


Subcellular localization of the deubiquitinating enzyme BAP1 is deterministic for its tumor suppressor activity. While the monoubiquitination of BAP1 by an atypical E2/E3-conjugated enzyme UBE2O and BAP1 auto-deubiquitination are known to regulate its nuclear localization, the molecular mechanism by which BAP1 is imported into the nucleus has remained elusive. Here, we demonstrated that transportin-1 (TNPO1, also known as Karyopherin β2 or Kapβ2) targets an atypical C-terminal proline-tyrosine nuclear localization signal (PY-NLS) motif of BAP1 and serves as the primary nuclear transporter of BAP1 to achieve its nuclear import. TNPO1 binding dissociates dimeric BAP1 and sequesters the monoubiquitination sites flanking the PY-NLS of BAP1 to counteract the function of UBE2O that retains BAP1 in the cytosol. Our findings shed light on how TNPO1 regulates the nuclear import, self-association, and monoubiquitination of BAP1 pertinent to oncogenesis.
Introduction
BRCA1-associated protein 1 (BAP1) is a multidomain deubiquitinase (DUB) with a ubiquitin C-terminal hydrolase (UCH) domain at its N-terminus (Jensen et al., 1998). Unlike the other three human UCH paralogs (UCHL1, UCHL3, and UCHL5), BAP1 harbors two putative nuclear localization signals (NLSs) within its C-terminal domain (CTD) and is found predominantly inside the nucleus (Ventii et al., 2008; Jensen et al., 1998). BAP1 is an integral part of the polycomb repressive DUB complex or the BAP1-core complexes encompassing several transcription factors and/or chromatin remodeling factors, including HCF-1, ASXL1/2/3, FOXK1/2, YY1, and KLF5 (Sahtoe et al., 2016; Qin et al., 2015; Ji et al., 2014; Baymaz et al., 2014; Misaghi et al., 2009; Yu et al., 2010). The DUB activity of BAP1 is implicated in diverse cellular pathways, including cell proliferation (Machida et al., 2009; Qin et al., 2015), cell cycle progression (Pan et al., 2015; Eletr and Wilkinson, 2011), cell death (He et al., 2019; Sime et al., 2018), and DNA repair (Ventii et al., 2008; Misaghi et al., 2009; Nishikawa et al., 2009). A large number of mutations in BAP1 have been linked to human malignancies, including malignant mesothelioma, uveal melanoma, clear cell renal cell carcinoma, and cutaneous carcinoma. Loss of the DUB activity of BAP1 is considered to be the main contributing factor of BAP1-dependent malignancies (Ventii et al., 2008; Harbour et al., 2010; Testa et al., 2011; Carbone et al., 2013). In addition to enzymatic dysfunction, cellular mislocalization of BAP1 is associated with multiple aggressive tumors. Loss of BAP1 within the nucleus was found in 50% of all uveal melanoma tissue specimens with increased risks of metastasis (Koopmans et al., 2014; Kalirai et al., 2014). A distinct focal perinuclear localization of BAP1 was found in 31% of all nuclear BAP1 negative uveal melanomas (Farquhar et al., 2018). In one study, 63% of malignant mesothelioma biopsy specimens exhibited loss of nuclear BAP1 (Carbone et al., 2016). Therefore, the absence of nuclear BAP1 is considered a prognostic hallmark for these aggressive cancers.
While BAP1 primarily functions as a transcription regulator, which requires its nuclear localization, Bononi et al. (2017) reported a novel function of cytoplasmic BAP1 that deubiquitinates and therefore stabilizes the type 3 inositol-1,4,5-trisphosphate receptor (IP3R3) at the mitochondria-associated ER membranes, thereby facilitating Ca2+-mediated mitochondrial apoptosis. The lower levels of BAP1 in heterozygous BAP1+/− carriers led to the reduction of IP3R3 and hence deficient mitochondrial apoptosis that potentiates oncogenesis (Bononi et al., 2017). This finding underscores the biological relevance of subcellular localization of BAP1. The nuclear localization of BAP1 can be regulated by a ubiquitin-conjugating enzyme, UBE2O, that displays both E2 and E3 ubiquitin ligase activity. UBE2O multi-monoubiquitinates BAP1, thereby promoting the export of BAP1 from the nucleus to the cytosol (Mashtalir et al., 2014; Ullah et al., 2019; Klemperer et al., 1989; Berleth and Pickart, 1996). However, comprehensive understanding with regard to the mechanism by which the trafficking of BAP1 between the nucleus and cytoplasm is regulated remains lacking and warrants further studies.
In this study, we demonstrated that transportin-1 (TNPO1) mediates the nuclear import of BAP1 by recognizing an atypical NLS, namely proline-tyrosine (PY)-NLS, within the CTD of BAP1. TNPO1 exhibits a strong binding affinity toward the NLS of BAP1 to outcompete other nuclear import factors. Our biophysical findings were verified by CRISPR/Cas9-based knockout (KO) of the TNPO1 gene that led to significantly increased cytoplasmic retention of BAP1, confirming that TNPO1 is the primary regulator of the nuclear import of BAP1. TNPO1 dissociates the CTD-mediated self-association of BAP to form a stoichiometric heterodimer. We further solved the crystal structure of TNPO1 in complex with the PY-NLS of BAP1 to demonstrate in conjunction with an in vitro monoubiquitination (mUb) assay that TNPO1 binding interferes with the UBE2O-mediated multi-mUb. Collectively, our findings established a versatile role of TNPO1 in regulating the nuclear import, oligomerization, and ubiquitination of BAP1.
Results
Identification of nuclear import factor of BAP1
BAP1 contains two distinct NLSs in its CTD, including a monopartite classical NLS (NLS1, 656KRKKFK661) and a bipartite classic NLS (NLS2, 699RRRQGVSIGRLHKQRKPDRRKRSR722). In addition, BAP1 harbors a nonclassical PY-NLS with both hydrophobic residue and basic residue-abundant epitopes (703GVSIGRLHKQRKPDRRKRSRPY724) that has a significant overlap with the sequences of NLS2 (Fig. 1 A).
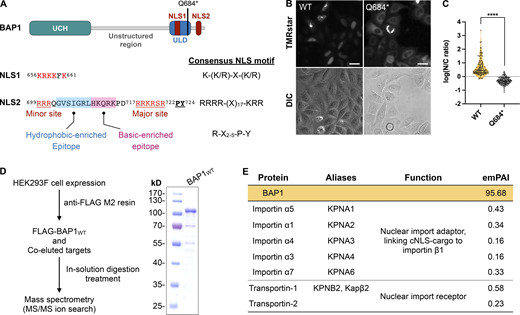
Identification of motifs and cellular factors governing the nuclear import of BAP1. (A) Domain structure and nuclear localization sequences of BAP1. BAP1 has two putative classic NLSs within the CTD of BAP1. NLS2 harbors a bipartite NLS and a PY-NLS. Two types of the N-terminal epitope of the PY-NLS motif, including hydrophobic and basic-enriched epitopes, are colored in light blue and light pink, respectively. (B) Live-cell imaging of BAP1 subcellular localization in HeLa cells expressing BAP1 wild-type (WT) and BAP1Q684* (Q684*). SNAP-tagged BAP1 variants were probed by TMRstar. DIC, differential interference contrast imaging. Scale bars = 25 μm. (C) Nuclear-to-cytoplasmic ratio for the C-terminal deletion BAP1Q684* compared with BAP1WT. P value was calculated by unpaired, two-tailed t test; ****, P < 0.0001. Sample sizes (n) of SNAP-BAP1WT- and SNAP-BAP1Q684*–expressed cells: 119 and 118, respectively. (D) Workflow of BAP1-associated protein identification. Purified FLAG-BAP1WT from affinity purification was verified by SDS-PAGE and stained with Coomassie blue. (E) Identification of nuclear import factors associated with BAP1 in the AP-MS analysis. Source data are available for this figure: SourceData F1.
