2020). However, the SAC is dispensable in yeast wherein Aurora-mediated correction of kinetochore-spindle attachments is sufficient to ensure fidelity of mitosis (Muñoz-Barrera et al., 2015). In animal cells, phosphorylation of the SKAC by Aurora kinase is essential to biorient chromosomes, enabling SKA kinetochore accumulation once biorientation is achieved (Redli et al., 2016). In T. gondii, SKA proteins were only detected at kinetochores after duplication of the centrosome, levels increasing upon formation of the diamond spindle at metaphase when kinetochore-microtubule attachments are most likely under greatest tension. Depletion of TgSKA1 and 2 led to a strong increase in mitotic index, in particular in cells with bipolar spindles and a metaphase-like arrested state, reminiscent of siRNA-mediated depletion of SKA components in HeLa cells. Localizations of SKA components were comparable during asexual divisions of the malaria parasite. In contrast, we were unable to identify a metaphase plate architecture during microgametogenesis that occurs upon mosquito stages of malaria parasite division. Kinetochore architecture with respect to tagged-SKA protein localization differs between asexual and sexual stages and we speculate this may correlate with requirements to satisfy specific biorientation checkpoints. The significance of differing SKA behavior remains to be explored, however SKA proteins enhance spindle attachment in animal cells (Helgeson et al., 2018) and it is quite possible kinetochores bind to the spindle with different strengths between apicomplexan modes of division.
Identification of AKiT7/MAD1 from kinetochore pulldowns and an apparent enrichment at kinetochores during nuclear divisions (at least by conventional fluorescence microscopy) does open the possibility of a conserved SAC response in Apicomplexa, with AKiT1/KNL1 an obvious suspect for loading of SAC protein. Suggestion of any SAC response in Apicomplexa raises some important questions regarding the existence of additional components, their recruitment to kinetochores and whether they enable a checkpoint comparable to that in other organisms. Neither AKiT1 nor AKiT7 bear motifs typically required for recruitment of SAC components (Kops et al., 2020), suggesting the SAC may well be wired differently or repurposed at apicomplexan kinetochores. Furthermore, kinases resembling Monopolar Spindle 1 and Polo-Like that are required for recruitment of SAC proteins to kinetochores have not been detected in Apicomplexa. However, a number of kinases distantly related to Aurora and Cyclin-dependent kinase families have been shown to be implicated in cell cycle control, in particular DNA synthesis and mitosis in both Plasmodium and Toxoplasma (Balestra et al., 2020; Hawkins et al., 2022; Morahan et al., 2020). Whether phosphoregulation has been repurposed at apicomplexan kinetochores and the effects such divergences have on chromosome segregation will likely broaden our views on the malleability of eukaryotic mitotic checkpoint control.
Materials and methods
Ethics statement
All animal experiments were conducted with the authorization number AB_GE18, according to the guidelines and regulations issued by the Swiss Federal Veterinary Office.
Generation of transgenic parasites targeting constructs
The oligonucleotides used to generate transgenic parasite lines are shown in Table S2.
For C-terminal tagging of P. berghei proteins using PlasmoGEM resources (http://plasmogem.umu.se/pbgem), 3xHA and mScarlet-I tagging constructs were generated using phage recombineering in Escherichia coli TSA strain with PlasmoGEM vectors using sequential recombineering and gateway steps (Pfander et al., 2011). For genes SKA2 and NUF2, the Zeocin-resistance/Phe-sensitivity cassette was introduced using oligonucleotides goi-recR1 × goi-recR2 for 3xHA tagging and goi mSc-F × goi mSc-R for mSc tagging vectors. Insertion of the GW cassette following gateway reaction was confirmed using primer pairs GW1 × goi-QCR1 and GW2 × goi-QCR2.
For C-terminal tagging of P. berghei proteins by pCP, constructs were newly derived from pOB277 (Patzewitz et al., 2013) in order to target endogenous loci by allele replacement instead of insertion. Briefly, a 588-bp fragment encompassing the coding sequence of 3xHA and DHFR flanked by KpnI and EcoRI was amplified from PlasmoGEM plasmid GW-R6K-3xHA using primers MB1048 and MB1049 and replaced the corresponding fragment in pOB277 to generate pCP-3xHA. The coding sequence for mNeonGreen (mNG) was codon-optimized for P. berghei (Yadav and Swati, 2012), flanked by AvrII and SacII sites and purchased from GeneArt (Data S5) and inserted upstream to 3xHA to generate pCP-mNG-3xHA. Sequences comprising ∼500 bp from the C-terminus of the coding sequence and ∼500 bp from the immediate 3′ UTR for genes SKA1, SKA3, and AKiTs 1–11 were cloned into KpnI and AvrII sites upstream to the mNG coding sequence, along with a NotI linearisation site between the targeting sequences.
