


Membrane contact sites are specialized platforms formed between most organelles that enable them to exchange metabolites and influence the dynamics of each other. The yeast vacuole is a degradative organelle equivalent to the lysosome in higher eukaryotes with important roles in ion homeostasis and metabolism. Using a high-content microscopy screen, we identified Ymr160w (Cvm1, for contact of the vacuole membrane 1) as a novel component of three different contact sites of the vacuole: with the nuclear endoplasmic reticulum, the mitochondria, and the peroxisomes. At the vacuole–mitochondria contact site, Cvm1 acts as a tether independently of previously known tethers. We show that changes in Cvm1 levels affect sphingolipid homeostasis, altering the levels of multiple sphingolipid classes and the response of sphingolipid-sensing signaling pathways. Furthermore, the contact sites formed by Cvm1 are induced upon a decrease in sphingolipid levels. Altogether, our work identifies a novel protein that forms multiple contact sites and supports a role of lysosomal contacts in sphingolipid homeostasis.
Introduction
The different membranes that compose a eukaryotic cell can establish proximity regions, formed by proteins or protein complexes that tether them together (Eisenberg-Bord et al., 2016). These areas are called membrane contact sites (MCSs) and do not represent an intermediary in a process of fusion, but rather serve a variety of specific functions (Prinz et al., 2019). These functions involve the application of forces that deform organelles or attach them to specific positions, or the exchange of metabolites and signals between the involved organelles.
MCSs are known to play important roles in the exchange of calcium and in the transport of membrane lipids (reviewed in Prinz et al., 2019), with recent work indicating that additional metabolites can also be exchanged at these platforms (Shai et al., 2018). The exchange of membrane lipids at MCSs is mediated by lipid transport proteins, which contain hydrophobic cavities to shield lipids from the hydrophilic environment, mediating their desorption from one membrane and their delivery to the other (Holthuis and Menon, 2014). The transfer of lipids at MCSs has been shown to be, among others, necessary for mitochondrial function (Kornmann et al., 2009), for the generation of sterol-rich lipid domains in the vacuole membrane (Murley et al., 2017), and for alleviating ceramide lipotoxicity in the ER (Liu et al., 2016).
Systematic screens designed to uncover all MCSs in the cell have highlighted the existence of proximity regions between every pair of organelles assessed (Kakimoto et al., 2018; Shai et al., 2018; Valm et al., 2017). However, many of these MCSs remain poorly understood, with no known functions or components. Therefore, it is very likely that the repertoire of functions that MCSs can play is not yet completely described.
One of the organelles identified to form multiple contact sites in Saccharomyces cerevisiae is the vacuole, equivalent to the lysosome in higher eukaryotes. This organelle is central to cellular metabolism as the final destination of the endocytic and autophagic pathways, degrading macromolecules and providing building blocks for the cell (Balderhaar and Ungermann, 2013). It also acts as a storage and detoxification organelle for many molecules and plays a central role in the sensing and signaling of nutrients, by hosting the TORC1 complex (Binda et al., 2009).
MCSs of the vacuole with the ER, peroxisomes, mitochondria, lipid droplets, and plasma membrane have been reported previously (Kakimoto et al., 2018; Shai et al., 2018). The contact of the vacuole with the nuclear ER, called the nuclear–vacuolar junction (NVJ), is one of the best-described contact sites to date. A main tether of this contact is formed by the ER transmembrane protein Nvj1, which interacts with the vacuole membrane protein Vac8 (Pan et al., 2000). The lipid transport proteins Lam6, Nvj2, and Osh1, and the enoyl reductase Tsc13, involved in the synthesis of very-long-chain fatty acids, are also localized to this structure (Elbaz-Alon et al., 2015; Hariri et al., 2017; Kohlwein et al., 2001; Levine and Munro, 2001; Murley et al., 2015). This suggests that the NVJ acts as a hub that coordinates lipid metabolism and transport.
A contact site between the vacuole and peroxisomes (PerVale) has been observed in S. cerevisiae through the use of a split-Venus–based reporter strain, but remains completely undescribed (Shai et al., 2018). In the methylotrophic yeast Hansenula polymorpha, this contact was observed by EM and shown to expand under conditions in which peroxisomes proliferate (Wu et al., 2019). The peroxisomal membrane protein Pex3 has been proposed as a component of this contact (Wu et al., 2019).
The vacuole also forms a contact site with the mitochondrial network called the vacuole and mitochondrial patch (vCLAMP). This structure is formed by the Vps39 subunit of the homotypic fusion and protein sorting (HOPS) tethering complex, independent of the rest of the complex (Elbaz-Alon et al., 2014; Hönscher et al., 2014). This protein bridges the two organelles by interacting with Ypt7 on the vacuole membrane and with the translocase of the outer membrane complex on the mitochondria (González Montoro et al., 2018; Lürick et al., 2017). This contact is present when cells grow in the presence of glucose but disassembles under respiratory conditions, likely through the phosphorylation of Vps39 (Hönscher et al., 2014). Because of the synthetic lethality of a VPS39 deletion with disruption of the ER-mitochondria encounter structure (ERMES) protein complex, this contact was proposed to be a pathway of lipid delivery to the mitochondria, redundant with the contact site between the ER and the mitochondrial outer membrane formed by ERMES. However, we showed that it is the HOPS function of Vps39 and not the vCLAMP function that is necessary when ERMES is lacking (González Montoro et al., 2018). Thus, the molecular function of the vCLAMP remains unknown, but cells devoid of vCLAMPs display impaired growth under specific stress conditions, such as the presence of ZnCl2 (González Montoro et al., 2018).
To uncover further components of the vCLAMP contact site, we designed and performed a high-content microscopy screen. Of the 24 hits identified, we focused on the uncharacterized protein Ymr160w, which we named “contacts of the vacuole membrane 1” (Cvm1). We show that Cvm1 not only is a novel component and a tether of the vCLAMP, but also forms part of two other MCSs of the vacuole: the PerVale and the NVJ. In all tested conditions, this protein localizes to the contact sites independently of previously described tethers. Finally, we show that Cvm1 plays a role in sphingolipid homeostasis and that Cvm1-mediated contact sites are induced upon chemical inhibition of sphingolipid biosynthesis.
Results
A high-content microscopy screen reveals candidates for vCLAMP residents and regulators
To find additional proteins involved in vCLAMP function and regulation, we performed a microscopy-based high-content screen. To identify all areas of proximity between vacuoles and mitochondria, and not necessarily those mediated by known tethering molecules, we used an unbiased bimolecular fluorescence complementation assay with the split-Venus protein (Kerppola, 2006). We tagged the zinc channel of the vacuole membrane, Zrc1, with the C-terminal fragment of the Venus protein and the transmembrane protein of the mitochondrial outer membrane, Tom70, with the N-terminal fragment of the Venus protein, with the tagged termini facing the cytosol. This results in reconstitution of the Venus molecule and thus fluorescence in all interfaces of close proximity between the vacuole and the mitochondria, i.e., contact sites (Fig. 1 A; Eisenberg-Bord et al., 2016; Shai et al., 2018). To uncover resident proteins and regulators, we took advantage of a recently generated collection of yeast strains in which each yeast protein is expressed under the control of the strong constitutive TEF2 promoter and tagged at its N-terminus with the mCherry fluorescent protein (Weill et al., 2018; Yofe et al., 2016). vCLAMP residents should colocalize with the reporter, while regulators should affect the shape, size, or intensity of the reporter signal. Using an automated mating procedure, we generated a collection of strains each expressing one overexpressed mCherry-tagged yeast protein and the split vCLAMP reporter and screened them by automated microscopy for colocalization and for their effect on the reporter (Fig. 1 B).

A high-content screen to uncover components and regulators of the vCLAMP contact site. (A) Schematic representation of the split-vCLAMP reporter strain. The strain contains the VC fragment of the split-Venus fused to the vacuolar transporter Zrc1 and the VN fragment fused to the outer mitochondrial transmembrane protein Tom70. The two fragments of the Venus protein can come into contact only when the two membranes are in very close apposition, as in a contact site. Reconstitution of the Venus protein emits a fluorescent signal, reporting on the contact site localization. (B) Schematic representation of the high-content microscopy screen. Yeast strains carrying the split-vCLAMP reporter were mated with a collection of strains each expressing one protein tagged with an mCherry fluorophore and under the strong TEF2 promoter. Haploid cells carrying the reporter and an overexpressed protein tagged with mCherry were analyzed by automated fluorescence microscopy and manually inspected for colocalization or changes in the abundance or morphology of the contact site. (C) Representative fluorescence microscopy images of the different categories of hits identified in the screen. All hits for each category are listed on the left. Images for each category correlate to the protein in bold. Scale bar represents 5 µm. (D) Representative fluorescence microscopy image of the colocalization between mCherry-Cvm1 expressed under the control of the TEF2 promoter and the split-vCLAMP reporter fluorescence, explained in A. Scale bar represents 5 µm. (E) The colocalization of Cvm1 with the split-vCLAMP signal occurs in the proximity of the vacuole and the mitochondrial network. Fluorescence microscopy analysis of mCherry-Cvm1 under the control of the TEF2 promoter, the split-vCLAMP reporter signal, Shm1-Halo stained with JF646 as a mitochondrial marker, and CMAC as vacuolar marker. Scale bar represents 5 µm. BF = Brightfield. (F) Diagram of the Cvm1 protein depicting the region predicted as homologous to α-β hydrolase fold proteins and the region predicted as intrinsically disordered.
A high-content screen to uncover components and regulators of the vCLAMP contact site. (A) Schematic representation of the split-vCLAMP reporter strain. The strain contains the VC fragment of the split-Venus fused to the vacuolar transporter Zrc1 and the VN fragment fused to the outer mitochondrial transmembrane protein Tom70. The two fragments of the Venus protein can come into contact only when the two membranes are in very close apposition, as in a contact site. Reconstitution of the Venus protein emits a fluorescent signal, reporting on the contact site localization. (B) Schematic representation of the high-content microscopy screen. Yeast strains carrying the split-vCLAMP reporter were mated with a collection of strains each expressing one protein tagged with an mCherry fluorophore and under the strong TEF2 promoter. Haploid cells carrying the reporter and an overexpressed protein tagged with mCherry were analyzed by automated fluorescence microscopy and manually inspected for colocalization or changes in the abundance or morphology of the contact site. (C) Representative fluorescence microscopy images of the different categories of hits identified in the screen. All hits for each category are listed on the left. Images for each category correlate to the protein in bold. Scale bar represents 5 µm. (D) Representative fluorescence microscopy image of the colocalization between mCherry-Cvm1 expressed under the control of the TEF2 promoter and the split-vCLAMP reporter fluorescence, explained in A. Scale bar represents 5 µm. (E) The colocalization of Cvm1 with the split-vCLAMP signal occurs in the proximity of the vacuole and the mitochondrial network. Fluorescence microscopy analysis of mCherry-Cvm1 under the control of the TEF2 promoter, the split-vCLAMP reporter signal, Shm1-Halo stained with JF646 as a mitochondrial marker, and CMAC as vacuolar marker. Scale bar represents 5 µm. BF = Brightfield. (F) Diagram of the Cvm1 protein depicting the region predicted as homologous to α-β hydrolase fold proteins and the region predicted as intrinsically disordered.
The screen allowed the identification of 24 proteins that either colocalized with the reporter, affected it, or both, which was further confirmed by low-throughput microscopy (Figs. 1 C and S1). Manual inspection of the list directed us to focus on the uncharacterized protein Ymr160w that showed extensive colocalization with the split-vCLAMP reporter (Fig. 1 D, white arrow) and also affected reporter strength when overexpressed—a potential characteristic of a tethering molecule. Not all the observed signal of Ymr160w colocalized with the reporter signal (Fig. 1 D, cyan arrow), suggesting that the protein might be present at other subcellular localizations in addition to the vCLAMP.

Representative fluorescence microscopy images for each hit of the high-content screen. Related to Fig. 1. (A–D) Split-vCLAMP signal in the absence of overexpressed mCherry-tagged proteins (A). Hits were characterized based on colocalization with split-vCLAMP reporter (B), colocalization and effect (C), and effect only (D). Scale bars represent 5 µm. CS = contact site; BF = Brightfield.
Representative fluorescence microscopy images for each hit of the high-content screen. Related to Fig. 1. (A–D) Split-vCLAMP signal in the absence of overexpressed mCherry-tagged proteins (A). Hits were characterized based on colocalization with split-vCLAMP reporter (B), colocalization and effect (C), and effect only (D). Scale bars represent 5 µm. CS = contact site; BF = Brightfield.
To verify that Ymr160w is indeed localized to the contact, we reimaged Ymr160w in strains expressing the reporter, a mitochondrial marker (Shm1-Halo), and a vacuolar marker (luminal staining with 7-amino-4-chloromethylcoumarin [CMAC]) and proved that this signal was observed in the interface between these two organelles (Fig. 1 E). Because Ymr160 is a bona fide vCLAMP protein, we named it Cvm1 (contacts of the vacuole membrane 1).