Identification of motifs and cellular factors governing the nuclear import of BAP1. (A) Domain structure and nuclear localization sequences of BAP1. BAP1 has two putative classic NLSs within the CTD of BAP1. NLS2 harbors a bipartite NLS and a PY-NLS. Two types of the N-terminal epitope of the PY-NLS motif, including hydrophobic and basic-enriched epitopes, are colored in light blue and light pink, respectively. (B) Live-cell imaging of BAP1 subcellular localization in HeLa cells expressing BAP1 wild-type (WT) and BAP1Q684* (Q684*). SNAP-tagged BAP1 variants were probed by TMRstar. DIC, differential interference contrast imaging. Scale bars = 25 μm. (C) Nuclear-to-cytoplasmic ratio for the C-terminal deletion BAP1Q684* compared with BAP1WT. P value was calculated by unpaired, two-tailed t test; ****, P < 0.0001. Sample sizes (n) of SNAP-BAP1WT- and SNAP-BAP1Q684*–expressed cells: 119 and 118, respectively. (D) Workflow of BAP1-associated protein identification. Purified FLAG-BAP1WT from affinity purification was verified by SDS-PAGE and stained with Coomassie blue. (E) Identification of nuclear import factors associated with BAP1 in the AP-MS analysis. Source data are available for this figure: SourceData F1.
To dissect the contributions of the individual NLSs in the context of nuclear import, we quantified the subcellular localizations of BAP1 in HeLa cells by live-cell imaging. In addition to WT BAP1 (hereafter BAP1WT), we constructed a BAP1 variant harboring a nonsense mutation Q684* (hereafter BAP1Q684*) that is prevalent in several cancers according to the COSMIC (https://cancer.sanger.ac.uk/cosmic) and cBioPortal (https://www.cbioportal.org) cancer databases. The C-terminal truncation caused by Q684* resulted in loss of the NLS2 while retaining the NLS1 (Fig. 1 A). A SNAP-tag was introduced to the N-termini of BAP1WT and BAP1Q684* for fluorescence imaging in live cells. Quantitation of the subcellular distributions of the BAP1 variants showed that BAP1WT predominately localized in the nucleus, whereas BAP1Q684* predominantly localized in the cytosol (Fig. 1, B and C), confirming an earlier observation that NLS2 is the essential NLS for nuclear import of BAP1 (Ventii et al., 2008).
To identify potential cellular factors pertinent to the nuclear import of BAP1, we transiently expressed an N-terminally FLAG-tagged BAP1 in HEK293 freestyle (HEK293F) cells as a bait to pull down BAP1-interacting proteins for mass spectrometry (MS)–based proteomic analysis (Fig. 1 D). We identified 1,149 BAP1-interacting proteins, including many well-established BAP1 interaction partners, namely HCF-1, O-GlcNAc transferase (OGT1), transcription factors Forkhead Box K1 and K2 (FOXK1 and FOXK2), and an atypical E2/E3 conjugated enzyme, UBE2O. In addition, we found VDAC1 and GRP75, which interact with IP3R3 to form a transient Ca2+-controlling machinery that is a substrate of BAP1 (Bononi et al., 2017). Five 14-3-3 protein isoforms (14-3-3-β, γ, θ, -ε, and -ζ) that interact with BAP1 to induce cell death in neuroblastoma were also identified (Table S1; Sime et al., 2018).
To focus on proteins involved in the nuclear localization of BAP1, we chose those that fall into the categories of “regulation of protein import into the nucleus,” “NLS-bearing protein import into nucleus,” “import into nucleus,” and “protein localization to nucleus” according to Gene Ontology analysis (Fig. 1 E and Table S1). We found five karyopherin-α paralogs, including importin α5 (KPNA1), α1 (KPNA2), α4 (KPNA3), α3 (KPNA4), and α7 (KPNA6), and seven karyopherin-β paralogs, including importin β1, importin-4 (IPO4), -5 (IPO5), -7 (IPO7), -9 (IPO9), TNPO1 (KPNB2, also known as Kapβ2), and transportin-2 (Fig. 1 E and Table S1). Importin-β generally requires importin-α as the adaptor for the initial protein cargo recognition to form the cargo:importin factors ternary complex (Pumroy and Cingolani, 2015). The importin-α/β system mediates the nuclear import of protein cargos that harbor classical NLSs (cNLSs). In contrast, transportins have an all-in-one function that engages both initial recognition and nuclear import of PY-NLS–containing cargos without the need for auxiliary adaptors (Lee et al., 2006; Twyffels et al., 2014). The cargo:nuclear import factor complexes subsequently pass through the nuclear pore complexes, traversing the nuclear envelope to the nucleus. Since BAP1 possesses recognition motifs for both the importin-α/β and transportins systems, the identification of multiple BAP1-associated nuclear import factors suggested some redundancy in the nuclear localization pathways for BAP1.
The nuclear import of BAP1 is primarily mediated by TNPO1
According to the exponentially modified protein abundance index (emPAI) of identified nuclear import factors from the proteomic results, we focused on three candidates with the highest emPAIs, namely TNPO1 (emPAI: 0.58), importin α5 (emPAI: 0.43), and importin α1 (emPAI: 0.34) to examine their physical interactions with BAP1 (Fig. 1 E). Recombinant importin-αs and TNPO1 were expressed in Escherichia coli and purified to homogeneity to investigate their interactions with the CTD of BAP1 (residues 596–729) fused with a maltose-binding protein (MBP) tag (hereafter MBP-BAP1CTD) by size-exclusion chromatography (SEC; Fig. 2). In the presence of two-fold excess karyopherins, MBP-BAP1CTD formed stable complexes with individual karyopherins and coeluted at earlier elution volumes than did isolated proteins (Fig. 2 A). The formation of the binary complexes was confirmed by SDS-PAGE, which showed distinct protein bands of the early elution peaks, corresponding to MBP-BAP1CTD and individual karyopherins (Fig. 2 B).