For placing P. berghei genes under blood-stage expression by pNP, constructs were newly derived from pCP. Briefly a 111-bp fragment encompassing the coding sequence of 3xHA was excised by KpnI and ApaI and replaced with a corresponding fragment amplified from PbGEM (http://plasmogem.umu.se/pbgem) plasmid GW-R6K-3xHA using MB1033 and MB1034 to generate pCP-3xHA_nostop. The CLAG9 promoter from pOB116 (Sebastian et al., 2012) was excised by XhoI and KpnI and placed into corresponding sites in pCP-3xHA_nostop to generate pNP-CLAG9-3xHA. Sequences comprising 502 bp from the C-terminus of the coding sequence and 501 bp from the immediate 5′ UTR for AKiT1 were cloned into ApaI and EcoRI sites downstream to the 3xHA coding sequence, along with a NotI linearisation site between the targeting sequences.
For C-terminal tagging of T. gondii, constructs were generated by KOD PCR (Brown et al., 2018). Genomic DNA extractions were performed with the Wizard SV genomic DNA purification kit (Promega). PCRs to generate specific gRNAs were performed with Q5 polymerase (New England Biolabs) while PCRs to generate specific knock-in constructs (mAID fusions and epitope tagging) were performed with KOD polymerase (Novagen). Specific gRNA were generated using the Q5 site-directed mutagenesis kit (New England Biolabs) on the pSAG1::Cas9-U6::sgUPRT vector (Shen et al., 2014).
P. berghei maintenance and transfection
P. berghei ANKA strain-derived clone 2.34 (Billker et al., 2004) together with derived transgenic lines were grown and maintained in CD1 outbred mice. 6–10 wk-old mice were obtained from Charles River Laboratories and females were used for all experiments. Mice were specific pathogen free (including Mycoplasma pulmonis) and subjected to regular pathogen monitoring by sentinel screening.
They were housed in individually ventilated cages furnished with a cardboard mouse house and Nestlet, maintained at 21 ± 2°C under a 12-h light/dark cycle and given commercially prepared autoclaved dry rodent diet and water ad libitum. The parasitaemia of infected animals was determined by microscopy of methanol-fixed and Giemsa-stained thin blood smears. For gametocyte production, parasites were grown in mice that had been phenyl hydrazine treated 3 d before infection. Exflagellation was induced in exflagellation medium (RPMI 1640 containing 25 mM Hepes, 4 mM sodium bicarbonate, 5% FCS, and 100 mM xanthurenic acid, pH 7.8). For gametocyte purification, parasites were harvested in suspended animation medium (SA; RPMI 1640 containing 25 mM Hepes, 5% FCS, and 4 mM sodium bicarbonate, pH 7.20) and separated from uninfected erythrocytes on a Histodenz/Nycodenz cushion made from 48% of a Histodenz/Nycodenz stock (27.6% [w/v] Histodenz/Nycodenz [Sigma/Alere Technologies] in 5.0 mM TrisHCl, 3.0 mM KCl, and 0.3 mM EDTA, pH 7.20) and 52% SA, final pH 7.2. Gametocytes were harvested from the interface.
Schizonts for transfection were purified from overnight in vitro culture on a Histodenz cushion made from 55% of the Histodenz/Nycodenz stock and 45% PBS. Parasites were harvested from the interface and collected by centrifugation at 500 g for 3 min, resuspended in 25 ml Amaxa Basic Parasite Nucleofector solution (Lonza) and added to 10 µg DNA dissolved in 10 μl H2O. Cells were electroporated using the FI-115 program of the Amaxa Nucleofector 4D. Transfected parasites were resuspended in 200 ml fresh RBCs and injected intraperitoneally into mice. Parasite selection with 0.07 mg/ml pyrimethamine (Sigma-Aldrich) in the drinking water (pH ∼ 4.5) was initiated 1 d after infection.