Cvm1 is a protein of 95 kD, with no predicted transmembrane domains (TMHMM2 and TOPCONS predictions) and no detected conserved domains in the databases Pfam or InterPro (Blum et al., 2021; Krogh et al., 2001; Mistry et al., 2021; Tsirigos et al., 2015). Analysis of the protein sequence with HHPred detected homology of the central region of the protein (amino acids 244–558) to several proteins of the α-β hydrolase fold, while the N-terminal region (up to amino acid 80–200 depending on the software) is predicted as intrinsically disordered by prediction tools such as MobiDB or DISOPRED3 (Fig. 1 F; Jones and Cozzetto, 2015; Piovesan et al., 2021; Zimmermann et al., 2018).
Cvm1 is a new vCLAMP-resident protein and tether
To confirm that Cvm1 is not simply affected by the split-vCLAMP synthetic reporter, we verified its localization in a strain without it. N-terminal GFP tagged Cvm1 under the control of the strong TEF1 promoter or the milder NOP1 promoter also shows striking enrichment in the region between mitochondria (Shm1-mKate2) and the vacuole (CMAC; Fig. 2, A and B, respectively, white arrows), in agreement with it being a vacuole–mitochondria contact site resident protein. At endogenous levels, Cvm1 localizes at the vacuole membrane, where it accumulates in patches (Fig. 2 C), some of which are located in the proximity of the mitochondria (white arrow) and some of which are not (cyan arrow). Importantly, analysis of cells overexpressing GFP-Cvm1 in the absence of the synthetic tether by electron tomography combined with immunogold labeling revealed that these cells have highly extended vCLAMPs to which Cvm1 is clearly localized (Fig. 2 D).
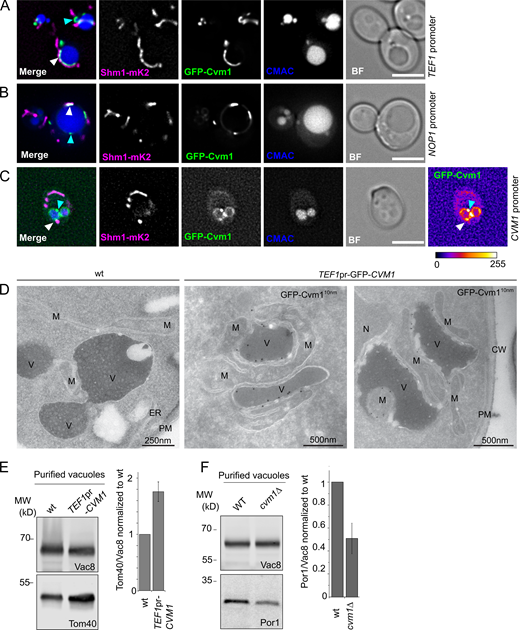
Cvm1 is a vCLAMP-resident protein and acts as a tether of the contact site. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the TEF1 promoter (A) or NOP1 promoter (B), Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. The GFP-Cvm1 signal is observed as accumulations in the regions where the mitochondrial network is apposed to the vacuole (white arrowheads). Additionally, some accumulations are observed which are away from the mitochondria, but always localize to the vacuolar rim (cyan arrowheads). Scale bar represents 5 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous CVM1 promoter in the endogenous chromosomal locus, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. The GFP-Cvm1 image is also shown with a fire look-up table (depicted below the image) to allow easier identification of accumulations. The GFP-Cvm1 signal is more homogeneous under the endogenous promoter than when Cvm1 is overexpressed. Strong accumulations can be observed, which occur away from the mitochondrial network (cyan arrowhead). Additionally, in regions of apposition of the mitochondrial network and the vacuole, some milder accumulations can be observed (white arrowhead). Scale bar represents 5 µm. (D) Ultrathin cryosections obtained from WT and overexpressed GFP-Cvm1 (TEF1 promoter) were immunogold-labeled for GFP. The presence of GFP-Cvm1 was detected at the interfaces between the vacuole (V) and the mitochondria (M), which were also extended in this strain. CW = cell wall; PM = plasma membrane. (E and F) Analysis of mitochondrial copurification in vacuole preparations. Vacuoles were purified from a WT and a deletion of CVM1 (E) and overexpression of CVM1 (F) under the control of the TEF1 promoter. Copurification of mitochondria was assessed from the levels of Por1 or Tom40 (mitochondrial markers) and Vac8 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graphs show mean ± SD of the ratio of Por1 or Tom40 to Vac8 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (E, n = 3; F, n = 4). Source data are available for this figure: SourceData F2.
Cvm1 is a vCLAMP-resident protein and acts as a tether of the contact site. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the TEF1 promoter (A) or NOP1 promoter (B), Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. The GFP-Cvm1 signal is observed as accumulations in the regions where the mitochondrial network is apposed to the vacuole (white arrowheads). Additionally, some accumulations are observed which are away from the mitochondria, but always localize to the vacuolar rim (cyan arrowheads). Scale bar represents 5 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous CVM1 promoter in the endogenous chromosomal locus, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. The GFP-Cvm1 image is also shown with a fire look-up table (depicted below the image) to allow easier identification of accumulations. The GFP-Cvm1 signal is more homogeneous under the endogenous promoter than when Cvm1 is overexpressed. Strong accumulations can be observed, which occur away from the mitochondrial network (cyan arrowhead). Additionally, in regions of apposition of the mitochondrial network and the vacuole, some milder accumulations can be observed (white arrowhead). Scale bar represents 5 µm. (D) Ultrathin cryosections obtained from WT and overexpressed GFP-Cvm1 (TEF1 promoter) were immunogold-labeled for GFP. The presence of GFP-Cvm1 was detected at the interfaces between the vacuole (V) and the mitochondria (M), which were also extended in this strain. CW = cell wall; PM = plasma membrane. (E and F) Analysis of mitochondrial copurification in vacuole preparations. Vacuoles were purified from a WT and a deletion of CVM1 (E) and overexpression of CVM1 (F) under the control of the TEF1 promoter. Copurification of mitochondria was assessed from the levels of Por1 or Tom40 (mitochondrial markers) and Vac8 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graphs show mean ± SD of the ratio of Por1 or Tom40 to Vac8 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (E, n = 3; F, n = 4). Source data are available for this figure: SourceData F2.
The extended contacts observed in the strain that overexpresses Cvm1 suggest that the protein can act as a tether of the contact site. To confirm this, we assessed the level of association of the two organelles by analyzing the copurification of mitochondria with isolated vacuoles (González Montoro et al., 2018). While the levels of the marker proteins of the different organelles in whole-cell lysate are not affected by the mutant genotypes (Fig. S2 A), vacuoles purified from a strain overexpressing Cvm1 contain higher levels of copurified mitochondrial material than vacuoles isolated from a WT strain, indicating that the two organelles are indeed more associated with each other (Fig. 2 E). Inversely, the deletion of Cvm1 results in less copurification of mitochondria with vacuoles (Fig. 2 F). The decreased tethering of the two organelles observed in the deletion strain shows that Cvm1 is indeed a tether of this contact site at endogenous levels.
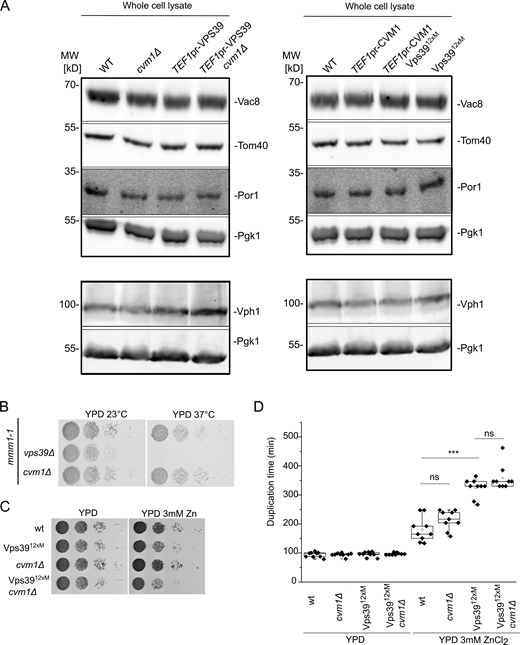
Cvm1 is not redundant with ERMES or the Vps39-mediated vCLAMP. Related to Figs. 2 and 3. (A) Analysis of the levels in whole-cell lysate of the marker proteins used to assess copurification of mitochondria with vacuoles in Fig. 2, E and F, and in Fig. 3, C and D. Pgk1 is used as a loading control. The levels of the marker proteins are not significantly affected by the different genotypes used. (B) Analysis of the genetic interaction of Cvm1 with the ERMES complex. Cells carrying the mmm1-1 temperature-sensitive allele, plus the indicated deletions or genomic modifications, were spotted as serial dilutions in YPD plates and grown at 23 or 37°C. (C) Analysis of the genetic interaction of Cvm1 with the Vps3912xM vCLAMP-impaired allele. Cells of the indicated genotypes were spotted as serial dilutions in YPD plates with or without 3 mM ZnCl2 and grown at 30°C. (D) Analysis of the genetic interaction of Cvm1 with the Vps3912xM vCLAMP-impaired allele. Cells of the indicated genotypes were diluted to OD600 = 0.1 in 96-well plates in YPD or YPD+ 3 mM ZnCl2 at 30°C. Absorbance at 600 nm was recorded using a plate reader during ∼16 h, and duplication times were calculated from the exponential phase of the growth curve. ***, P < 0.001. Source data are available for this figure: SourceData FS2.
Cvm1 is not redundant with ERMES or the Vps39-mediated vCLAMP. Related to Figs. 2 and 3. (A) Analysis of the levels in whole-cell lysate of the marker proteins used to assess copurification of mitochondria with vacuoles in Fig. 2, E and F, and in Fig. 3, C and D. Pgk1 is used as a loading control. The levels of the marker proteins are not significantly affected by the different genotypes used. (B) Analysis of the genetic interaction of Cvm1 with the ERMES complex. Cells carrying the mmm1-1 temperature-sensitive allele, plus the indicated deletions or genomic modifications, were spotted as serial dilutions in YPD plates and grown at 23 or 37°C. (C) Analysis of the genetic interaction of Cvm1 with the Vps3912xM vCLAMP-impaired allele. Cells of the indicated genotypes were spotted as serial dilutions in YPD plates with or without 3 mM ZnCl2 and grown at 30°C. (D) Analysis of the genetic interaction of Cvm1 with the Vps3912xM vCLAMP-impaired allele. Cells of the indicated genotypes were diluted to OD600 = 0.1 in 96-well plates in YPD or YPD+ 3 mM ZnCl2 at 30°C. Absorbance at 600 nm was recorded using a plate reader during ∼16 h, and duplication times were calculated from the exponential phase of the growth curve. ***, P < 0.001. Source data are available for this figure: SourceData FS2.
Cvm1 forms a parallel tethering complex to previously described vCLAMP tethers
Having established Cvm1 as a tether, it was important to understand how it functions relative to the previously described tethering complex of the vCLAMP, formed by Ypt7, Vps39, and Tom40 (Elbaz-Alon et al., 2014; González Montoro et al., 2018; Hönscher et al., 2014). Co-overexpression of Cvm1 and Vps39 showed that they formed accumulations at the vacuole–mitochondria interface that mostly exclude each other (yellow or cyan arrows in Fig. 3 A; 73% of Cvm1 structures and 56% of Vps39 structures). However, some regions where both proteins accumulated together could be observed (region marked with a yellow and a cyan arrow in Fig. 3 A; 27% of Cvm1 structures and 43% of Vps39 structures). While the observed exclusion could be an artifact of the overexpression, caused by artificial protein crowding, it most likely indicates that the two proteins are not part of the same tethering complex and may even form distinct contact areas between the two organelles. The protein Vps13 has been described to localize at vCLAMPs (Lang et al., 2015) but shows no overlap with the vCLAMPs formed by Vps39 (González Montoro et al., 2018). Assessment of the localization of Cvm1 with respect to Vps13 also showed that the two proteins do not colocalize (Fig. 3 B).