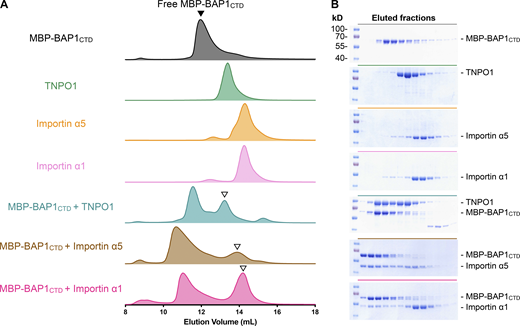
Physical interactions between BAP1 and TNPO1, importin α5, and importin α1. (A) SECs of free MBP-BAP1CTD (gray, black closed triangle), TNPO1 (green), importin α5 (orange), importin α1 (light pink), and MBP-BAP1CTD preincubated with TNPO1 (cyan), importin α5 (brown), and importin α1 (dark pink) in descending order. Free MBP-BAP1CTD was mixed with excess TNPO1, importin α5, and importin α1, resulting in the elution peak of binary complex at earlier elution volume and a lagging one that corresponds to the free nuclear import factor (open triangle). (B) SDS-PAGE analyses of the compositions of the elution peaks. The gels were stained with Coomassie blue. Source data are available for this figure: SourceData F2.
Physical interactions between BAP1 and TNPO1, importin α5, and importin α1. (A) SECs of free MBP-BAP1CTD (gray, black closed triangle), TNPO1 (green), importin α5 (orange), importin α1 (light pink), and MBP-BAP1CTD preincubated with TNPO1 (cyan), importin α5 (brown), and importin α1 (dark pink) in descending order. Free MBP-BAP1CTD was mixed with excess TNPO1, importin α5, and importin α1, resulting in the elution peak of binary complex at earlier elution volume and a lagging one that corresponds to the free nuclear import factor (open triangle). (B) SDS-PAGE analyses of the compositions of the elution peaks. The gels were stained with Coomassie blue. Source data are available for this figure: SourceData F2.
To quantify the binding affinities of individual karyopherins to BAP1CTD, we synthesized a FITC-labeled peptide corresponding to residues 699–729 of BAP1 (hereafter FITC-BAP1699–729) for fluorescence polarization (FP) analysis to quantify the binding affinities of individual karyopherins to BAP1CTD. Among the three karyopherins, TNPO1 exhibited the lowest dissociation constant (Kd) of 19.6 nM, whereas the Kd values of importin α5 and α1 were 0.67 μM and >7.2 μM, respectively, corresponding to a 34-fold and >360-fold increase compared with that of TNPO1 (Fig. 3). The results confirmed the physical interactions between BAP1 and the three karyopherins. The results also suggested that the NLS of BAP1 may be preferentially recognized by TNPO1 over importin α5 and α1 for nuclear import.

Functional assessment of BAP1 binding to TNPO1. TNPO1 exhibits the strongest binding affinity to BAP1 NLS among three import factors. Kd values were derived from the FP analyses as a function of binding protein concentration by fitting to a one-site binding model. Error bars are indicated as 95% confidence intervals based on technical triplicates (n = 3).
Functional assessment of BAP1 binding to TNPO1. TNPO1 exhibits the strongest binding affinity to BAP1 NLS among three import factors. Kd values were derived from the FP analyses as a function of binding protein concentration by fitting to a one-site binding model. Error bars are indicated as 95% confidence intervals based on technical triplicates (n = 3).
To validate our biophysical findings in cells, we individually knocked out the TNPO1 (encoding TNPO1), KPNA1 (importin α5), and KPNA2 (importin α1) genes using CRISPR-Cas9 genome editing technology in HEK293 cells (Fig. S1). We performed immunofluorescence microscopy to assess the nuclear localization of endogenous BAP1 or ectopically expressed SNAP-BAP1 in these karyopherin-KO cells. Of the three KO cell lines, TNPO1-KO cells exhibited the most significant BAP1 retention in the cytosol (Fig. 4 A). A small portion of nuclear BAP1 signals in the KPNA1-KO cells was observed. In contrast, most BAP1 was found in the nuclei of the KPNA2-KO cells. Quantitation of the nucleus/cytoplasm fraction of BAP1 indicated the least amount of nuclear BAP1 in TNPO1-KO cells (0.21), compared with KPNA1-KO cells (0.44) and KPNA2-KO cells (0.64; Fig. 4 B). These findings confirmed our hypothesis of the dominant role of TNPO1 in BAP1 nuclear localization, while importin α5 and α1 may provide functional redundancy to TNPO1.
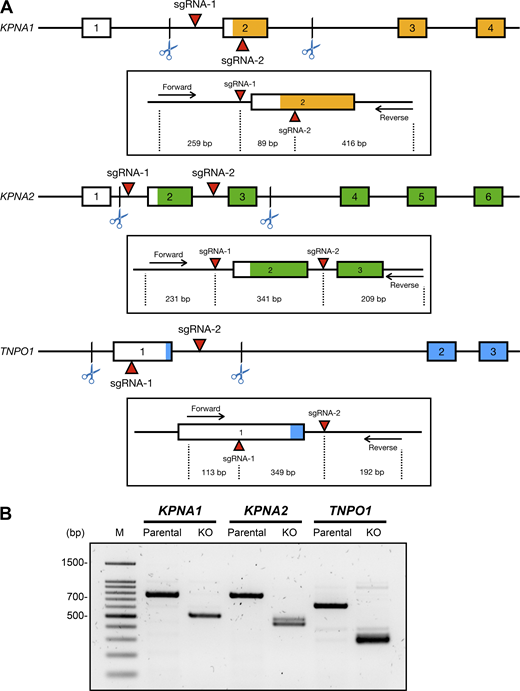
KO of TNPO1, importin α5, and importin α1 gene in HEK293 cells. (A) Schematic illustration of CRISPR/Cas9-mediated genome editing of KPNA1 (importin α5), KPNA2 (importin α1), and TNPO1 genes by introducing the double-cut KO strategy. The sgRNA target positions of each gene are indicated by red arrows. The insets indicate the expected sizes of PCR fragments generated by specific forward/reverse primers. (B) Detection of CRISPR-Cas9–dependent KPNA1, KPNA2, and TNPO1 deletions, in comparison with the parental, was confirmed by PCR-based genotyping. Source data are available for this figure: SourceData FS1.
KO of TNPO1, importin α5, and importin α1 gene in HEK293 cells. (A) Schematic illustration of CRISPR/Cas9-mediated genome editing of KPNA1 (importin α5), KPNA2 (importin α1), and TNPO1 genes by introducing the double-cut KO strategy. The sgRNA target positions of each gene are indicated by red arrows. The insets indicate the expected sizes of PCR fragments generated by specific forward/reverse primers. (B) Detection of CRISPR-Cas9–dependent KPNA1, KPNA2, and TNPO1 deletions, in comparison with the parental, was confirmed by PCR-based genotyping. Source data are available for this figure: SourceData FS1.