T. gondii maintenance and transfection
T. gondii tachyzoites were grown in human foreskin fibroblasts (HFFs; American Type Culture Collection-CRL 1634) maintained in DMEM (Gibco) supplemented with 5% FCS, 2 mM glutamine, and 25 μg/ml gentamicin. Absence of Mycoplasma contamination was checked regularly by immunofluorescence. All mAiD fusion strains were generated in a Tir1 expressing cell line (Brown et al., 2017) and depletion of protein achieved by incubation with 500 µM of indole-3-acetic acid (IAA).
Freshly egressed tachyzoites were transfected by electroporation (Soldati and Boothroyd, 1993). For each transfection, 40 µg of specific gRNA was used to target the 3′ UTR of the gene of interest. Mycophenolic acid (25 mg/ml) and xanthine (50 mg/ml) or pyrimethamine (1 µg/ml) were employed to select resistant parasites carrying the HXGPRT and DHFR cassette, respectively.
For assessment of protein depletion by plaque assays, HFFs were infected with fresh parasites and grown for 7 d before fixation with PFA/GA. After fixation, HFFs were washed with PBS and the host cells monolayer was stained with crystal violet.
For morphological analysis of cells depleted for kinetochore components, parasites were processed for immunofluorescence or U-ExM (described below; Table S5 for stainings). The mitotic index of cells was calculated in both auxin-induced, noninduced and parental cell lines.
Immunoblotting
Immunoblotting was used to confirm expression of tagged proteins. Actively dividing cells were washed in PBS and resuspended at between 1–5 × 105 cells μl−1. Lysis was in Laemmli buffer (2% w/v SDS, 0.4 M 2-mercaptoethanol, 10% glycerol, and 50 mM Tris-HCl, pH 7.2) at 95°C for 5 min. Lysates containing around 5 × 106 cells were separated on 4–20% polyacrylamide gels (Invitrogen) in running buffer (25 mM Tris, 250 mM glycine, and 0.1% w/v SDS). Proteins were electrophoretically transferred to 0.45 μm pore-size nitrocellulose membrane at 1.6V cm−1 for 14 h in transfer buffer (25 mM Tris, 192 mM glycine, 0.02% w/v SDS, and 10–20% methanol). Membranes were blocked and in 5% w/v milk powder in TBS-T (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween-20) for 1 h. Membranes were incubated in primary antibody (Table S5) in 1% w/v milk in TBS-T for 1 h and washed in TBS-T. Detection was by secondary peroxidase-conjugated antibody (Table S5), in 1% w/v milk in TBS-T for 1 h. Membranes were washed and detected by chemiluminescence with Western Lightning ECL (PerkinElmer) and exposure to photographic film.
Protein localization
For localization of tagged proteins in P. berghei by native fluorescence, cells from different proliferative stages during the parasite lifecycle were mounted in exflagellation medium (RPMI 1640 containing 25 mM Hepes, 4 mM sodium bicarbonate, 5% FCS, and 100 mM xanthurenic acid, pH 7.8), and imaged using an inverted Zeiss Axio Observer Z1 microscope fitted with an Axiocam 506 mono 14-bit camera and Plan Apochromat 63×/1.4 Oil DIC III objective.
For immunofluorescence of T. gondii, parasites were inoculated on an HFF monolayer with coverslips in 24-well plates, grown for 16–30 h depending on the experiment, and fixed with cold methanol (−20°C) for 7 min. Coverslips were then washed with PBS and blocked for 20 min in PBS-BSA 5%. Primary and secondary antibodies (Table S5) were then incubated sequentially for 1 h each in PBS-BSA 2%. Three washes of 5 min each were performed between primary and secondary antibodies incubations, using PBS. Coverslips were mounted in VECTASHIELD Antifade Mounting Medium with DAPI and imaged using the inverted Zeiss Axio Observer.
For U-ExM, cells were sedimented on poly-D-lysine (A-003-E; Sigma) coverslips (150 μl/coverslip) during 10 min at room temperature (RT) and fixed in −20°C methanol during 7 min and prepared for U-ExM as previously published (Bertiaux et al., 2021). Briefly, coverslips were incubated for 5 h in 2× 1.4% AA/2% FA mix at 37°C prior gelation in APS/Temed/Monomer solution (19% sodium acrylate, 10% AA, and 0.1% BIS-AA in PBS 10X) during 1 h at 37°C. Gels were denatured at 95°C for 1 h. Gels were incubated in double distilled H2O (ddH2O) overnight for expansion. The following day, gels were washed in PBS before incubation with primary antibodies (Table S5) for 3 h at 37°C. Gels were washed in PBS-Tween 0.1% prior incubation with secondary antibodies (Table S5) for 3 h at 37°C. Gels were washed in PBS-Tween 0.1%. Gels were incubated in ddH2O for a second round of expansion before imaging. For NHS-ester staining, directly after antibody stainings gels were incubated in NHS-Ester (catalog number: 46402; Thermo Fisher Scientific) 10 μg/ml in PBS for 1 h and 30 min at RT on a rocking platform and washed 3× in PBS before overnight expansion. Confocal microscopy was performed on an Inverted Leica DMi8 with an HC PL Apo 100×/1.40 oil immersion objective fitted with a Leica DFC7000T camera.