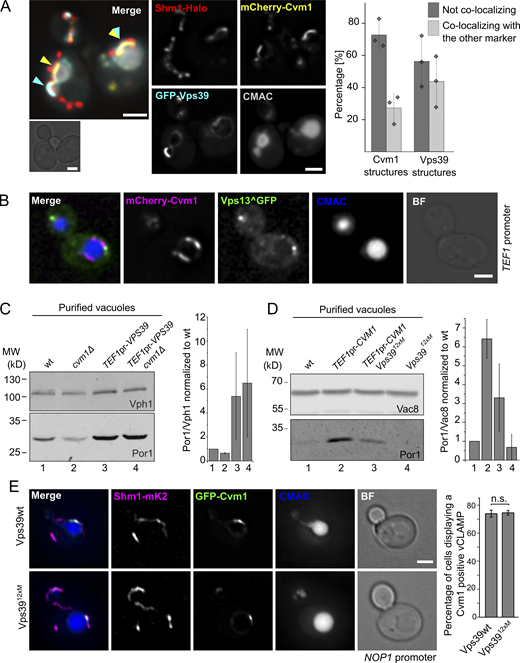
Relationship of Cvm1 with other vCLAMP components. (A) Vps39 and Cvm1 stain mostly distinct areas of the vCLAMP contact site. Representative images and quantification of a fluorescence microscopy analysis of the localization of mCherry-Cvm1 and GFP-Vps39, both under the control of the TEF1 promoter, Shm1-Halo stained with JF646 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Both the signal of Vps39 and Cvm1 accumulate in the vacuole–mitochondria interface, but they mostly exclude each other (cyan and yellow arrowheads). However, some regions show double labeling (overlapping cyan and yellow arrowheads). All scale bars represent 2 µm. The bar graph shows the percentage of Vps39 or Cvm1 structures that colocalize or not with the other marker; bars are mean from three independent experiments, shown as individual dots. Error bars represent SD. (B) Cvm1 and Vps13 do not colocalize. Fluorescence microscopy analysis of the localization of mCherry-Cvm1 under the control of the TEF1 promoter, Vps13 internally tagged with GFP, and CMAC staining as a vacuolar marker. No colocalization was observed between the signals of Vps13 and Cvm1. Scale bar represents 2 µm. BF = Brightfield. (C) Analysis of the dependence of Vps39 on Cvm1 to generate extended vCLAMPs. The mitochondrial copurification in vacuole preparations was analyzed. Vacuoles were purified from the indicated strains, and the copurification of mitochondria was assessed from the levels of Por1 (mitochondrial marker) and Vph1 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graph shows mean ± SD of the ratio of Por1/Vph1 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (n = 2). (D) Analysis of the dependence of Cvm1 on Vps39 to generate extended vCLAMPs. The mitochondrial copurification in vacuole preparations was analyzed. Vacuoles were purified from the indicated strains, and the copurification of mitochondria was assessed from the levels of Por1 (mitochondrial marker) and Vac8 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graph shows mean ± SD of the ratio of Por1/Vac8 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (n = 4). (E) Cvm1-mediated vCLAMPs are still formed when Vps39 vCLAMPs are impaired. Fluorescence microscopy analysis of a strain expressing GFP-Cvm1 under the control of the NOP1 promoter, Shm1-mKate2 as a mitochondrial marker, and labeled with CMAC as a vacuole lumen marker. Cvm1 forms accumulations in the interface between the vacuole and the mitochondria in the presence of both the WT Vps39 allele and the Vps3912xM allele, which is impaired for vCLAMP formation. Scale bar represents 2 µm. The bar graph displays mean ± SD of the percentage of cells showing Cvm1-positive vCLAMPs in both strains. Source data are available for this figure: SourceData F3.
Relationship of Cvm1 with other vCLAMP components. (A) Vps39 and Cvm1 stain mostly distinct areas of the vCLAMP contact site. Representative images and quantification of a fluorescence microscopy analysis of the localization of mCherry-Cvm1 and GFP-Vps39, both under the control of the TEF1 promoter, Shm1-Halo stained with JF646 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Both the signal of Vps39 and Cvm1 accumulate in the vacuole–mitochondria interface, but they mostly exclude each other (cyan and yellow arrowheads). However, some regions show double labeling (overlapping cyan and yellow arrowheads). All scale bars represent 2 µm. The bar graph shows the percentage of Vps39 or Cvm1 structures that colocalize or not with the other marker; bars are mean from three independent experiments, shown as individual dots. Error bars represent SD. (B) Cvm1 and Vps13 do not colocalize. Fluorescence microscopy analysis of the localization of mCherry-Cvm1 under the control of the TEF1 promoter, Vps13 internally tagged with GFP, and CMAC staining as a vacuolar marker. No colocalization was observed between the signals of Vps13 and Cvm1. Scale bar represents 2 µm. BF = Brightfield. (C) Analysis of the dependence of Vps39 on Cvm1 to generate extended vCLAMPs. The mitochondrial copurification in vacuole preparations was analyzed. Vacuoles were purified from the indicated strains, and the copurification of mitochondria was assessed from the levels of Por1 (mitochondrial marker) and Vph1 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graph shows mean ± SD of the ratio of Por1/Vph1 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (n = 2). (D) Analysis of the dependence of Cvm1 on Vps39 to generate extended vCLAMPs. The mitochondrial copurification in vacuole preparations was analyzed. Vacuoles were purified from the indicated strains, and the copurification of mitochondria was assessed from the levels of Por1 (mitochondrial marker) and Vac8 (vacuolar marker) in the purified vacuole fraction by Western blot. The bar graph shows mean ± SD of the ratio of Por1/Vac8 in the vacuole fraction normalized to the ratio for the WT sample in each experiment (n = 4). (E) Cvm1-mediated vCLAMPs are still formed when Vps39 vCLAMPs are impaired. Fluorescence microscopy analysis of a strain expressing GFP-Cvm1 under the control of the NOP1 promoter, Shm1-mKate2 as a mitochondrial marker, and labeled with CMAC as a vacuole lumen marker. Cvm1 forms accumulations in the interface between the vacuole and the mitochondria in the presence of both the WT Vps39 allele and the Vps3912xM allele, which is impaired for vCLAMP formation. Scale bar represents 2 µm. The bar graph displays mean ± SD of the percentage of cells showing Cvm1-positive vCLAMPs in both strains. Source data are available for this figure: SourceData F3.
Since both Vps39 and Cvm1 can act as tethers of the contact site, we assayed whether they require each other to establish the contact. Overexpression of Vps39 was still able to expand the contact site in the absence of Cvm1 (Fig. 3 C), which indicates that Vps39-induced vCLAMP tethers are independent of Cvm1. To test the converse dependence, we used a mutated version of Vps39, Vps3912xM, which inhibits vCLAMP formation but is functional in the context of the HOPS complex, and thus does not affect vacuole morphology (González Montoro et al., 2018). Overexpression of Cvm1 in the context of the Vps3912xM produces an increase of copurified mitochondria, but to a lesser extent than in the presence of the WT Vps39 (Fig. 3 D). The levels of the proteins used as markers of the organelles are not affected by the mutant genotypes (Fig. S2 A). Such a phenotype could either represent a sum of the two phenotypes or be an indication that Cvm1 requires an initial proximity between the two membranes formed by Vps39 to establish the contacts. Consistent with Cvm1 acting independently of Vps39, GFP-Cvm1 expressed under the control of the NOP1 promoter still formed accumulations in the interface of mitochondria and the vacuole in the presence of the Vps3912xM vCLAMP-impaired allele, to the same extent as in cells containing the WT Vps39 allele (Fig. 3 E).
The vCLAMP was initially suggested to be redundant in function with the MCS formed by the ERMES complex between the ER and the mitochondrial network (Elbaz-Alon et al., 2014; Hönscher et al., 2014; Kornmann et al., 2009). However, we later showed that this was not the case, and the synthetic sickness between ERMES subunits and Vps39 was due to its role in vesicular trafficking and not in vCLAMPs (González Montoro et al., 2018). We tested for genetic interactions anyway between deletion of CVM1 and disruption of the ERMES complex through a thermosensitive version of Mmm1 and found no synthetic sickness (Fig. S2 B). Disruption of CVM1 also showed no negative genetic interaction with the Vps3912xM allele that cannot engage in vCLAMP formation (Fig. S2, C and D; González Montoro et al., 2018). Even though the molecular function of the Vps39-mediated vCLAMP is unknown, strains carrying the vCLAMP-impaired Vps39 allele display diminished growth in the presence of ZnCl2 (González Montoro et al., 2018; Fig. S2, C and D). Deletion of CVM1 in this background did not produce a more pronounced growth defect, suggesting that the two tethers have independent functions (Fig. S2, C and D).
Taken together, our results show that Cvm1 is a novel tethering component of the vacuole–mitochondria contact site. Cvm1 localizes to this interface independently of previously described tethers and is not functionally redundant with the Vps39-vCLAMP tether or the ERMES complex. Our data indicate that at least two flavors of vCLAMP exist, one formed by Vps39 and one formed by Cvm1.
Cvm1 resides at multiple contact sites
Interestingly, we observed many patches of Cvm1 accumulation on the vacuolar membrane that were not in proximity to mitochondria (Fig. 2, A–C, cyan arrows; 42% of the accumulation patches of GFP-Cvm1 for the strain expressing Cvm1 under the control of the NOP1 promoter). This suggested that Cvm1 could mediate contacts of the vacuole with additional organelles or structures. To identify these structures, we performed colocalization experiments of Cvm1 with multiple organelle markers. We observed no significant colocalization with markers of the Golgi complex (Mnn9), trans-Golgi network (Sec7), lipid droplets (Erg6), late endosomes (Vps8), or plasma membrane (Pma1; Fig. S3, A–E). However, we often observed colocalization of Cvm1 enrichments with the perinuclear ER, observed with the marker protein Sec63 (Fig. 4 A) and with the peroxisomal protein Pex3 (Fig. 5 A).

Cvm1 does not co-localize with markers of the lipid droplets, endosomes, the Golgi complex, the trans-Golgi network, or the plasma membrane. Related to Fig. 4. (A–D) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter with markers of different organelles. Erg6-mKate2 was used as a marker for lipid droplets, Vps8-Halo as a marker of late endosomes, Sec7-Halo as a marker of the trans-Golgi network/early endosomes, and Mnn9-Halo as a marker of the early Golgi complex. All strains containing a Halo-tagged protein were labeled with the JF646 ligand. Lipid droplets were imaged in logarithmic and stationary phase, because their morphology differs in these two growth phases. No significant colocalization was observed between GFP-Cvm1 and any of the markers. Scale bars represent 2 µm. BF = Brightfield. (E) Fluorescence microscopy analysis of the localization of mCherry-Cvm1 under the control of the TEF1 promoter and Pma1-GFP as a marker of the plasma membrane. No significant colocalization was observed between the two signals. Scale bar represents 2 µm. (F) Colocalization of Cvm1 with Vac8. Fluorescence microscopy images of a strain expressing GFP-Cvm1 under the control of the NOP1 promoter and Vac8-mKate2. Both proteins localize along the vacuole membrane. Some regions of enrichment of Cvm1 are also enriched in Vac8 compared with the rest of the vacuole membrane. Scale bar represents 2 µm.
Cvm1 does not co-localize with markers of the lipid droplets, endosomes, the Golgi complex, the trans-Golgi network, or the plasma membrane. Related to Fig. 4. (A–D) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter with markers of different organelles. Erg6-mKate2 was used as a marker for lipid droplets, Vps8-Halo as a marker of late endosomes, Sec7-Halo as a marker of the trans-Golgi network/early endosomes, and Mnn9-Halo as a marker of the early Golgi complex. All strains containing a Halo-tagged protein were labeled with the JF646 ligand. Lipid droplets were imaged in logarithmic and stationary phase, because their morphology differs in these two growth phases. No significant colocalization was observed between GFP-Cvm1 and any of the markers. Scale bars represent 2 µm. BF = Brightfield. (E) Fluorescence microscopy analysis of the localization of mCherry-Cvm1 under the control of the TEF1 promoter and Pma1-GFP as a marker of the plasma membrane. No significant colocalization was observed between the two signals. Scale bar represents 2 µm. (F) Colocalization of Cvm1 with Vac8. Fluorescence microscopy images of a strain expressing GFP-Cvm1 under the control of the NOP1 promoter and Vac8-mKate2. Both proteins localize along the vacuole membrane. Some regions of enrichment of Cvm1 are also enriched in Vac8 compared with the rest of the vacuole membrane. Scale bar represents 2 µm.