TNPO1 is the main regulator of BAP1 nuclear import. (A) Immunofluorescence of subcellular localization of endogenous BAP1 or ectopically expressed SNAP-tag fused BAP1 in TNPO1-, KPNA1 (encoding importin α5)-, and KPNA2 (importin α1)-KO cells. Scale bar = 10 μm. (B) Quantitative analysis of the nuclear/cytoplasmic fraction of ectopically expressed BAP1 in TNPO1-, KPNA1-, and KPNA2-KO HEK293 cells. The bar chart shows the mean and SEM of three independent experiments, counting ∼250 cells in each experiment; data analyzed by two-way ANOVA and processed to multiple comparisons using Bonferroni posttest. **, adjusted P < 0.01; ***, adjusted P < 0.001 from two-way ANOVA and Bonferroni posttest.
TNPO1 is the main regulator of BAP1 nuclear import. (A) Immunofluorescence of subcellular localization of endogenous BAP1 or ectopically expressed SNAP-tag fused BAP1 in TNPO1-, KPNA1 (encoding importin α5)-, and KPNA2 (importin α1)-KO cells. Scale bar = 10 μm. (B) Quantitative analysis of the nuclear/cytoplasmic fraction of ectopically expressed BAP1 in TNPO1-, KPNA1-, and KPNA2-KO HEK293 cells. The bar chart shows the mean and SEM of three independent experiments, counting ∼250 cells in each experiment; data analyzed by two-way ANOVA and processed to multiple comparisons using Bonferroni posttest. **, adjusted P < 0.01; ***, adjusted P < 0.001 from two-way ANOVA and Bonferroni posttest.
TNPO1 regulates CTD-mediated self-association of BAP1
To evaluate the binding stoichiometry between TNPO1 and BAP1, we used SEC coupled with multiangle light scattering (MALS). MBP-BAP1CTD by itself displayed protein concentration–dependent oligomerization with an apparent mol wt of 92.0–109.8 kD on increasing protein concentration ≤60 µM (Fig. 5 A). As the theoretical mol wt of an MBP-BAP1CTD monomer is 59 kD, the results suggested a dynamic monomer-dimer equilibrium for MBP-BAP1CTD. Indeed, the C-terminal UCH37-like domain (ULD)-mediated dimerization is found in a BAP1 ortholog from Drosophila melanogaster, Calypso (Foglizzo et al., 2018). The NLS motif of Calypso is located beyond residue 430, and AlphaFold cannot confidently model its structure (Jumper et al., 2021; Varadi et al., 2022). Likewise, the two NLS motifs of BAP1 are connected to the ULD through a very long disordered loop, and their structures and relative positions with respect to the remaining folded structure cannot be reliably predicted by AlphaFold. While structural information of BAP1 is currently lacking, BAP1 and Calypso share the same coiled-coil ULD that could mediate homodimerization of BAP1 (Fig. S2; Foglizzo et al., 2018). SEC-MALS analysis of isolated TNPO1 was strictly monomeric, with a mol wt of 91.2 ± 5.1 kD (the theoretical mol wt is 98.1 kD). Preincubation of MBP-BAP1CTD with TNPO1 showed an increased apparent mol wt of 136.2 ± 8.1 kD for the main elution peak, which corresponded to a 1:1 binary complex (Fig. 5 B). The results illustrated the ability of TNPO1 to dissociate homodimeric MBP-BAP1CTD to form a heterodimeric TNPO1:MBP-BAP1CTD complex with a 1:1 binding stoichiometry.

TNPO1 regulates the CTD-mediated dimerization of BAP1. (A) SEC-MALS analysis of free MBP-BAP1CTD as a function of protein concentration. (B) Binary complex formation between MBP-BAP1CTD and TNPO1. The SEC-MALS analyses of the binary complex, free TNPO1, and free MBP-BAP1CTD are shown in descending order. The mol wt values derived from SEC-MALS are shown in insets.
TNPO1 regulates the CTD-mediated dimerization of BAP1. (A) SEC-MALS analysis of free MBP-BAP1CTD as a function of protein concentration. (B) Binary complex formation between MBP-BAP1CTD and TNPO1. The SEC-MALS analyses of the binary complex, free TNPO1, and free MBP-BAP1CTD are shown in descending order. The mol wt values derived from SEC-MALS are shown in insets.

BAP1 and its Drosophila ortholog Calypso share a highly conserved C-terminal region. Sequences of the C-terminal region of BAP1 and the Drosophila ortholog Calypso were aligned using T-coffee/Expresso and visualized by Esprit 3.0 (Robert and Gouet, 2014). Schematic representation of the secondary structure shown on the top of alignment corresponds to the solved structure of Calypso. The boxes in red indicate completely conserved residues, and boxes in yellow indicate the residues have similar physiochemical properties.
BAP1 and its Drosophila ortholog Calypso share a highly conserved C-terminal region. Sequences of the C-terminal region of BAP1 and the Drosophila ortholog Calypso were aligned using T-coffee/Expresso and visualized by Esprit 3.0 (Robert and Gouet, 2014). Schematic representation of the secondary structure shown on the top of alignment corresponds to the solved structure of Calypso. The boxes in red indicate completely conserved residues, and boxes in yellow indicate the residues have similar physiochemical properties.
TNPO1 regulates the UBE2O-dependent mUb of BAP1
To investigate the structural basis of how TNPO1 targets BAP1, we crystallized TNPO1 in complex with the residues 706–724 of BAP1 harboring the PY-NLS (hereafter BAP1PY-NLS) and solved the crystal structure to a resolution of 3.7 Å (Table 1). TNPO1 is composed of 20 HEAT repeats forming a superhelical architecture (Chook and Blobel, 1999; Cansizoglu and Chook, 2007; Lee et al., 2006). Each HEAT repeat comprises two antiparallel α-helices (Fig. S3 A). The crystal structure of the TNPO1: BAP1PY-NLS complex illustrated how HEAT repeats 8–12 (ranging from residues 300–550) of TNPO1 form a crevice to accommodate BAP1PY-NLS (Fig. 6, A and B; and Fig. S3 B). The proline-tyrosine residues (P723 and Y724) of BAP1 made extensive contacts with the hydrophobic swath of TNPO1 encompassing residues A380, A381, L419, A425, I457, and W460 (Fig. 6 B). The refined structural model showed that R720 of BAP1 could participate in tripartite interactions with the side chains of T506, E509, and D543 of TNPO1. Additionally, the side chain hydroxyl group of S721 of BAP1 could be hydrogen bonded to the side chain of E498 of TNPO1 (Fig. 6 B). Due to the intrinsic disorder of the flanking residues of BAP1PY-NLS, including K711, K714, K725, and K727 that have been reported to be monoubiquitinated by UBE2O (Mashtalir et al., 2014), their atomic structures could not be resolved in the crystal structure (Fig. 6 A). Nevertheless, their close proximity to the PY-NLS motif that was deeply buried inside the TNPO1 crevice suggests these mUb sites would be sterically sequestered by TNPO1 binding (Fig. 6, A and B).