All images of fluorescent proteins were captured at room temperature with equal exposure settings. Images for level comparison were processed with the same alterations to minimum and maximum display levels. For analysis of relative positions of kinetochore components, peak locations of tagged proteins of focus centroids in each channel were calculated. Analysis was performed in Fiji (Schindelin et al., 2012) and the statistical programming package R (http://r-project.org). Distances were always measured from dual staining experiments. The expansion factor of each gel was after applied to the value to obtain the real distance.
Immunopurification
For the purification of kinetochore complexes, gametocytes were purified from 5 to 10 ml P. berghei infected mouse blood. Cells were washed twice in HKMEG (150 mM KCl, 150 mM glucose, 25 mM Hepes, 4 mM MgCl2, and 1 mM EGTA, pH 7.8) containing 100× protease inhibitor cocktail and 20 μM MG132. Sequential cross-linking followed the procedure in D’Archivio and Wickstead, 2017. Briefly, cells were treated with 0% (“no”), 0.1% (“low”), or 1% (“high”) formaldehyde in HKMEG for 10 min, quenched with 10 ml of 1 M glycine, and lysed in HKMEG containing 1% (vol/vol) Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT, 100 × protease inhibitor cocktail, and 20 μM MG132. Lysate was sonicated in an ultrasonic water bath for 20 min applied for 50% of the cycle and cleared by centrifugation at >20,000 g for 30 min. The soluble fraction was then allowed to bind to affinity-purified rat anti-HA antibody (Sigma) attached to paramagnetic beads (Dynabeads protein G; Invitrogen) for 4 h. Beads were washed in HKMEG containing 0.1% (vol/vol) Nonidet P-40, 0.5 mM dithiothreitol. Beads were resuspended in 100 μl of 6 M urea in 50 mM ammonium bicarbonate (AB). 2 μl of 50 mM dithioerythritol (DTE) were added and the reduction was carried out at 37°C for 1 h. Alkylation was performed by adding 2 μl of 400 mM iodoacetamide for 1 h at room temperature in the dark. Urea was reduced to 1 M by addition of 500 ml AB and overnight digestion was performed at 37°C with 5 ml of freshly prepared 0.2 mg/ml trypsin (Promega) in AB. Supernatants were collected and completely dried under speed vacuum. Samples were then desalted with a C18 microspin column (Harvard Apparatus) according to manufacturer’s instructions, completely dried under speed vacuum and stored at −20°C.
Mass spectrometry
Protein identification followed the procedure in Balestra et al., 2020. Briefly, samples were diluted in 20 μl loading buffer (5% acetonitrile and 0.1% formic acid [FA]) and 2 μl were injected onto the column. LC-ESI-MS/MS was performed either on a Q-Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC 1,000 liquid chromatography system (Thermo Fisher Scientific) or an Orbitrap Fusion Lumos Tribrid mass Spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC 1,200 liquid chromatography system (Thermo Fisher Scientific). Peptides were trapped on an Acclaim pepmap100, 3 µm C18, 75 µm × 20 mm nano trap column (Thermo Fisher Scientific) and separated on a 75 µm × 250 mm (Q-Exactive) or 500 mm (Orbitrap Fusion Lumos), 2 µm C18, 100 A˚ Easy-Spray column (Thermo Fisher Scientific). The analytical separation used a gradient of H2O/0.1% FA (solvent A) and CH3CN/0.1% FA (solvent B). The gradient was run as follows: 0–5 min 95% A and 5% B, then to 65% A and 35% B for 60 min, then to 10% A and 90% B for 10 min, and finally for 15 min at 10% A and 90% B. Flow rate was 250 nl/min for a total run time of 90 min. Data-dependent analysis (DDA) was performed on the Q-Exactive Plus with MS1 full scan at a resolution of 70,000 full width at half maximum (FWHM) followed by MS2 scans on up to 15 selected precursors. MS1 was performed with an automatic gain control (AGC) target of 3 × 106, a maximum injection time of 100 ms and a scan range from 400 to 2,000 m/z. MS2 was performed at a resolution of 17,500 FWHM with an AGC target at 1 × 105 and a maximum injection time of 50 ms. Isolation window was set at 1.6 m/z and 27% normalized collision energy was used for higher energy collisional dissociation (HCD). DDA was performed on the Orbitrap Fusion Lumos with MS1 full scan at a resolution of 120,000 FWHM followed by as many subsequent MS2 scans on selected precursors as possible within a 3-s maximum cycle time. MS1 was performed in the Orbitrap with an AGC target of 4 × 105, a maximum injection time of 50 ms and a scan range from 400 to 2,000 m/z. MS2 was performed in the Ion Trap with a rapid scan rate, an AGC target of 1 × 104 and a maximum injection time of 35 ms. Isolation window was set at 1.2 m/z and 30% normalized collision energy was used for higher energy collisional dissociation.