Cvm1 localizes at the NVJ. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter (A) or the endogenous CVM1 promoter (B), Sec63-Halo stained with JF646 as a marker of the ER, and CMAC staining as a vacuolar marker. GFP-Cvm1 signal can be observed along the vacuole membrane, and accumulations of Cvm1 are observed in regions where the vacuole is closely apposed to the ER. Scale bar represents 2 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of a split-NVJ reporter strain with mCherry-Cvm1 expressed under the control of the ADH1 promoter and CMAC as vacuolar staining. The split-NVJ reporter contains the VC fragment fused to the vacuolar protein Zrc1 and the VN fragment fused to the ER protein Sec63. Strong accumulations of Cvm1 do not colocalize with the reporter, but weaker accumulations of Cvm1 can be observed in regions positive for the reporter. mCherry-Cvm1 signal is shown with a Fire look-up table to make the enrichments of Cvm1 easier to observe. A bar showing the correspondence between intensity levels and color is shown below. Scale bar represents 2 µm. (D) Colocalization of Cvm1 with the NVJ marker Nvj1. Fluorescence microscopy images of a strain expressing GFP-Cvm1 under the control of the endogenous CVM1 promoter and Nvj1-mKate2 as a marker of the NVJ contact site. (1–3) Cvm1 localizes along the vacuole membrane. Some cells show enrichment of Cvm1 with the NVJ (1), whereas others show enrichment in a portion of the contact (2) or no enrichment (3), as shown by the line profiles along the vacuole membrane. The bar graph to the right shows the frequency of observation of the different phenotypes. Scale bars represent 2 µm. (E) Enrichment of Cvm1 in the vacuole–ER interface does not depend on Nvj1. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter, Sec63-Halo stained with the JF646 ligand as a marker of the ER, and CMAC staining as a vacuolar marker. The experiment was performed in a strain containing the endogenous Nvj1 or a deletion of the gene. Accumulations of Cvm1 in regions of colocalization with the ER can still be observed in the absence of Nvj1.
Cvm1 localizes at the NVJ. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter (A) or the endogenous CVM1 promoter (B), Sec63-Halo stained with JF646 as a marker of the ER, and CMAC staining as a vacuolar marker. GFP-Cvm1 signal can be observed along the vacuole membrane, and accumulations of Cvm1 are observed in regions where the vacuole is closely apposed to the ER. Scale bar represents 2 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of a split-NVJ reporter strain with mCherry-Cvm1 expressed under the control of the ADH1 promoter and CMAC as vacuolar staining. The split-NVJ reporter contains the VC fragment fused to the vacuolar protein Zrc1 and the VN fragment fused to the ER protein Sec63. Strong accumulations of Cvm1 do not colocalize with the reporter, but weaker accumulations of Cvm1 can be observed in regions positive for the reporter. mCherry-Cvm1 signal is shown with a Fire look-up table to make the enrichments of Cvm1 easier to observe. A bar showing the correspondence between intensity levels and color is shown below. Scale bar represents 2 µm. (D) Colocalization of Cvm1 with the NVJ marker Nvj1. Fluorescence microscopy images of a strain expressing GFP-Cvm1 under the control of the endogenous CVM1 promoter and Nvj1-mKate2 as a marker of the NVJ contact site. (1–3) Cvm1 localizes along the vacuole membrane. Some cells show enrichment of Cvm1 with the NVJ (1), whereas others show enrichment in a portion of the contact (2) or no enrichment (3), as shown by the line profiles along the vacuole membrane. The bar graph to the right shows the frequency of observation of the different phenotypes. Scale bars represent 2 µm. (E) Enrichment of Cvm1 in the vacuole–ER interface does not depend on Nvj1. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter, Sec63-Halo stained with the JF646 ligand as a marker of the ER, and CMAC staining as a vacuolar marker. The experiment was performed in a strain containing the endogenous Nvj1 or a deletion of the gene. Accumulations of Cvm1 in regions of colocalization with the ER can still be observed in the absence of Nvj1.

Cvm1 localizes at the peroxisome–vacuole contact site. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter, Pex3-mKate2, or mCherry-SKL as peroxisomal markers, and CMAC staining as a vacuolar marker. Accumulations of GFP-Cvm1 signal can be observed on peroxisomes that are apposed to the vacuole. No signal of GFP-Cvm1 was observed on peroxisomes away from the vacuole. Additionally, GFP-Cvm1 signal can be observed on other areas of the vacuole. Scale bar represents 2 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of a split-PerVale reporter strain with mCherry-Cvm1 expressed under the control of the TEF1 promoter and CMAC as a vacuolar staining. The split-PerVale reporter contains the VC fragment fused to the vacuolar protein Zrc1 and the VN fragment fused to the peroxisomal protein Pex25. Accumulations of Cvm1 sometimes colocalize with the signal of the split-PerVale reporter. Scale bar represents 2 µm. (D) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. GFP-Cvm1 signal can be observed on the vacuolar membrane. Rarely, accumulations are observed next to peroxisomes that are apposed to the vacuole (see quantification in E). Scale bar represents 2 µm. (E) Quantification of the percentage of cells in which accumulations of Cvm1 are observed next to mitochondria, peroxisomes, or the nuclear ER, when Cvm1 is expressed under the control of either the NOP1 promoter or the endogenous CVM1 promoter. Bars represent average ± SD of three independent experiments, shown as individual dots. For each experiment, ≥50 cells were counted per condition. (F) Plot showing the number of accumulations of Cvm1 on the vacuole membrane per cell (circles), when Cvm1 is expressed under the control of the CVM1 promoter or the NOP1 promoter. The average for each of three independent experiments is shown as a diamond; ≥40 cells were counted per experiment and condition. (G) Plot showing the number of Cvm1 accumulations per cell (circles) in proximity of either mitochondria, peroxisomes, or the nuclear ER, when Cvm1 is expressed under the control of the NOP1 promoter. The average for each of three independent experiment is shown as a diamond; ≥70 cells were counted per experiment and condition. (H) Fluorescence microscopy analysis of a GFP-Cvm1 under the control of the NOP1 promoter with Pex3-mKate2 as a peroxisomal marker, Nvj1-Halo labeled with JF646, and CMAC as a vacuolar staining. 9 ± 8% of the structures containing GFP-Cvm1 and Pex3-mKate2 were found in the proximity of Nvj1-Halo (n = 3 independent experiments). Scale bars represent 2 µm. (I) Fluorescence microscopy analysis of a GFP-Cvm1 under the control of the NOP1 promoter with Nvj1-mKate2, Tom70-Halo labeled with JF646 as a mitochondrial marker, and CMAC as a vacuolar staining. 10 ± 4% of the structures containing GFP-Cvm1 and Tom70-Halo were found in the proximity of Nvj1-mKate2 (n = 3 independent experiments). Scale bars represent 2 µm.
Cvm1 localizes at the peroxisome–vacuole contact site. (A and B) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the NOP1 promoter, Pex3-mKate2, or mCherry-SKL as peroxisomal markers, and CMAC staining as a vacuolar marker. Accumulations of GFP-Cvm1 signal can be observed on peroxisomes that are apposed to the vacuole. No signal of GFP-Cvm1 was observed on peroxisomes away from the vacuole. Additionally, GFP-Cvm1 signal can be observed on other areas of the vacuole. Scale bar represents 2 µm. BF = Brightfield. (C) Fluorescence microscopy analysis of a split-PerVale reporter strain with mCherry-Cvm1 expressed under the control of the TEF1 promoter and CMAC as a vacuolar staining. The split-PerVale reporter contains the VC fragment fused to the vacuolar protein Zrc1 and the VN fragment fused to the peroxisomal protein Pex25. Accumulations of Cvm1 sometimes colocalize with the signal of the split-PerVale reporter. Scale bar represents 2 µm. (D) Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. GFP-Cvm1 signal can be observed on the vacuolar membrane. Rarely, accumulations are observed next to peroxisomes that are apposed to the vacuole (see quantification in E). Scale bar represents 2 µm. (E) Quantification of the percentage of cells in which accumulations of Cvm1 are observed next to mitochondria, peroxisomes, or the nuclear ER, when Cvm1 is expressed under the control of either the NOP1 promoter or the endogenous CVM1 promoter. Bars represent average ± SD of three independent experiments, shown as individual dots. For each experiment, ≥50 cells were counted per condition. (F) Plot showing the number of accumulations of Cvm1 on the vacuole membrane per cell (circles), when Cvm1 is expressed under the control of the CVM1 promoter or the NOP1 promoter. The average for each of three independent experiments is shown as a diamond; ≥40 cells were counted per experiment and condition. (G) Plot showing the number of Cvm1 accumulations per cell (circles) in proximity of either mitochondria, peroxisomes, or the nuclear ER, when Cvm1 is expressed under the control of the NOP1 promoter. The average for each of three independent experiment is shown as a diamond; ≥70 cells were counted per experiment and condition. (H) Fluorescence microscopy analysis of a GFP-Cvm1 under the control of the NOP1 promoter with Pex3-mKate2 as a peroxisomal marker, Nvj1-Halo labeled with JF646, and CMAC as a vacuolar staining. 9 ± 8% of the structures containing GFP-Cvm1 and Pex3-mKate2 were found in the proximity of Nvj1-Halo (n = 3 independent experiments). Scale bars represent 2 µm. (I) Fluorescence microscopy analysis of a GFP-Cvm1 under the control of the NOP1 promoter with Nvj1-mKate2, Tom70-Halo labeled with JF646 as a mitochondrial marker, and CMAC as a vacuolar staining. 10 ± 4% of the structures containing GFP-Cvm1 and Tom70-Halo were found in the proximity of Nvj1-mKate2 (n = 3 independent experiments). Scale bars represent 2 µm.
Both endogenous and overexpressed Cvm1 showed enrichment patches on the vacuole membrane that colocalized with the perinuclear ER (Fig. 4, A and B, respectively). To confirm that these regions are places of real apposition of the membranes below the resolution of light microscopy, we used a split-Venus–based reporter strain for the ER–vacuole contact site, similar to the one we used for the screen for the vCLAMP (Shai et al., 2018). Overexpressed mCherry-Cvm1 showed enrichment in the areas marked by this reporter compared with the rest of the vacuolar membrane, even though these were not the strongest accumulations observed, which likely represent vCLAMPs (Fig. 4 C).
The contact site between the nuclear ER and the vacuole, called the NVJ, is well described. The protein Nvj1 forms a tether at this contact site by its interactions with the vacuolar protein Vac8 and is exclusively located there (Pan et al., 2000). We thus compared Cvm1 localization with that of Nvj1. We observed a range of phenotypes, which included Cvm1 accumulating in the whole structure marked by Nvj1 (Fig. 4 D, line profile 1), accumulating in only part of the structure (line profile 2), or showing no accumulation in this contact site (line profile 3). Quantification of these phenotypes showed that most cells show some degree of accumulation of Cvm1 in the NVJ area: 39% of cells show enrichment in the whole NVJ, 27% of cells show enrichment in part of the structure, and 6% of cells show an accumulation adjacent to the NVJ (Fig. 4 D). Furthermore, some Cvm1 accumulations colocalized with accumulations of Vac8 on the vacuolar membrane (Fig. S3 F). Because the tether formed by Nvj1 and Vac8 is characterized as the principal tether of this structure, we tested if Cvm1 requires Nvj1 to localize to the NVJ. However, Cvm1 was still observed to form accumulations that colocalize with Sec63 in an nvj1Δ strain, indicating that this is not the case (Fig. 4 E). Cvm1 can thus be a part of the NVJ, but does not require Nvj1 to localize in this subcellular niche.
To confirm the localization of Cvm1 to the PerVales, we colocalized it with the peroxisomal marker mCherry-SKL, which is targeted to the peroxisome matrix by the peroxisomal targeting sequence of amino acids SKL at the C-terminus. We could again observe accumulations of Cvm1 in the interface between some peroxisomes and the vacuole, labeled with CMAC (Fig. 5 B). It should be noted that colocalization of Cvm1 with peroxisomes occurred only in the vicinity of the vacuole. To confirm that these proximity regions are indeed contact sites, we used a split-Venus–based reporter for the vacuole–peroxisome contact site (Shai et al., 2018). Colocalization of Cvm1 with the contact site reporter was observed (Fig. 5 C), confirming the presence of Cvm1 in this interface. However, not every contact site signal was positive for Cvm1, indicating the existence of more than one type of PerVale in the cell.
Quantification of the frequency of localization of Cvm1 at these different subcellular sites showed that at endogenous levels, 62% of cells contain Cvm1 enrichment in the NVJ, while the accumulation of Cvm1 in proximity to either mitochondria or peroxisomes is very rare (3% of cells in both cases; Fig. 5 E). Overexpression of Cvm1 from the NOP1 promoter resulted in all contact sites becoming more frequent. In particular, the contact site with mitochondria was now observed in 79% of cells (Fig. 5 E). Under the control of the endogenous promoter, most cells contain only one accumulation of Cvm1 in the vacuole membrane, while overexpression of Cvm1 from the NOP1 promoter resulted in most cells displaying two to three accumulations (Fig. 5 F). Thus, we analyzed how the multiple accumulations caused by overexpression were distributed among the different contact sites and observed that most accumulations represent contact with the mitochondria or the ER, whereas accumulations close to the peroxisomes represent a small portion. Although most cells contain either one or no contacts with a specific organelle, we could observe cells with up to four contacts of the same type (Fig. 5 G).