Data collection and refinement statistics (molecular replacement)
| TNPO1 in complex with BAP1PY-NLS (Protein Data Bank accession no. 7VPW) | |
|---|---|
| Data collection | |
| Space group | P22121 |
| Cell dimensions | |
| a, b, c (Å) | 68.90, 129.69, 170.21 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 103.158–3.763 |
| Rmerge | 0.1602 (1.24)a |
| I/σI | 7.33 (2.54) |
| Completeness (%) | 98.79 (94.43) |
| Redundancy | 7.2 (7.2) |
| Refinement | |
| Resolution (Å) | 63.863–3.763 (3.897–3.763) |
| No. reflections | 15,849 (1,472) |
| Rwork/Rfree | 0.2293/0.2584 |
| No. atoms | |
| Protein | 6,705 |
| B-factors | |
| Protein | 87.26 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.805 |
| MolProbity score | 1.38 |
| Clashscore | 3.26 |
| Ramachandran plot | |
| Favored (%) | 96.17 |
| Allowed (%) | 3.83 |
| Outliners (%) | 0 |
Statistics for the highest-resolution shell are shown in parentheses.

Detailed view of the TNPO1:BAP1 706–724 complex with corresponding electron density. (A and B) Cartoon and stick representation of a single HEAT repeat from the residues 518–557 of TNPO1 (A) and the BAP1706–724 peptide (B) are shown with corresponding 2mFo-Fc composite omit maps contoured at 1σ. The corresponding 2mFo-Fc maps of the highlighted regions have limited the display to a zone within the range (r) of 3 Å of selected atoms by using the function “volume zone” in UCSF ChimeraX.
Detailed view of the TNPO1:BAP1 706–724 complex with corresponding electron density. (A and B) Cartoon and stick representation of a single HEAT repeat from the residues 518–557 of TNPO1 (A) and the BAP1706–724 peptide (B) are shown with corresponding 2mFo-Fc composite omit maps contoured at 1σ. The corresponding 2mFo-Fc maps of the highlighted regions have limited the display to a zone within the range (r) of 3 Å of selected atoms by using the function “volume zone” in UCSF ChimeraX.
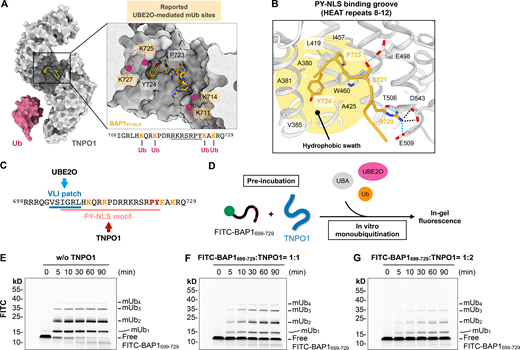
TNPO1 sterically antagonizes UBE2O-mediated mUb of BAP1. (A) Crystal structure of TNPO1 in complex with BAP1PY-NLS. Due to the intrinsic flexibility, only residues 718–724 were resolved, shown in sticks (highlighted by underlining). The position of four lysine residues reported to be monoubiquitinated by UBE2O are tentatively indicated by pink spheres linked by a dashed line. Ub, ubiquitin. (B) Zoomed-in view of the PY-NLS binding groove of TNPO1 revealing the interaction between TNPO1 and BAP1PY-NLS. The highlighted region colored in yellow represents the hydrophobic swatch in the HEAT repeats 8–12 of TNPO1, which accommodates the proline-tyrosine residues (723PY724) of BAP1PY-NLS. The dashed lines indicate salt bridge (black) and hydrogen bond (light blue). (C) Overlapping binding motifs of UBE2O and TNPO1 within the NLS region of BAP1 (residues 699–729). Reported mUb sites are in orange. (D) Schematic representation of TNPO1 competition assay design. (E–G) In vitro mUb assays of FITC-labeled BAP1699–729 peptide in the absence (E) and presence of one equivalent (F) and two equivalents (G) of TNPO1. Detection of mUb was visualized by in-gel fluorescence. Source data are available for this figure: SourceData F6.
TNPO1 sterically antagonizes UBE2O-mediated mUb of BAP1. (A) Crystal structure of TNPO1 in complex with BAP1PY-NLS. Due to the intrinsic flexibility, only residues 718–724 were resolved, shown in sticks (highlighted by underlining). The position of four lysine residues reported to be monoubiquitinated by UBE2O are tentatively indicated by pink spheres linked by a dashed line. Ub, ubiquitin. (B) Zoomed-in view of the PY-NLS binding groove of TNPO1 revealing the interaction between TNPO1 and BAP1PY-NLS. The highlighted region colored in yellow represents the hydrophobic swatch in the HEAT repeats 8–12 of TNPO1, which accommodates the proline-tyrosine residues (723PY724) of BAP1PY-NLS. The dashed lines indicate salt bridge (black) and hydrogen bond (light blue). (C) Overlapping binding motifs of UBE2O and TNPO1 within the NLS region of BAP1 (residues 699–729). Reported mUb sites are in orange. (D) Schematic representation of TNPO1 competition assay design. (E–G) In vitro mUb assays of FITC-labeled BAP1699–729 peptide in the absence (E) and presence of one equivalent (F) and two equivalents (G) of TNPO1. Detection of mUb was visualized by in-gel fluorescence. Source data are available for this figure: SourceData F6.
To test whether TNPO1 binding interferes with the mUb of the NLS of BAP1, we generated recombinant UBE2O to establish an in vitro mUb assay with residues 699–729 of BAP1 (hereafter BAP1699–729), which include the VLI patch and PY-NLS motif (Fig. 6 C). The use of fluorescein-labeled WT ubiquitin (fluorescein-UbWT) and the K0 ubiquitin mutant, in which all seven endogenous lysine residues are replaced by arginine residues (fluorescein-UbK0), resulted in similar ubiquitin ladders resolved by SDS-PAGE (Fig. S4), indicating that the multiple bands corresponded to different numbers of mUb sites rather than different lengths of polyubiquitin on a single ubiquitination site. Site-specific MS analysis within the NLS2 was hampered by the abundance of arginine (R) and lysine (K) residues within this segment, resulting in short and redundant peptide digests, as trypsin and Arg-C both recognize the R and K for proteolysis. Nevertheless, we could observe up to four of the five lysine residues (K711, K714, K719, K725, and K727) within the BAP1699–729 peptide that were monoubiquitinated by in-gel fluorescence imaging of the monoubiquitinated products (Fig. S4). Having established the multi-mUb of BAP1699–729 by UBE2O, we asked whether TNPO1 binding to BAP1699–729 would down-regulate the ubiquitin ligase activity of UBE2O. Preincubation of the FITC-labeled BAP1699–729 with one or two molar ratios of TNPO1 for 1 h before initiating the mUb by UBE2O resulted in a substantially reduced level of multi-mUb of BAP1699–729 in a TNPO1 dose–dependent manner (Fig. 6, E–G; and Fig. S5). Our structural and biochemical findings collectively demonstrated that TNPO1 binding to the NLS of BAP1 renders the mUb by UBE2O, which has direct implications in promoting BAP1 nuclear localization.