Label-free quantitation
Label-free quantitation was followed (D’Archivio and Wickstead, 2017) and was performed on .MGF data files using the Central Proteomics Facilities Pipeline (cpfp.sourceforge.io). Data were searched with X!Tandem and OMS SA engines against a custom, nonredundant protein database of predicted protein sequences from ANKA 2.34 strain including exogenous protein sequences (fluorescent proteins, drug selection markers, and exogenous proteins expressed in the parental cells) and common contaminating peptides. Possible modification of peptides by N-terminal acetylation, phosphorylation (P), ubiquitination (Ub), carbamidomethylation (C), oxidation (M), and deamidation (N/Q) was permitted in searches. Peptide identifications were validated with PeptideProphet and ProteinProphet (Nesvizhskii et al., 2003) and lists compiled at the peptide and protein level. iProphet was used to combine search engine identifications and refine identifications and probabilities. Normalized spectral index quantitation (SINQ) was applied to the grouped metasearches to give protein-level quantitation between labelled samples and controls (Trudgian et al., 2011), and implemented by the Central Proteomics Facilities Pipeline. SINQ values are summed intensities of matched fragment ions for all spectra assigned to a peptide (identified by ProteinProphet), normalized for differences in protein loading between datasets and for individual protein length. Proteins with 1 detected peptide and an estimated false discovery rate of ≤1% relative to a target-decoy database were considered. A total of 780 distinguishable Plasmodium proteins were detected across all experiments. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD028595. Processed data are also provided in Table S3.
Enrichment and principal component analyses were performed in the statistical programming package R (http://r-project.org). Quantitative values were analyzed as either log-transformed SINQ values (for principal component analysis) or log-transformed ratio of sample SINQ value versus control immunopurification (enrichment analysis).
Bioinformatic analyses
Searches were based on predicted protein datasets for 90 alveolate organisms from genomes and transcriptomes (Table S1), in addition to kinetochore protein sequences identified previously (van Hooff et al., 2017). For transcriptomes, ORFs were predicted using TransDecoder. Initial profiles for each identified apicomplexan kinetochore protein were aligned using MAFFT (Katoh et al., 2005), the L-INS-i strategy, trimmed to conserved regions with trimAl, modelled by HMMER3 (Eddy, 2009) and searched to find similar sequences in all predicted proteomes and kinetochore sequences. Hits were added iteratively and used to create new profiles until no new sequences were identified. HMMs for all apicomplexan orthologous groups were also generated and defined by OrthoFinder (Emms and Kelly, 2019), using the default settings (MAFFT for alignments and FastTree for tree inference). Alignments were visualized and modified using Jalview (Waterhouse et al., 2009). Profile–profile comparisons were performed using HH-suite3 (Söding, 2005).
For phylogenies, trimmed alignments were used to infer maximum likelihood phylogenies using IQ-TREE2 (Minh et al., 2020). Trees were visualized in FigTree (http://tree.bio.ed.ac.uk/software/figtree) and Phytools (Revell, 2012), as part of the statistical programming package R (http://r-project.org).