We conclude that the main localization of Cvm1 at endogenous levels and standard growth conditions is the NVJ. The localization of Cvm1 to the vacuole–peroxisome and vacuole–mitochondria contact sites, which in our case was evidenced by overexpression, may be induced under specific conditions (Fig. 5, E–G). It must be taken into account, however, that quantification of the contact sites by fluorescence microscopy requires an accumulation of the protein in the interfaces. Endogenous contact sites might exist that are not identifiable by this method. Indeed, even though this kind of structure was very rare for vCLAMPs under endogenous levels, we know from our biochemical approach that there is a significant contribution of Cvm1 to this contact site at endogenous levels (Fig. 2 F).
Because the contacts with the mitochondria and peroxisomes are produced by overexpression of Cvm1, we wondered if they are formed in the vicinity of the NVJ, its main localization under endogenous levels, as part of a three-way contact site. We thus imaged GFP-Cvm1 under the control of the NOP1 promoter with Nvj1 and a marker of either peroxisomes or mitochondria. We observed that the contact sites are formed mostly away from the NVJ (Fig. 5, H and I) and could only be found in the proximity of this structure in ∼10% of the cases.
Cvm1 is involved in sphingolipid metabolism
According to a high-throughput chemogenomic screen, the main condition producing a fitness defect in a strain lacking CVM1 is the addition of the drug myriocin (Hillenmeyer et al., 2008). This drug is an inhibitor of the serine palmitoyltransferase (SPT), the enzyme complex catalyzing the first and rate-limiting step in sphingolipid biosynthesis (Fig. 6 A). We confirmed that the deletion of CVM1 causes hypersensitivity to myriocin and that the effect is specific for the lack of this gene, and not due to the modifications in the genome, as the effect is suppressed by transformation of the strain with a plasmid encoding for Cvm1 (Fig. 6 B). We then tested the effect of the overexpression of Cvm1 on myriocin sensitivity and found that these strains are strongly resistant (Fig. 6 C). Using the myriocin sensitivity phenotype of cvm1Δ cells, we confirmed the functionality of N-terminal tagged Cvm1 as used throughout this study (Fig. S4 A). In contrast, lack of the lipid transfer protein Vps13 or a mutation in Vps39 that inhibits vCLAMP formation produces no changes in the sensitivity to myriocin (Fig. S4 B).

Cvm1 plays a role in sphingolipid homeostasis. (A) Diagram of the S. cerevisiae sphingolipid biosynthesis pathway, the enzymes involved, and the steps inhibited by the drugs myriocin and AbA. LCB, long-chain base. (B) Cells lacking Cvm1 are hypersensitive to myriocin. A WT strain, a strain carrying a CVM1 deletion, or three different clones carrying a CVM1 deletion transformed with a plasmid carrying CVM1 were spotted as seriated dilutions on YPD medium with or without 1 μM myriocin. (C) Cells with overexpressed CVM1 under the control of the TEF1 promoter are hyperresistant to myriocin. WT and CVM1 overexpression strains were spotted as seriated dilutions on YPD medium with or without 1.5 µM myriocin. (D) Cells lacking or overexpressing Cvm1 are hypersensitive to AbA. A WT strain, a CVM1 deletion strain, and a strain overexpressing CVM1 From the TEF1 promoter were spotted as seriated dilutions on YPD medium with or without 70 nM AbA. (E) MS-based lipidomics of whole-cell lysates of WT, Δcvm1, and CVM1 overexpression strains. Samples were normalized according to protein levels and an internal standard. For each lipid class, a ratio to the WT strain is shown. Five independent purifications were measured; bar graphs represent average ± SD, and the values for the independent samples are shown as diamonds. Samples that deviate significantly from the WT (P < 0.05) are marked with an asterisk. PA, phosphatidic acid; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; DG, diacylglycerol; TAG, triacylglycerol. (F and G) Targeted multiple-reaction monitoring MS lipid measurements of dihidrosphingosine (DHS), phytosphingosine (PHS), and ceramides (PHC) in whole-cell lysates of WT, Δcvm1 (F), and CVM1 (G) overexpression strains. Bars represent mean ± SD from four independent samples which are also shown as diamonds. Samples that deviate significantly from the WT (P < 0.05) are marked with an asterisk.
Cvm1 plays a role in sphingolipid homeostasis. (A) Diagram of the S. cerevisiae sphingolipid biosynthesis pathway, the enzymes involved, and the steps inhibited by the drugs myriocin and AbA. LCB, long-chain base. (B) Cells lacking Cvm1 are hypersensitive to myriocin. A WT strain, a strain carrying a CVM1 deletion, or three different clones carrying a CVM1 deletion transformed with a plasmid carrying CVM1 were spotted as seriated dilutions on YPD medium with or without 1 μM myriocin. (C) Cells with overexpressed CVM1 under the control of the TEF1 promoter are hyperresistant to myriocin. WT and CVM1 overexpression strains were spotted as seriated dilutions on YPD medium with or without 1.5 µM myriocin. (D) Cells lacking or overexpressing Cvm1 are hypersensitive to AbA. A WT strain, a CVM1 deletion strain, and a strain overexpressing CVM1 From the TEF1 promoter were spotted as seriated dilutions on YPD medium with or without 70 nM AbA. (E) MS-based lipidomics of whole-cell lysates of WT, Δcvm1, and CVM1 overexpression strains. Samples were normalized according to protein levels and an internal standard. For each lipid class, a ratio to the WT strain is shown. Five independent purifications were measured; bar graphs represent average ± SD, and the values for the independent samples are shown as diamonds. Samples that deviate significantly from the WT (P < 0.05) are marked with an asterisk. PA, phosphatidic acid; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; DG, diacylglycerol; TAG, triacylglycerol. (F and G) Targeted multiple-reaction monitoring MS lipid measurements of dihidrosphingosine (DHS), phytosphingosine (PHS), and ceramides (PHC) in whole-cell lysates of WT, Δcvm1 (F), and CVM1 (G) overexpression strains. Bars represent mean ± SD from four independent samples which are also shown as diamonds. Samples that deviate significantly from the WT (P < 0.05) are marked with an asterisk.

Extended lipid analyses of strains lacking or overexpressing Cvm1. Related to Fig. 6. (A) Cvm1 tagged in its N-terminus is functional as assessed by the myriocin sensitivity phenotype. WT and GFP tagged Cvm1 strains were spotted as seriated dilutions on YPD plates or YPD plates containing 1 μM myriocin. (B) Strains carrying the Vps3912xM vCLAMP-impaired mutant or a deletion of VPS13Δ growth normally on myriocin. WT, CVM1 deletion, VPS13 deletion, and VPS3912xM strains were spotted as seriated dilutions on YPD medium with or without 1 µM myriocin. (C and D) Targeted MS-based lipid measurement of different phytoceramide species from whole-cell lysates of WT, Δcvm1, and strains overexpressing CVM1, related to Fig. 6, F and G. The ratios of the peak areas normalized to the WT are shown for the different measured phytoceramide species; bars represent mean ± SD for four independent samples. Samples that differ significantly from the WT (P < 0.05) are marked with an asterisk. (E) Cells with overexpressed CVM1 under the control of the TEF1 promoter are hypersensitive to terbinafine. WT, CVM1 overexpression, and CVM1 deletion strains were spotted as seriated dilutions on YPD medium with or without 50 µg/ml terbinafine. (F) Cells with overexpressed CVM1 under the control of the TEF1 promoter show decreased ergosterol levels. Ergosterol levels (µg ergosterol/mg protein) of WT, Δcvm1, and CVM1 overexpression strains were measured using the Amplex Red Cholesterol-Assay-Kit (Invitrogen). Bars represent mean ± SD for three independent measurements. *, P < 0.05.
Extended lipid analyses of strains lacking or overexpressing Cvm1. Related to Fig. 6. (A) Cvm1 tagged in its N-terminus is functional as assessed by the myriocin sensitivity phenotype. WT and GFP tagged Cvm1 strains were spotted as seriated dilutions on YPD plates or YPD plates containing 1 μM myriocin. (B) Strains carrying the Vps3912xM vCLAMP-impaired mutant or a deletion of VPS13Δ growth normally on myriocin. WT, CVM1 deletion, VPS13 deletion, and VPS3912xM strains were spotted as seriated dilutions on YPD medium with or without 1 µM myriocin. (C and D) Targeted MS-based lipid measurement of different phytoceramide species from whole-cell lysates of WT, Δcvm1, and strains overexpressing CVM1, related to Fig. 6, F and G. The ratios of the peak areas normalized to the WT are shown for the different measured phytoceramide species; bars represent mean ± SD for four independent samples. Samples that differ significantly from the WT (P < 0.05) are marked with an asterisk. (E) Cells with overexpressed CVM1 under the control of the TEF1 promoter are hypersensitive to terbinafine. WT, CVM1 overexpression, and CVM1 deletion strains were spotted as seriated dilutions on YPD medium with or without 50 µg/ml terbinafine. (F) Cells with overexpressed CVM1 under the control of the TEF1 promoter show decreased ergosterol levels. Ergosterol levels (µg ergosterol/mg protein) of WT, Δcvm1, and CVM1 overexpression strains were measured using the Amplex Red Cholesterol-Assay-Kit (Invitrogen). Bars represent mean ± SD for three independent measurements. *, P < 0.05.
To affect sphingolipid biosynthesis at a different step, we used the drug Aureobasidin A (AbA), an inhibitor of the inositol phosphorylceramide (IPC) synthase Aur1, which thus blocks the production of all S. cerevisiae complex sphingolipids (Fig. 6 A). We found that deletion of CVM1 also causes hypersensitivity to AbA. Interestingly, overexpression of Cvm1 from the strong TEF1 promoter also causes hypersensitivity to Aba (Fig. 6 D).
These results suggest an involvement of Cvm1 in sphingolipid metabolism. In agreement with this, high-throughput genetic studies have reported a negative genetic interaction of cvm1Δ with deletion of TSC3, the regulatory subunit of the SPT, and with a temperature-sensitive allele of a subunit of the SPT, lcb2-19 (Costanzo et al., 2016). Thus, we performed mass spectrometry (MS)–based lipidomics of the cvm1Δ strain and a strain overexpressing CVM1 from the strong TEF1 promoter. While most lipid classes do not differ significantly from the control strain in these mutants, complex sphingolipids are significantly decreased in the strain that overexpresses Cvm1 (Fig. 6 E), confirming that Cvm1 affects sphingolipids. Because our lipidomics method does not detect the sphingolipid biosynthetic intermediates, long-chain bases, and ceramides, we used a targeted method to measure them. We observed a significant decrease in ceramide levels in the deletion strain and a significant increase in the overexpression strain (Fig. 6, F and G). All ceramide species are affected by the changes in Cvm1 levels, but the highly hydroxylated phytoceramide 18:0 26:0:5 (ceramide D) is particularly depleted in the deletion strain (Fig. S4, C and D).
Because sterols and sphingolipids are closely related to each other with respect to their role in biological membranes, we also addressed the effect of Cvm1 levels on ergosterol. We observed that deletion of CVM1 had no effect on resistance to the drug terbinafine, an inhibitor of the squalene epoxidase, but overexpression of Cvm1 did result in hypersensitivity to this drug (Fig. S4 E). Consistent with these results, direct measurement of ergosterol levels in cells showed a reduction in cells that overexpress Cvm1 but no effect in the deletion strain (Fig. S4 F).
Sphingolipids are tightly regulated within cells through signaling feedback loops that sense the levels of complex sphingolipids and adjust the activity of different steps of the biosynthetic pathway accordingly (reviewed in Olson et al., 2016; Roelants et al., 2017). If Cvm1 levels affect sphingolipid homeostasis, a change in the response of such pathways should be observed. One of these pathways relies on the relocalization of the proteins Slm1 and Slm2 in the plasma membrane in response to changes in complex sphingolipid levels. While Slm1 normally localizes to eisosomes, it exits these membrane compartments when plasma membrane sphingolipid levels are decreased (Berchtold et al., 2012; Roelants et al., 2011; Sun et al., 2012). Indeed, we observed that Slm1 colocalizes almost completely with the eisosome component Pil1 in control cells, but has reduced localization to eisosomes in cells that lack Cvm1, similarly to the effect caused by addition of myriocin (Fig. 7, A and B). Additionally, the levels of complex sphingolipids regulate eisosome numbers, by phosphorylation of the eisosome component Pil1 (Walther et al., 2007). Consistent with reduced complex sphingolipid levels, the cvm1Δ strain has fewer eisosomes than a WT strain (Fig. 7, A and C). This and other signaling pathways are integrated and result in changes in the phosphorylation state of both Orm1 and Orm2 proteins, two negative regulators of the SPT complex (Gururaj et al., 2013; Roelants et al., 2011). We thus addressed if changes in Cvm1 levels affect the phosphorylation state of Orm1. Indeed, overexpression of Cvm1 results in higher phosphorylation of Orm1 at steady state (Fig. 7 D). These results indicate that the changes in sphingolipid levels caused by altering the amount of Cvm1 are relevant for the cell, as they are sensed and affect the output of signaling pathways that respond to sphingolipids. The activation of these pathways also suggests that without these homeostatic responses, the effect of Cvm1 loss would be even greater. Taken together, these results support a role of Cvm1 in sphingolipid homeostasis.