Establishment of UBE2O-dependent in vitro monoubiquitination using the unlabeled NLS peptide of BAP1 and different fluorescein-labeled ubiquitins. (A and B) In vitro monoubiquitination of BAP1699–729 peptide treated with fluorescein-labeled WT ubiquitin (fluorescein-UbWT; A) or ubiquitin variant (fluorescein-UbK0, all lysine residues to alanine residues; B) detected by in-gel fluorescence with FITC channel and SDS-PAGE analyses with Coomassie blue staining. Source data are available for this figure: SourceData FS4.
Establishment of UBE2O-dependent in vitro monoubiquitination using the unlabeled NLS peptide of BAP1 and different fluorescein-labeled ubiquitins. (A and B) In vitro monoubiquitination of BAP1699–729 peptide treated with fluorescein-labeled WT ubiquitin (fluorescein-UbWT; A) or ubiquitin variant (fluorescein-UbK0, all lysine residues to alanine residues; B) detected by in-gel fluorescence with FITC channel and SDS-PAGE analyses with Coomassie blue staining. Source data are available for this figure: SourceData FS4.
In vitro monoubiquitination of FITC-labeled BAP1699–729in the absence of TNPO1 and the presence of TNPO1 with different molar ratios. (A–C) Uncropped gel images corresponding to Fig. 6, D–F, with the detection contents of all protein materials used in the reaction by Coomassie blue staining. Source data are available for this figure: SourceData FS5.
In vitro monoubiquitination of FITC-labeled BAP1699–729in the absence of TNPO1 and the presence of TNPO1 with different molar ratios. (A–C) Uncropped gel images corresponding to Fig. 6, D–F, with the detection contents of all protein materials used in the reaction by Coomassie blue staining. Source data are available for this figure: SourceData FS5.
Discussion
DUB activity and nuclear localization are essential for the tumor suppressor activity of BAP1 (Ventii et al., 2008; Zhang et al., 2018). Although BAP1 is the first reported nuclear UCH, the molecular basis by which its nuclear import is regulated remains sparsely understood. The presence of the two putative NLS motifs, namely the monopartite classical NLS1 and the bipartite classical NLS2, suggests that nuclear import of BAP1 is potentially mediated by the canonical importin α/β system. Nonetheless, a nonclassical PY-NLS motif within the NLS2 reveals the implication of TNPO1 as an alternative nuclear import route. In this study, we demonstrated by comparative analysis of the subcellular localization of BAP1WT and BAP1Q684* that the NLS2, but not the NLS1, is responsible for nuclear import (Fig. 1, B and C). Through immunoprecipitation using transiently expressed BAP1 as bait, potential BAP1-binding proteins were identified by subsequent MS-based proteomic analysis. In particular, we identified importin α1, importin α5, and TNPO1 as prominent BAP1 nuclear import factors. Their physical interactions with BAP1 were verified by SEC, and their binding affinities to BAP1 were quantified by FP measurements, which established the strongest binding affinity of TNPO1 to the BAP1 NLS compared with importin α1 and α5 (Figs. 2 and 3). Importantly, the immunofluorescence imaging of karyopherin-KO cells led us to conclude that BAP1 nuclear import is chiefly regulated by TNPO1 (Fig. 4).
UBE2O is an atypical E2/E3 hybrid ubiquitin-conjugating enzyme (Berleth and Pickart, 1996; Ullah et al., 2019). UBE2O is reported to primarily monoubiquitinate the NLS region of BAP1 by recognizing a conserved patch of aliphatic hydrophobic amino acids, known as the VLI patch, leading to the cytoplasmic sequestration of BAP1 (Mashtalir et al., 2014). Indeed, UBE2O was found to associate with BAP1 in our affinity purification (AP)-MS analysis (Table S1), in line with previous reports (Sowa et al., 2009; Hauri et al., 2016; Mashtalir et al., 2014). The underlying mechanism of cytoplasmic retention of BAP1 was considered to result from the ubiquitination-mediated nuclear export (Mashtalir et al., 2014). However, several lines of evidence indicate that UBE2O primarily localizes in the cytoplasm (Sen Nkwe et al., 2020; Chen et al., 2018; Mashtalir et al., 2014). Since UBE2O harbors two functional NLS signals, Mashtalir et al. (2014) suggested that the mUb of the BAP1 NLS within the nucleus might be mediated by a small portion of UBE2O in the nucleus. In addition, they found that the mUb of BAP1 can be reverted by the auto-deubiquitination of BAP1 (Mashtalir et al., 2014). Hence, the opposing functions of UBE2O-mediated mUb and auto-deubiquitination activity of BAP1 require delicate spatial and temporal regulations in the context of the ubiquitination-mediated BAP1 nuclear export. Considering the predominantly cytoplasmic distribution of UBE2O that can monoubiquitinate cytoplasmic BAP1, thereby affecting its nuclear import (Mashtalir et al., 2014), our mUb assay provides strong evidence to indicate the dominant role of TNPO1 in suppressing the function of UBE2O and facilitating the nuclear import of BAP1 (Fig. 6). The crystal structure of TNPO1 in complex with BAP1PY-NLS provided the structural basis of how the PY-NLS motif of BAP1 is recognized by TNPO1 and how the complex formation results in steric hindrance to prevent UBE2O-mediated mUb of BAP1 NLS (Fig. 6).
In summary, we verified and demonstrated the essential role of TNPO1 in regulating the nuclear import of BAP1 by CRISPR/Cas9-based KO study (Figs. 1 and 4). We provided the structural basis of how TNPO1 recognizes BAP1 through binding to the atypical PY-NLS motif within its CTD (Figs. 2, 3, 4, 5, and 6). TNPO1 dissociates the CTD-mediated dimerization of BAP1 to form a stable binary complex (Fig. 5). Upon complex formation, TNPO1 sterically prevents UBE2O from monoubiquitinating the NLS of BAP1 (Fig. 6). This work therefore sheds light on the mechanism by which TNPO1 governs the nuclear import of the important tumor suppressor BAP1 to execute its function of transcription regulation.
Materials and methods
Molecular cloning, protein expression, and purification
The open reading frames of BAP1 and UBE2O were obtained from Addgene (pDEST-FLAG-HA-BAP1, plasmid #22539, and pcDNA3.1 3xFLAG-TEV-UBE2O, plasmid #105718). A cancer-associated nonsense mutation Q684* (residues 1–683) was generated with specific primers: forward, 5′-AATTGGATCCGCCACCATGGACTACAAGGATGACG-3′; and reverse, 5′-AATTGCGGCCGCTTATTAAGCCAGCATGGAGATAAAGGT-3′.
The DNA sequences corresponding to individual BAP1 constructs were subcloned into the pcDNA3.4_TOPO expression vector (Invitrogen) with a FLAG-tag at the N-terminus for purification. The cancer-associated nonsense mutation, Q684*, is documented in the Catalogue of Somatic Mutations in Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic) and cBioPortal (https://www.cbioportal.org).