AF2 (Varadi et al., 2022) was operated through ColabFold (Mirdita et al., 2022). For each full-length and/or domain sequence we predicted, we initiated the homology search with a diverse custom-made alignment of apicomplexan homologs, which was supplemented using an MMseqs2 searches against large local sequence databases (e.g., Unigene30). Most models were run max_recycles: 12. Because of a length limit using ColabFold of 1400aa, we truncated proteins above that length into parts. P. falciparum 3D7 protein structures are already available in the AlphaFold EBI database v2 (http://alphafold.ebi.ac.uk). All predictions performed using AF2 can be found in Data S4. DALI webserver (Holm et al., 2006) was used for structural comparisons to RWD domains, using the all-against-all function (ekhidna2.biocenter.helsinki.fi/dali).
Online supplemental material
Fig. S1 shows generation and validation of tagged NUF2 and SKA components in Plasmodium and Toxoplasma. Fig. S2 shows cross-linking IP of NUF2 identifies novel Plasmodium kinetochore proteins. Fig. S3 shows AKiT1 is a component of the Plasmodium kinetochore. Fig. S4 shows AKiT1 is required for kinetochore segregation in Toxoplasma. Fig. S5 shows relative protein abundance following immunoprecipitation under limited cross-linking and mass spectrometry. Fig. S6 shows AKiTs are novel identified components of the Plasmodium kinetochore. Fig. S7 shows AKiT3 and AKiT6 bear similarity of the head domain of MIS12. Fig. S8 shows T. gondii gene TGME49_209880 is incorrectly predicted and harbors a bona fide AKiT9 at its C-terminus. Fig. S9 shows CENP-C is required for T. gondii proliferation. Data S1, S2, and S3 shows maximum likelihood inferences for STU2 (S1), AKiT1 (S2) and the CUPIN domains of CENP-C (S3). Data S4 shows compressed file containing all data pertaining AlphaFold2-predicted structures used in this study. Data S5 shows mNeonGreen. Fasta file for mNeonGreen, codon-optimised for P. berghei and purchased from GeneArt. Table S1 shows sources for detection of putative homologs of apicomplexan kinetochore proteins. Strategy and nomenclature follows as described in (Koreny et al., 2021). Table S2 shows oligonucleotide primers used for generation of P. berghei and T. gondii transgenic parasites. Table S3 shows label-free semi quantitative mass spectrometry of P. berghei kinetochore proteins. Table S4 shows apicomplexan kinetochore proteins described in this study. Gene essentiality was determined by mutagenesis screens deposited on PlasmoDB for rodent and human malaria parasites (Bushell et al., 2017; Zhang et al., 2018) and ToxoDB for T. gondii (Sidik et al., 2016). T. gondii proteinsubcellular compartmentalizations as determined by HyperLOPIT (Barylyuk et al., 2020) are also shown. Table S5 shows antibodies used for immunodetection and subcellular localization studies of tagged AKiTs.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary materials. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD028595.
Acknowledgments
Mass spectrometry and initial quantitation were performed at the Proteomics Core Facility, University of Geneva (www.unige.ch/medecine/proteomique). We thank in particular the service of Alexandre Hainard, Patrizia Arboit, and Carla Pasquarello Mosimann in preparation and running of mass spectrometry samples. Microscopy was performed at the Bioimaging Core Facility, University of Geneva (www.unige.ch/medecine/bioimaging/en/bioimaging-core-facility), and we thank Olivier Brun and François Prodon for their technical assistance. We thank Natacha Klages for technical assistance. We thank Mathieu Gissot for generously providing us with the TgChromo1 antibody (Gissot et al., 2012). For kindly providing us with sequence data and assemblies, we thank Varsha Mathur (Mathur et al., 2019; Mathur et al., 2021) and Jan Janouškovec (Janouskovec et al., 2019). We also thank Bill Wickstead and Patrick Meraldi for general discussions and critical reading of the manuscript.
This work was supported by the Swiss National Science Foundation grant 31003A_179321 and 310030_208151 M. Brochet and 310245753030_185325 D. Soldati-Favre. M. Brochet is an INSERM and EMBO young investigator.
The authors declare no competing financial interests.
Author contributions: Conceptualization: L.A. Brusini, M. Brochet. Methodology: L.A. Brusini. Software: L.A. Brusini, E.C. Tromer. Formal analysis: L.A. Brusini, E.C. Tromer. Investigation: L.A. Brusini, N. Dos Santos Pacheco, E.C. Tromer. Original draft preparation: L.A. Brusini. Review and editing: L.A. Brusini, N. Dos Santos Pacheco, E.C. Tromer, M. Brochet, D. Soldati-Favre. Supervision: M. Brochet. Funding acquisition: M. Brochet, D. Soldati-Favre. All authors contributed to the article and approved the submitted version.