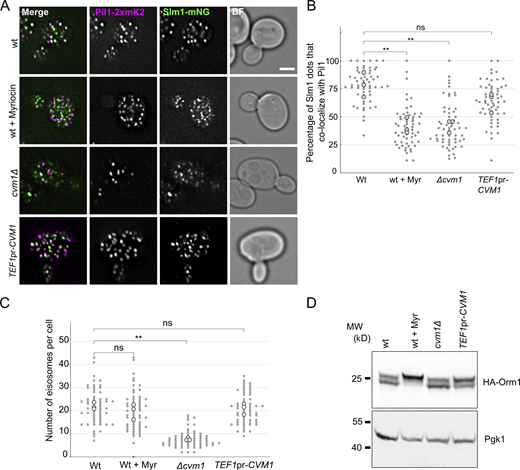
Cvm1 levels affect sphingolipid-sensing pathways. (A) Colocalization of Slm1 with Pil1. Colocalization of Slm1-mNeonGreen with Pil1-2xmKate2 was analyzed in WT, cvm1Δ, and TEF1pr-CVM1 strains. As a control, the WT strain was incubated with 5 μM myriocin for 45 min. Scale bar represents 2μm. BF = Brightfield. (B) Plot showing the quantification of the experiment shown in A. For each image, a manual threshold was applied for each channel, and the percentage of Slm1 structures that colocalized with Pil1 structures was determined for each cell. Each dot represents one cell, and the bigger dots represent the mean for each independent experiment. 20 cells were quantified for each condition and experiment, and the experiment was performed three times. (C) Plot showing the quantification of eisosomes per cell from the experiment shown in A. The number of eisosomes per cell assessed as Pil1-2xmKate2–positive structures was determined. Each dot represents one cell, and the bigger dots represent the mean for each independent experiment. 20 cells were quantified for each condition and experiment, and the experiment was performed three times. (D) Analysis of the phosphorylation-dependent upshift of Orm1. Whole-cell lysates from the indicated strains were used to analyze the upshift of HA-tagged Orm1 by Western blot. As a control, the WT strain was grown for 1 h in YPD + 5 µM myriocin. Pgk1 is used as a loading control. **, P < 0.01. Source data are available for this figure: SourceData F7.
Cvm1 levels affect sphingolipid-sensing pathways. (A) Colocalization of Slm1 with Pil1. Colocalization of Slm1-mNeonGreen with Pil1-2xmKate2 was analyzed in WT, cvm1Δ, and TEF1pr-CVM1 strains. As a control, the WT strain was incubated with 5 μM myriocin for 45 min. Scale bar represents 2μm. BF = Brightfield. (B) Plot showing the quantification of the experiment shown in A. For each image, a manual threshold was applied for each channel, and the percentage of Slm1 structures that colocalized with Pil1 structures was determined for each cell. Each dot represents one cell, and the bigger dots represent the mean for each independent experiment. 20 cells were quantified for each condition and experiment, and the experiment was performed three times. (C) Plot showing the quantification of eisosomes per cell from the experiment shown in A. The number of eisosomes per cell assessed as Pil1-2xmKate2–positive structures was determined. Each dot represents one cell, and the bigger dots represent the mean for each independent experiment. 20 cells were quantified for each condition and experiment, and the experiment was performed three times. (D) Analysis of the phosphorylation-dependent upshift of Orm1. Whole-cell lysates from the indicated strains were used to analyze the upshift of HA-tagged Orm1 by Western blot. As a control, the WT strain was grown for 1 h in YPD + 5 µM myriocin. Pgk1 is used as a loading control. **, P < 0.01. Source data are available for this figure: SourceData F7.
A decrease in complex sphingolipid levels induces Cvm1-mediated contact sites
Because Cvm1 levels determine growth rate upon depletion of sphingolipids by addition of myriocin or AbA (Fig. 6, B and C), and changes in Cvm1 amounts affect the levels of sphingolipids (Fig. 6, E–G), we investigated if Cvm1 is also regulated by sphingolipid levels, further supporting a functional link. To do this, we visualized the localization of the protein under sphingolipid depletion. At endogenous levels, Cvm1 localizes mostly homogeneously to the vacuole membrane, with slight accumulations near the mitochondrial network and enrichment at the NVJ in some cells (Figs. 2 C, 4 D, and 8 A). However, incubation with myriocin for 60 min or AbA for 30 min caused Cvm1 to accumulate in the NVJ in practically all cells (Fig. 8, A and B; and Fig. S5 A). In addition, the enrichment in this structure was much more pronounced. To quantify this effect, the ratio between the mean intensity value of GFP-Cvm1 in the area marked by Nvj1 and the mean intensity value along the rest of the vacuole membrane was measured. Our results show that under control conditions, some cells show enrichment of Cvm1 in the NVJ (ratio >1), while others show a homogeneous partitioning (ratio ∼1; Fig. 8 C, mean enrichment factor for yeast extract, peptone, dextrose [YPD] condition = 1.23), consistent with what we described before (Fig. 4 D). However, inhibition of the sphingolipid biosynthesis pathway at both the level of the SPT (+ myriocin) or the level of Aur1 (+ AbA), causes Cvm1 to enrich strongly in the NVJ (mean enrichment factor: myriocin = 2.26, AbA = 2.45; Fig. 8, A–C; and Fig. S5 A).

Cvm1-mediated contacts are induced by a decrease in complex sphingolipid levels. (A) Cvm1 accumulates at the NVJ upon depletion of sphingolipids. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Nvj1-mKate2 as a NVJ marker, and CMAC staining as a vacuolar marker. Cells were grown in rich media (YPAD) and incubated with 5 μM myriocin for 60 min where indicated. Under normal growth conditions, Cvm1 accumulates at the NVJ in some cells. After incubation with myriocin, the accumulations of Cvm1 at the NVJ become more pronounced and happen in a higher number of cells. Scale bar represents 2 μm. Dotted lines mark cell outlines. (B) Quantification of the effect of myriocin and AbA on the localization of GFP-Cvm1 to the NVJ; representative images are shown in A and Fig. S5 A. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging. The percentage of cells in which Cvm1 was enriched at the NVJ was quantified for each growth condition. The bars represent the mean ± SD for three independent measurements. (C) Quantification of enrichment of GFP-Cvm1 at the NVJ upon treatment with myriocin and AbA. For each cell, an NVJ enrichment factor was calculated as the mean intensity value of GFP-Cvm1 along a line profile within the NVJ, divided by the mean intensity value in a line profile along the rest of the vacuolar membrane. Each dot represents one cell, and the bigger circles represent the mean for each of three independent experiments. For each experiment and condition, at least 40 cells were analyzed. (D) Cvm1 accumulates more frequently at PerVale upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. Cells were grown in YPAD and incubated with 5 μM myriocin for 60 min where indicated. Scale bar represents 2 μm. (E) Quantification of the effect of myriocin and AbA on the distribution of GFP-Cvm1. Cells were grown in YPAD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging (related to D and Fig. S5 B). A Cvm1-positive PerVale was considered as an accumulation of GFP-Cvm1 fluorescence in the place where a peroxisome (labeled with Pex3-mKate2) was apposed to the vacuole (labeled with CMAC). A percentage was determined for each experiment, and the bars represent the mean of the three experiments; 50 cells were quantified per condition and experiment. (F) Cvm1 accumulations at the vCLAMP are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min where indicated. Scale bar represents 2 μm. (G) Quantification of effect of myriocin and AbA on the distribution of GFP-Cvm1. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging (related to F and Fig. S5 C). A Cvm1-positive vCLAMP was considered as an accumulation of GFP-Cvm1 fluorescence in the place where the mitochondria (labeled with Shm1-mKate2) was apposed to the vacuole (labeled with CMAC). A percentage was determined for each experiment, and the bars represent the mean of the three experiments; error bars represent SD; 50 cells were quantified per condition and experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Cvm1-mediated contacts are induced by a decrease in complex sphingolipid levels. (A) Cvm1 accumulates at the NVJ upon depletion of sphingolipids. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Nvj1-mKate2 as a NVJ marker, and CMAC staining as a vacuolar marker. Cells were grown in rich media (YPAD) and incubated with 5 μM myriocin for 60 min where indicated. Under normal growth conditions, Cvm1 accumulates at the NVJ in some cells. After incubation with myriocin, the accumulations of Cvm1 at the NVJ become more pronounced and happen in a higher number of cells. Scale bar represents 2 μm. Dotted lines mark cell outlines. (B) Quantification of the effect of myriocin and AbA on the localization of GFP-Cvm1 to the NVJ; representative images are shown in A and Fig. S5 A. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging. The percentage of cells in which Cvm1 was enriched at the NVJ was quantified for each growth condition. The bars represent the mean ± SD for three independent measurements. (C) Quantification of enrichment of GFP-Cvm1 at the NVJ upon treatment with myriocin and AbA. For each cell, an NVJ enrichment factor was calculated as the mean intensity value of GFP-Cvm1 along a line profile within the NVJ, divided by the mean intensity value in a line profile along the rest of the vacuolar membrane. Each dot represents one cell, and the bigger circles represent the mean for each of three independent experiments. For each experiment and condition, at least 40 cells were analyzed. (D) Cvm1 accumulates more frequently at PerVale upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. Cells were grown in YPAD and incubated with 5 μM myriocin for 60 min where indicated. Scale bar represents 2 μm. (E) Quantification of the effect of myriocin and AbA on the distribution of GFP-Cvm1. Cells were grown in YPAD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging (related to D and Fig. S5 B). A Cvm1-positive PerVale was considered as an accumulation of GFP-Cvm1 fluorescence in the place where a peroxisome (labeled with Pex3-mKate2) was apposed to the vacuole (labeled with CMAC). A percentage was determined for each experiment, and the bars represent the mean of the three experiments; 50 cells were quantified per condition and experiment. (F) Cvm1 accumulations at the vCLAMP are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min where indicated. Scale bar represents 2 μm. (G) Quantification of effect of myriocin and AbA on the distribution of GFP-Cvm1. Cells were grown in YPD and incubated with 5 μM myriocin for 60 min or 250 nM AbA for 30 min before imaging (related to F and Fig. S5 C). A Cvm1-positive vCLAMP was considered as an accumulation of GFP-Cvm1 fluorescence in the place where the mitochondria (labeled with Shm1-mKate2) was apposed to the vacuole (labeled with CMAC). A percentage was determined for each experiment, and the bars represent the mean of the three experiments; error bars represent SD; 50 cells were quantified per condition and experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
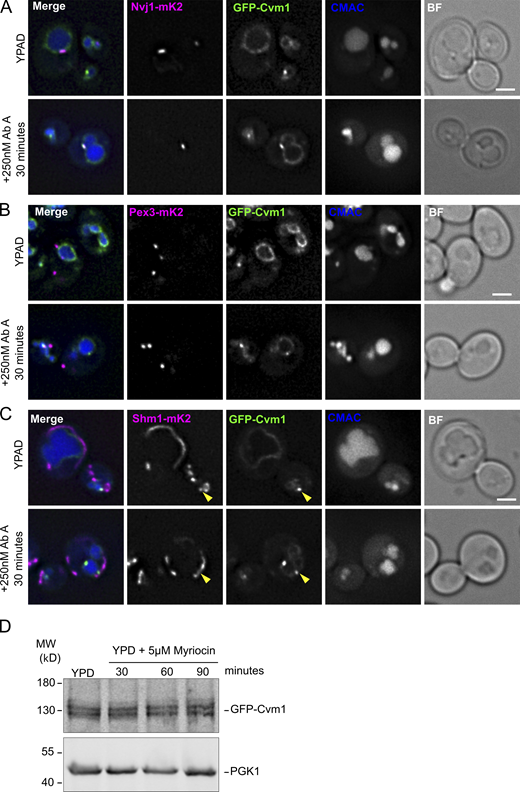
Cvm1-mediated contacts are induced by a decrease in complex sphingolipids, but protein levels of Cvm1 are not altered. Related to Fig. 8. (A) Cvm1 accumulates at the NVJ upon depletion of sphingolipids. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Nvj1-mKate2 as a NVJ marker, and CMAC staining as a vacuolar marker. Cells were grown in rich media (YPAD) and incubated with 250 nM AbA for 30 min where indicated. Under normal growth conditions, Cvm1 is accumulated in the NVJ in some cells and not in others. After incubation with AbA, the accumulations of Cvm1 at the NVJ become more pronounced and happen in more cells. Scale bar represents 2 μm. BF = Brightfield. (B) Cvm1 accumulations at the PerVale are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. Cells were grown in YPAD and incubated with 5 μM AbA for 60 min where indicated. Scale bar represents 2 μm. (C) Cvm1 accumulations at the vCLAMP are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Cells were grown in YPD and incubated with 5 μM AbA for 60 min where indicated. The yellow arrowheads indicate accumulations of Cvm1 on the vacuole membrane that co-localize with the mitochondrial network. Scale bar represents 2 μm. (D) Cvm1 protein levels are not affected by myriocin treatment. Whole-cell lysates of a strain containing GFP-Cvm1 under the control of its endogenous promoter were normalized by total protein amount and analyzed by SDS-PAGE and Western blot. The levels of GFP-Cvm1 do not change significantly throughout the treatment with the drug. Pgk1 was used as a loading control. Source data are available for this figure: SourceData FS5.