The plasmids corresponding to the BAP1 variants and UBE2O were individually transfected into HEK293 freestyle cells (Invitrogen) with polyethylenimine (linear, 25 kD; Polysciences) at a DNA/polyethylenimine ratio of 1:2. The transfected cells were incubated at 37°C and 8% CO2 for 48–72 h. The cells were lysed using a Dounce homogenizer (Wheaton) in 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% IEGAL CA-630 (Sigma-Aldrich), and 1 mM PMSF with 25–30 strokes. The cell lysate was filtered through a 0.22-μm cutoff membrane and incubated with anti-FLAG M2 antibody resins (Sigma-Aldrich) at 4°C for 4 h. The resins were iteratively washed with a wash buffer (50 mM Tris-HCl, pH 7.6, and 150 mM NaCl). The target protein was eluted with 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 300 ng/µl 3× FLAG peptide. All recombinant proteins were concentrated and loaded into a SEC column (superpose 6 increase 10/300 GL; GE Healthcare) in SEC buffer (50 mM Tris-HCl , pH 7.6, 200 mM NaCl, 10% [vol/vol] glycerol, 0.2 mM tris(2-carboxyethyl)phosphine, 2 mM Mg[OAc]2, and 1 mM EGTA).
The CTD (residues 596–729) of BAP1 was subcloned into the pET-MBP vector, which encodes for a His6-MBP fusion tag at the N-terminus, followed by a TEV protease cleavage site for tag removal (hereafter His6-MBP-BAP1CTD): forward, 5′-AATTGGATCCGGAAAACCTGTATTTTCAGGGCGGCAGCAGCAGCCCAGTGGAGAAG-3′; and reverse, 5′-AATTCTCGAGTTATCACTGGCGCTTGGCC-3′. Full-length TNPO1 (also known as Kapβ2) was a kind gift of Dr. Yuh Min Chook (University of Texas Southwestern Medical Center at Dallas, Dallas, TX). An acidic loop corresponding to residues 337–367 was replaced by a GGSGGSG linker for optimized crystallization (Yoshizawa et al., 2018).
Specific primers for In-Fusion HD Cloning kit, Takara Bio, were forward, 5′-TGGTTCTGGCGGCTCTGGTGATACAATTTCTGACTGGAATC-3′; and reverse, 5′-GAGCCGCCAGAACCACCCCGTATATCCTGTTCACTATCAG-3′.
Full-length importin α5 and importin α1 were obtained from Addgene (pCMVTNT-T7-KPNA1/plasmid #26677 and pCMVTNT-T7-KPNA2/plasmid #26678). The N-terminal importin β-binding (IBB) domains of importin α1 and α5 were excluded from the recombinant protein expression constructs, as the IBB domain is not required for cargo recognition.
For KPNA1 (importin α5, residues 66–512): forward-1, 5′-GGGCGGCAGCGAAGAAGAAACAGAAGAAGAAGTTATGTCAGATGG-3′; forward-2, 5′-AATTGGATCCGAAAACCTGTATTTTCAGGGCGGCAGCGAAGAAG-3′; reverse-1, 5′-AATTCTCGAGTCAAATAAGATCAAAGGCCTTTTGGTAGATCTCC-3′; and reverse-2, 5′-AATTGCGGCCGCTCAAATAAGATCAAAGGCCTTTTGGTAGATCT-3′.
For KPNA2 (importin α1, residues 60–529): forward-1, 5′-CAGGGCGGCAGCCTGAAGAGGAGAAATGTAAGCTCATTTCC-3′; forward-2, 5′-AATTGGATCCGAAAACCTGTATTTTCAGGGCGGCAGCCTG-3′; reverse-1, 5′-AATTCTCGAGTCAGAAAGTGTAGCCTTCAGAGGTAG-3′; reverse-2, 5′-AATTGCGGCCGCTCAGAAAGTGTAGCCTTCAGAGGTAG-3′.
They were subcloned into the pGEX-4T3 vector for recombinant protein expression. All nuclear import factors were fused with an N-terminal GST tag followed by a TEV cleavage site. His6-MBP-BAP1CTD, GST-TNPO1, and GST-importin-αs were expressed in E. coli strain BL21(DE3). All cultures were grown to OD600 = 0.4–0.6, and expression was induced with 0.1 mM IPTG at 16°C for 20 h.
A tandem purification strategy was used for His6-MBP-BAP1CTD purification. First, the cells were lysed in 50 mM Tris-HCl, pH 7.6, 1 M NaCl, 10% (vol/vol) glycerol, and 0.2 mM TCEP. After centrifugation, the supernatant was loaded into a MBPTrap HP 5 ml column (GE Healthcare), and target protein was eluted by the same buffer with 10 mM maltose. Next, the eluted sample was further purified by a HisTrap HP 5-ml column (GE Healthcare) in 50 mM Tris-HCl, pH 7.6, 1 M NaCl, 10% (vol/vol) glycerol, and 0.2 mM TCEP and eluted with elution buffer (50 mM Tris-HCl, pH 7.6, 1 M NaCl, 10% (vol/vol) glycerol, 0.2 mM TCEP, and 400 mM imidazole).
For GST-tagged import factors, the cells were lysed in 50 mM Tris-HCl (pH 7.6), 200 mM NaCl, 20% (vol/vol) glycerol, 0.2 mM TCEP, and 1 mM EDTA. Cell lysates were incubated with Glutathione Sepharose 4B resin (GE Healthcare) at 4°C for 4 h. On-column digestion for removing the GST-tag was achieved by TEV protease at room temperature for 2 h. Tag-free recombinant import factors were collected in the flow-through. His6-MBP-BAP1CTD and untagged import factors were subsequently purified by SEC using a Superdex 200 16/60 GL column (GE Healthcare) in 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 10% (vol/vol) glycerol, 0.2 mM TCEP, 2 mM Mg(OAc)2, and 1 mM EGTA. The purity of the recombinant proteins was confirmed by SDS-PAGE.
Subcellular localization and phenotypic profiling of BAP1 variants
HeLa-Kyoto cells were regularly maintained in DMEM with 10% FBS and 1% penicillin/streptomycin and plated in the chamber-slide (µ-Slide; Ibidi) before transfection. The DNA sequences corresponding to BAP1 variants were subcloned into the pSNAP-tag vector, with an N-terminal SNAP-tag for fluorescence labeling.
Plasmids of SNAP-tagged BAP1 variants were transfected into cells using X-tremeGENE HP reagent (Roche). 2 d after transfection, exogeneous SNAP-tagged BAP1s were live-labeled with SNAP-Cell TMR-star (New England BioLabs), and nuclei were stained with Hoechest33342 (Thermo Fisher Scientific) for 30 min. Labeled cells were transferred into the fresh medium and used for epifluorescent live-cell microscopy. Images were taken using a Leica DMI6000 microscope with an HCX PL FL 20×/0.40-NA CORR objective lens and an Andor Luca R EMCCD sensor. Integral fluorescent intensities of labeled BAP1 variants in the separate cellular compartments were processed and counted based on double-stained monochrome images using MetaMorph Multi-Wavelength cell scoring software application (Molecular Devices).