Cvm1-mediated contacts are induced by a decrease in complex sphingolipids, but protein levels of Cvm1 are not altered. Related to Fig. 8. (A) Cvm1 accumulates at the NVJ upon depletion of sphingolipids. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Nvj1-mKate2 as a NVJ marker, and CMAC staining as a vacuolar marker. Cells were grown in rich media (YPAD) and incubated with 250 nM AbA for 30 min where indicated. Under normal growth conditions, Cvm1 is accumulated in the NVJ in some cells and not in others. After incubation with AbA, the accumulations of Cvm1 at the NVJ become more pronounced and happen in more cells. Scale bar represents 2 μm. BF = Brightfield. (B) Cvm1 accumulations at the PerVale are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Pex3-mKate2 as a peroxisomal marker, and CMAC staining as a vacuolar marker. Cells were grown in YPAD and incubated with 5 μM AbA for 60 min where indicated. Scale bar represents 2 μm. (C) Cvm1 accumulations at the vCLAMP are more frequent upon decreasing sphingolipid levels. Fluorescence microscopy analysis of the localization of GFP-Cvm1 under the control of the endogenous promoter, Shm1-mKate2 as a mitochondrial marker, and CMAC staining as a vacuolar marker. Cells were grown in YPD and incubated with 5 μM AbA for 60 min where indicated. The yellow arrowheads indicate accumulations of Cvm1 on the vacuole membrane that co-localize with the mitochondrial network. Scale bar represents 2 μm. (D) Cvm1 protein levels are not affected by myriocin treatment. Whole-cell lysates of a strain containing GFP-Cvm1 under the control of its endogenous promoter were normalized by total protein amount and analyzed by SDS-PAGE and Western blot. The levels of GFP-Cvm1 do not change significantly throughout the treatment with the drug. Pgk1 was used as a loading control. Source data are available for this figure: SourceData FS5.
Interestingly, the localization of Cvm1 at the vCLAMP and PerVale contact sites was also enhanced by treatment of the cells with these drugs. At endogenous levels, Cvm1 was rarely enriched in places of apposition of peroxisomes to the vacuole (observed in 3% of cells; Fig. 8, D and E). However, after incubation with myriocin or AbA, these contacts were observed in a much higher proportion of cells (21% for myriocin, 13% for AbA; Fig. 8, D and E; and Fig. S5 B). Likewise, the accumulations of Cvm1 in the proximity of the mitochondrial network were observed more often after treatment with the drugs, even though they remain a rare observation even under these conditions (Fig. 8, F and G; and Fig. S5 C). The enhancement of the contact sites is a result of relocalization of the protein and not increased protein levels in response to myriocin treatment (Fig. S5 D).
These results indicate that the contact sites formed by Cvm1 on the vacuole membrane are enhanced when sphingolipid levels decrease. In line with this, the levels of Cvm1 determine growth rate under sphingolipid-limiting conditions.
Discussion
The role of MCSs in mediating communication between organelles and maintaining cellular homeostasis is increasingly recognized. In this work, we used an unbiased screening approach as a starting point to discover new proteins of the vCLAMP contact. This allowed us to identify Cvm1, a so-far-uncharacterized protein, as a novel component of three different MCSs involving the vacuole. The localization of Cvm1 to multiple MCSs suggests that it performs a function specific to the contact site environment, shared by these three interfaces. Recently, other proteins have also been shown to localize to multiple MCSs (Bean et al., 2018; Elbaz-Alon et al., 2015; Murley et al., 2015; Park et al., 2016), suggesting that cells can use a limited repertoire of proteins with MCS-specific activities to functionalize the whole network of contacts. This poses the interesting question of what mechanism allows for these multiple localizations, as well as the regulation of the distribution of a limited pool of protein among them. In the case of the lipid transport protein Vps13, this is achieved by competing adaptor proteins in the different contact sites (Bean et al., 2018).
Taking advantage of several approaches, we show that Cvm1 functions as a tether of the vCLAMP, and that this tethering capacity plays a role in the extent of the contact even at endogenous levels of the protein. However, this does not mean that Cvm1 is only a tether. Many proteins use the binding to both membranes as a way to localize to a contact site, while performing additional functions. The involvement of Cvm1 in sphingolipid homeostasis suggests that it aids in lipid transport through contact sites, either directly or by recruiting additional factors.
Cvm1 localizes to some but not all vCLAMPs, NVJs, and PerVales. This indicates the presence of heterogeneity within these contact sites, with varying proteomes. MCSs have already been implicated in generating heterogeneity within organelle populations, such as lipid droplets, which may provide functional flexibility to organelles (Eisenberg-Bord et al., 2018; Teixeira et al., 2018). This shows a further level of heterogeneity, however: the subpopulation of an organelle that contacts another compartment may do so through contacts of different proteomes and functionalities. Likewise, we saw that Cvm1 does not colocalize with any known component of the vCLAMP. This highlights the importance of using methods that simply detect the proximity between the membranes to define contact sites (e.g., EM, split fluorescent protein reporters) and not just known tethers or components.
Cvm1 localization to the NVJ, which is the most prominent localization under endogenous Cvm1 levels, strengthens the role of this contact site as a regulatory hub for lipid flux and metabolism. Different studies have shown a role of the NVJ in regulating the biosynthesis of storage lipids and the biogenesis of lipid droplets (Barbosa et al., 2015; Eisenberg-Bord et al., 2018; Hariri et al., 2017; Teixeira et al., 2018). Interestingly, two other proteins involved in sphingolipid homeostasis have also been shown to localize to the NVJ, the enzyme Tsc13, required for very-long-chain fatty acid synthesis (Kohlwein et al., 2001), and the protein Nvj2, a ceramide transporter (Liu et al., 2016). Taken together, these findings suggest that the NVJ is also a hub for the coordination of sphingolipid homeostasis.
Sphingolipids are essential as membrane components, especially to generate the densely packed outer layer of the plasma membrane together with sterols. Unlike other lipid classes, sphingolipids cannot be stored in cells, making their biosynthesis and turnover a key point of regulation to maintain membrane homeostasis. This is exemplified by the signaling network formed by the kinases TORC2 and Ypk1/Ypk2, which regulates different enzymes in the biosynthetic pathway, including the SPT and the ceramide synthases, upon changes in plasma membrane sphingolipid levels (Berchtold et al., 2012; Muir et al., 2014; Roelants et al., 2011). Our data show that MCSs of the endolysosomal system also play a role in the regulation of sphingolipid levels, likely by controlling the mobilization of sphingolipids from one compartment to the other. A role of the endolysosomal system in sphingolipid homeostasis has been previously shown, as affecting endosome to Golgi vesicular trafficking produces a dysregulation of sphingolipid species (Fröhlich et al., 2015), and vesicular transport along the endolysosomal pathway becomes essential when synthesis of complex sphingolipids is affected (Voynova et al., 2015). Our data strengthen this connection and suggest that the endolysosomal pathway connects to other organelles to exchange sphingolipids through MCSs. Accordingly, it was recently shown that Niemann-Pick type C protein 1 plays a role in the formation of lysosomal MCSs (Höglinger et al., 2019; Meneses-Salas et al., 2020). Mutations in this protein are the most common cause of Niemann-Pick type C disease, a hallmark of which is the accumulation of sphingolipids in cells (Newton et al., 2018). This suggests that endolysosomal contact sites play a conserved role in the regulation of sphingolipids, with a pathological outcome when they are affected.
Furthermore, the contact sites mediated by Cvm1 increase and recruit larger amounts of the protein upon a drop in sphingolipid levels. This indicates that regulatory mechanisms exist that control the abundance of this structure as a way to counterbalance a defective cellular sphingolipid composition and maintain homeostasis. This adds to several other observations of MCSs being dynamic and regulated. The NVJ is one of the clearest examples of this. It is a contact site that extends in size in response to nutrient availability, and many of its known components localize there only under specific conditions, such as changes in carbon source or amino acid availability, or ceramide toxicity in the ER (Hariri et al., 2017; Lang et al., 2015; Liu et al., 2016). We now show that a new component of the NVJ responds to complex sphingolipid levels for its localization. How the machinery sensing and transducing this imbalance relates to the already known sphingolipid homeostatic mechanisms is an important question for the future, as well as the molecular mechanisms that result in contact site expansion or reduction.
Materials and methods
Plasmids, strains, and molecular biology
S. cerevisiae strains were based on either BY4741 or SEY6210. Genetic manipulation in the yeast S. cerevisiae was carried out by homologous recombination of PCR-amplified cassettes as described in Janke et al. (2004). The yeast strains and plasmids used are listed in Tables S1 and S2, respectively.
The CVM1 complementing plasmid was generated by PCR amplification of the CVM1 open reading frame, plus 519 base pairs upstream and 464 base pairs downstream from S. cerevisiae genomic DNA, using the primers oAL428, 5′-AAAAGTCGACCTAGCTGGGAATGTTC-3′, and oAL429, 5′-TTTTGGATCCCCGCCACCCATTGGAG-3′. The PCR product was digested with BamHI and SalI restriction enzymes and ligated into the same sites into a pRS315-based plasmid containing the KanMX resistance cassette (Sikorski and Hieter, 1989).
High-content screen for contact site proteins
To identify potential contact site proteins, a reporter for the mitochondria–vacuole contact site (Tom70-VN Zrc1-VC; Shai et al., 2018) was integrated into a collection of 1,165 strains that have an overexpression promoter (TEF2pr) and an N-terminal mCherry tag from the SWAT TEF2pr-mCherry library (Weill et al., 2018; Yofe et al., 2016). The library strains were selected to represent all cases where protein localization is annotated as “punctate,” as this is the most common localization for contact site proteins. To combine the two traits, a query strain expressing the reporter was crossed into the library strains using the synthetic genetic array method (Cohen and Schuldiner, 2011; Tong and Boone, 2006).
All yeast manipulations were performed in high-density format (384–1,536 strains per plate) using a RoToR bench-top colony array instrument (Singer Instruments). In short, cells were mated on rich medium plates, and diploids were selected in synthetic medium lacking histidine and containing Geneticin (200 µg/ml, G418; Formedium) and Nourseothricin (200 µg/ml, NAT; WERNER BioAgents). Sporulation was induced by transferring cells to nitrogen starvation media plates for 8 d. Haploid cells were selected in SD-Leu (for MAT α selection) and -Arg-Lys with toxic amino-acid derivatives L-canavanine (50 mg/liter; Sigma-Aldrich) and thialysine (50 mg/liter, S-AEC; Sigma-Aldrich) to select against diploids. Finally, haploid cells containing the combination of manipulations desired were selected by depleting the medium of His and adding G418 and NAT. Selected strains were verified by microscopy and confirmed by PCR.
The resulting library was screened using an automated microscopy setup. Cells were transferred from agar plates into 384-well plates for growth in liquid medium using the RoToR arrayer. Liquid cultures were grown in a LiCONiC incubator overnight at 30°C in SD-His. A JANUS liquid handler (PerkinElmer) connected to the incubator was used to dilute the strains to an OD600 of ∼0.2, and plates were incubated at 30°C for 4 h. Strains were then transferred by the liquid handler into glass-bottom 384-well microscope plates (Matrical Bioscience) coated with concanavalin A (Sigma-Aldrich). After 20 min, wells were washed twice with SD-Riboflavin medium to remove nonadherent cells and to obtain a cell monolayer. Plates were then transferred to an Olympus automated inverted fluorescent microscope system using a robotic swap arm (Hamilton). Cells were imaged in SD-Riboflavin at 18–20°C using a 60× air lens (NA 0.9) and with an ORCA-ER charge-coupled device camera (Hamamatsu), using ScanR software. Images were acquired in two channels: GFP (excitation filter 490/20 nm, emission filter 535/50 nm) and mCherry (excitation filter 572/35 nm, emission filter 632/60 nm). After acquisition, images were manually reviewed using ImageJ (National Institutes of Health). Hits from the library were reimaged using a VisiScope Confocal Cell Explorer system composed of a Zeiss Yokogawa spinning disk scanning unit (CSU-W1) coupled with an inverted Olympus IX83 microscope. Single-focal-plane images were acquired with a 60× oil lens and captured using a PCO-Edge sCMOS camera, controlled by VisiView software (GFP/Venus [488 nm], RFP/mCherry [561 nm]). Images were transferred to ImageJ for linear adjustments to contrast and brightness.
Low-throughput microscopy
Cells were grown to logarithmic phase in rich media (YPD; or YPAD: yeast extract, peptone, adenine, dextrose) or synthetic medium, supplemented with essential amino acids (SDC). The vacuolar membrane was stained by adding 30 μM FM4-64 (Thermo Fisher Scientific) for 30 min, followed by washing and incubation in medium without dye for 1 h. The acidic lumen of the vacuole was stained by adding 20 µM CMAC dye (Invitrogen) for 15 min, followed by one washing step in SDC medium. Proteins tagged with the HaloTag were labeled with the Janelia Fluor 646 ligand (Promega). The yeast cells (0.5 OD units) were incubated with 2.5 µM of Janelia Fluor 646 for 30 min, followed by 10 washing steps with SDC medium (Day et al., 2018). Where indicated, cells were incubated with 5 μM myriocin from Mycelia sterilia (Sigma-Aldrich) or 250 nM AbA (TaKaRa Bio) before imaging.
Cells were imaged live in SDC medium at 23°C on an Olympus IX-71 inverted microscope equipped with 100× NA 1.49 and 60× NA 1.40 objectives; a sCMOS camera (PCO); an InsightSSI illumination system; 4′,6-diamidino-2-phenylindole, GFP, mCherry, and Cy5 filters; and SoftWoRx software (Applied Precision). We used z-stacks with 200-, 250-, or 350-nm spacing for constrained-iterative deconvolution (SoftWoRx). All microscopy image processing and quantification was performed using ImageJ. One plane of the z-stack is shown in each figure.
For the quantification of enrichment of GFP-Cvm1 in the NVJ, all cells in which the Nvj1-mKate2 signal was visible were taken into account. A line profile was drawn along the region positive for Nvj1-mKate2 and a second one along the rest of the vacuole membrane. The enrichment factor was calculated as the mean intensity value for GFP-Cvm1 along the first line profile divided by the mean intensity value in the second. The experiment was performed three times, and for each experiment and condition, ≥40 cells were analyzed. The single-cell data, the means for each experiment, and P values for a t test calculated using the mean for each experiment are shown in a SuperPlot, according to (Lord et al., 2020).
Growth tests
For growth test in solid medium, cells were grown to saturation in liquid YPD, diluted in YPD, grown to exponential phase, and diluted to an OD600 of 0.25. Serial dilutions (1:10) were spotted onto plates with different media and imaged after 1–4 d depending on the medium.
Immuno-EM
Cells were grown in YPG (0.5% glucose) to exponential phase and then embedded for the Tokuyasu procedure as previously described (Griffith et al., 2008). Ultrathin cryosections were immunogold-labeled using a rabbit anti-GFP (ab290; Abcam, see Table S3 for details), followed by detection with protein A-gold 10-nm conjugate (Cell Microscopy Center, University Medical Center Utrecht, The Netherlands). Cell sections were examined using an 80-KV transmission electron microscope (FEI-Cm100bio).
MS-based lipidomics
All yeast strains were grown in YPD for ≥24 h in mid-log phase. At an OD of 0.8, 25 OD units of cells were collected, washed with 155 mM ammonium formate, frozen in liquid nitrogen, and afterwards stored at −80°C. Cells were lysed in 155 mM ammonium formate by mechanical disruption with glass beads using a FastPrep Homogenizer (MP Biomedicals). The protein concentration of each sample was determined using the Bradford reagent (Bio-Rad).
Measurement of all lipid classes
Lipids were extracted from whole-cell lysates containing 400 μg of protein by two-step chloroform:methanol extraction. The first extraction was performed with 15:1 chloroform/methanol. The organic phase was extracted and dried. With the remaining aqueous phase, a second extraction was performed with 2:1 chloroform/methanol. The dried lipid extracts were then dissolved in A:B buffer (1:1; mobile phase A: 50:50 ACN/H20, 10 mM NH4HCO2, and 0.1% HCO2H; mobile phase B: 10:88:2 ACN/IPA/H2O, 2 mM NH4HCO2, and 0.02% HCO2H) and injected into the HPLC for analysis in positive (first extract) and negative (second extract) mode. Lipid extracts were separated on an Accucore C18 column (2.1 × 150 mm, 2.6 µm; Thermo Fisher Scientific) connected to a Nexera XR HPLC (Shimadzu). A binary solvent system was used (mobile phases A and B as described above) in a 20-min gradient at a flow rate of 300 μl/min with the column temperature kept constant at 35°C. The HPLC was connected online to a Q-Exactive Plus mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific). Mass spectra were acquired in data-dependent mode to automatically switch between full scan MS and ≤10 data-dependent MS/MS scans. The maximum injection time for full scans was 100 ms, and resolution was adjusted to 70,000 at m/z 200 and a mass range of 200–1,200 m/z in negative and positive mode. The 10 most intense ions from the survey scan were selected and fragmented with high-energy collision dissociation with a normalized collision energy of 30. Target values for MS/MS were set at 100,000 with a maximum injection time of 50 ms at a resolution of 35,000 at m/z 200.
Peaks were analyzed using the Lipid Search algorithm (Thermo Fisher Scientific). Peaks were defined through raw files, product ion, and precursor ion accurate masses. Candidate molecular species were identified by database (>1,000,000 entries) search ion adducts. Mass tolerance was set to 5 ppm for the precursor mass. Samples were aligned within a time window, and results were combined in a single report. For quantification of complex sphingolipids, Skyline software was used (Pino et al., 2020). An internal standard for lysophosphatidylethanolamine (17:1) was spiked in before extraction and used for normalization. Peak areas for every lipid class were summed and normalized by the average value for the control strain. The measured lipid classes were phosphatidic acid, phosphatidylglycerol, phosphatidylinositol, phosphatidylserine, phosphatidylethanolamine, phosphatidylcholine, diacylglycerol, triacylglycerol, IPC, and mannosyl-IPC. Five independent purifications were measured, bar graphs represent mean ± SD, and the individual data points are also depicted.
Targeted measurement of long-chain bases and ceramides
Lipids were extracted from whole-cell lysates containing 200 μg protein by chloroform:methanol (2:1) extraction. Dried lipid extracts were dissolved in A:B buffer (65:35%; mobile phase A: 50:50 ACN/H2O, 10 mM NH4HCO2, 0.1% HCO2H; mobile phase and B: 10:88:2 ACN/IPA/H2O, 2 mM NH4HCO2, 0.02% HCO2H) and analyzed by LC-MS/MS.
Lipid extracts were separated on an Accurcore C30 column (2.1 × 150 mm, 2.6 µm; Thermo Fisher Scientific) connected to a Nexera XR HPLC (Shimadzu). A binary solvent system was used (mobile phases A and B as described above) in a 6-min gradient at a flow rate of 0.4 ml/min with the column temperature kept constant at 40°C. The lipid samples were analyzed by a QTRAP 5500 LC-MS/MS system (SCIEX), with an IonDrive TurboV source. The data were acquired in positive, multiple reaction monitoring mode with 60-s detection windows.
The peaks were analyzed using SciexOS software. Before extraction, sphingosine (d17:1; Avanti Polar Lipids) was spiked in as an internal standard for normalization. The area peaks of the different lipid classes, dihydrosphingosine, phytosphingosine, and phytoceramide, were summed and normalized by the average of the WT sample. Five independent purifications were measured. Bar graphs represent mean ± SD, and the individual data points are also depicted.
Sterol measurement using the Amplex Red cholesterol assay
The total cellular ergosterol levels were determined using the Amplex Red Cholesterol-Assay-Kit (Invitrogen) following the manufacturer’s protocol. Briefly, 30 OD units of cells resuspended in 500 μl lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% SDS) were mechanically lysed with glass beads in a Fast Prep Homogenizer (MP Biomedicals). The protein concentration of each sample was determined using the Bradford reagent (Bio-Rad), and whole-cell lysates equivalent to 6 µg protein were used. Fluorescence was measured with a SpectraMax M3 (Molecular Devices) microplate reader with an excitation of 540 nm and an emission detection of 590 nm. The determinations were performed in triplicate for each strain.
Statistical analysis
Statistical analysis was based on t tests when two conditions were compared and ANOVA when more than two conditions were compared: *, P < 0.05; **, P < 0.01; ***, P < 0.001. For microscopy experiments, the statistical comparisons were performed among the means of individual experiments, not based on data points for individual cells.
Vacuole purification, whole-cell lysate preparation, SDS-PAGE, and Western blot
Vacuoles were isolated as described in Haas (1995). Protein concentration was measured by Bradford method (Bio-Rad), and the same protein amount was loaded onto SDS-PAGE for analysis.
To prepare whole-cell lysates, proteins were extracted by an alkaline extraction protocol. Briefly, 4 OD units of cells in mid-log phase were harvested and snap frozen in liquid nitrogen. The pellet was resuspended in 500 μl lysis solution (0.2 M NaOH and 0.2% β-mercaptoethanol) and incubated on ice for 15 min. Then, 75 μl of 100% TCA was added, and the solution was mixed and incubated for 15 min on ice, followed by 15-min centrifugation at 20,000 g at 4°C. The supernatant was discarded, and the pellet was resuspended in 1 ml ice-cold acetone by sonication. After centrifugation, the supernatant was discarded, and the pellet was dried at room temperature. The pellet was resuspended in 1× Laemmli buffer (4% SDS, 0.05% bromophenol blue, 0.0625 M Tris, pH 7.4, 2.5% β-mercaptoethanol, and 10% glycerol) by agitating at 80°C and sonication.
Proteins were separated by SDS-PAGE in 10% Bis-Tris acrylamide/bisacrylamide gels and transferred to a nitrocellulose membrane (GE Healthcare) by established protocols. After transfer, the membranes were blocked for 30 min in PBS and 5% milk. Subsequently, the primary antibody was added and incubated for 1.5 h at room temperature or overnight at 4°C with gentle shaking. Membranes were washed four times with PBS for 5 min, followed by incubation with a 1:20,000 dilution of a fluorescent dye-coupled secondary antibody (Thermo Fisher Scientific/Invitrogen). Fluorescent detection of the signal was performed in a Li-Cor Odyssey scanner. Western blot signal was quantified using ImageJ. The different antibodies used are listed in Table S3.
Online supplemental material
Fig. S1 shows representative fluorescence microscopy images for each hit of the high-content screen; related to Fig. 1. Fig. S2 is related to Figs. 2 and 3. Fig. S3 is related to Fig. 4. Fig. S4 is related to Fig. 6. Fig. S5 is related to Fig. 8. Table S1 shows S. cerevisiae strains used in this study. Table S2 shows plasmids used in this study. Table S3 shows antibodies used in this study.
Acknowledgments
We thank members of the different involved groups for helpful discussions. We are very grateful to Christian Ungermann for support, space, and discussions. We are thankful to Aldana González Montoro for advice and help with statistical analyses and plots. We are thankful to Ralf Erdmann and to Judith Müller for providing the peroxisome marker plasmids. We thank Amir Fadel for help with the high-throughput screening. Light microscopy was performed in the iBiOS facility of the University of Osnabrück; mass spectrometry was carried out in the Mass Spectrometry facility of CellNanOs and University of Osnabrück; and EM imaging was performed at the University Medical Center Groningen microscopy and imaging center.
This work was funded by a Young Investigator Fund grant from the Schering and Fritz Thyssen foundations (20.18.0.029MN) and a Deutsche Forchungsgemeinschaft Grant within the SFB944 to A. González Montoro, as well as by a Deutsche Forchungsgemeinschaft grant within the SFB1190 and a Volkswagen Foundation Life Grant (93092MS) to M. Schuldiner. A. González Montoro is a recipient of a European Molecular Biology Organization Advanced Long-term Fellowship (aALTF 609-2018). M. Schuldiner is an incumbent of the Dr. Gilbert Omenn and Martha Darling Professorial Chair in Molecular Genetics. M. Mari is supported by an ALW Open Programme grant (ALWOP.355) of the Dutch Research Council. I. Gomes Castro is the recipient of a European Molecular Biology Organization Long-term Fellowship (ALTF-580-2017).
The authors declare no competing financial interests.
Author contributions: Investigation: D.D. Bisinski, I. Gomes Castro, M. Mari, A. González Montoro. Methodology: D.D. Bisinski, I. Gomes Castro, M. Mari, S. Walter, F. Fröhlich, A. González Montoro. Formal analysis: D.D. Bisinski, I. Gomes Castro, F. Fröhlich, A. González Montoro. Visualization: D.D. Bisinski, I. Gomes Castro, A. González Montoro. Conceptualization: D. Bisinski, I. Gomes Castro, M. Schuldiner, A. González Montoro. Funding acquisition: M. Schuldiner, A. González Montoro. Writing—original draft: A. González Montoro. Writing—review and editing: All authors.