Immunofluorescence staining of exogenous and endogenous BAP1
For detecting endogenous BAP1, parental, TNPO1-, KPNA1-, and KPNA2-KO lines were fixed with 4% formalin, stained with mouse anti-BAP1 antibody (1:100, clone #C-4, sc-28383; Santa Cruz Biotechnology), and anti-mouse Alexa Fluor 594 antibody (1:2,000, A21203; Invitrogen), and mounted in DAPI-containing mounting medium (AB104139; Abcam). For detecting exogenous BAP1, HEK293T cells were transfected with SNAP-tagged BAP1 using X-tremeGENE HP reagent (Roche) for 2 d, and SNAP-tagged BAP1s were live-labeled with SNAP-Cell TMR-star (New England BioLabs) overnight. Labeled cells were fixed with 4% formalin and mounted in DAPI-containing mounting medium (Abcam). Immunofluorescence images were taken using a Leica DMI6000 microscope with an HCX PL FL 63×/1.4-NA objective lens and an Andor Luca R EMCCD sensor. Image acquisition was performed with the application of MetaMorph Multi-Wavelength acquisition software (Molecular Devices). Phenotypic distribution of BAP1 was manually counted in three independent assays, with ≥250 cells counted in each experiment.
Cas9 ribonucleoprotein (RNP) nucleofection
Recombinant Cas9 protein and sgRNA were prepared as previously described (Huang et al., 2021). Briefly, the plasmid pMJ915 (plasmid #69090; Addgene), encoding recombinant Streptococcus pyogenes Cas9, was transformed into E. coli strain BL21(DE3) for protein expression by IPTG induction. The Cas9 protein was first purified from E. coli lysate using an Ni-NTA affinity column. After removing the N-terminal His6-MBP-tag with TEV protease, the untagged Cas9 protein was purified by ion exchange chromatography using a Heparin column, followed by SEC using a Superdex 200 16/60 gel filtration column. The purified Cas9 protein was stored in Cas9 RNP buffer (20 mM Hepes, 150 mM KCl, 10% glycerol, and 1 mM β-mercaptoethanol, pH 7.5). sgRNAs were synthesized from DNA templates by in vitro transcription using T7 polymerase and were purified by electrophoresis in urea-PAGE to extract the full-length sgRNA. sgRNAs were then treated with calf intestinal alkaline phosphatase to remove the immunogenic 5′ triphosphate moiety to reduce intracellular immune response (Wienert et al., 2018; Kim et al., 2018). Each target gene was disrupted by a double-cutting strategy in which two Cas9 RNP complexes were co-introduced to induce two double-strand breaks in the gene coding region. The sgRNA target sequences are as follows: KPNA1 KO sgRNA-1, 5′-CCACTGTACTCCAGTCCGGG-3′; KPNA1 KO sgRNA-2, 5′-ATCCCGATGAGATGCGCAGG-3′; KPNA2 KO sgRNA-1, 5′-TCAGCC-CTTTAGAGGGAAGG-3′; KPNA2 KO sgRNA-2, 5′-TTGGAAGGGGTGAAAACTG-3′; TNPO1 KO sgRNA-1, 5′-AGGAAAATTGCTGCCTGGGT-3′; and TNPO1 KO sgRNA-2, 5′-CCCGGCCGTTTGAAGCCGAG-3′.
BFP-derived 127-bp single-stranded oligodeoxynucleotides (ssODNs) were prepared by dissolving Ultramer DNA Oligo (Integrated DNA Technologies) in molecular-grade water and adjusted to 100 μM for editing enhancement (Richardson et al., 2016). HEK293T cells were subcultured for 48 h before nucleofection. Cas9 RNP was assembled by swirling mixing Cas9 protein into sgRNA solution followed by incubation at 37°C for 15 min (Huang et al., 2021). The double-cut Cas9 RNP mixture was defined as 20 μM and prepared by combining an equal amount of sgRNA-1 and sgRNA-2 Cas9 RNP. All nucleofection experiments were performed using the 4D-Nucleofector X Unit (Lonza). Each nucleofection reaction contained 2 × 105 cells in 20 μl of nucleofection buffer, 5 μl of double-cut Cas9 RNP mixture, and 1 μl of 100 μM BFP-derived 127-bp ssODN. The HEK293T cells were pulsed with SF kit (Lonza) using the program CM-130. After nucleofection, the pulsed cells were allowed to recover in the prewarmed medium at 37°C for 5 min and transferred to 6-well plates for subsequent analysis.
Genomic PCR amplification and editing analysis
The pulsed cells were harvested 48 h after nucleofection. The cells were pelleted at 300 g for 5 min, washed once with DPBS, and resuspended in QuickExtract DNA Extraction Solution (Lucigen). Genomic DNA extraction was performed at 65°C for 15 min, 98°C for 5 min, and on ice for 10 min. The following primers were used for genomic PCR: KPNA1 forward, 5′-CCAGCTACTCAGGAAACTGAAGTG-3′; KPNA1 reverse, 5′-CAGCAACACTTTAGTGGTCACTTGTG-3′; KPNA2 forward, 5′-GATGGAGTATGGAAAAAGAATGTAGAAG-3′; KPNA2 reverse, 5′-GTTTTCCTGCAGCGGAGAAGTAGCATCATCAGG-3′; TNPO1 forward, 5′-GAGTGCGAGCACTTGTGAG-3′; and TNPO1 reverse, 5′-GCTTAACAGGCGAGGCAAAC-3′
The targeted loci were amplified from extracted genomic DNA by KAPA HiFi HotStart PCR Kit (KAPA Biosystems). PCR products were resolved by 2% TAE agarose gel, poststained with SYBR Safe DNA Gel Stain (Thermo Fisher Scientific), and visualized by iBright FL1000 imaging system (Thermo Fisher Scientific).
Establish homozygous gene KO HEK293T single cell clone
The cells were collected 48 h after nucleofection. The cells were pelleted at 300 g for 5 min, washed once with DPBS, and resuspended in ice-cold FACS buffer (DPBS supplemented with 2% FBS, 25 mM HEPES, and 0.5 mM EDTA) containing 0.1 μg/ml DAPI solution (Thermo Fisher Scientific). Resuspended cells were then passed through a 35-µm cell strainer and collected into a 5-ml polypropylene test tube on ice in the dark until sorting. FACS was performed using a FACSAria IIIu Cell Sorter (BD Biosciences) by the flow cytometry core facility at the Institute of Biomedical Sciences, Academia Sinica, Taiwan. Single cells were sorted into a 96-well plate cultivated in complete DMEM. 96-well plates were incubated at 37°C for ≥10 d until single-cell colonies were formed. Single-cell colonies were picked up and analyzed by genomic PCR amplification to check the KO results. Homozygous KO clones were then expanded and cryopreserved until further experiments.
SEC-MALS analysis
The absolute mol wt values of MBP-BAP1CTD and TNPO1 were determined by SEC-MALS using a Wyatt Dawn Heleos II MALS detector (Wyatt Technology Corp.) coupled to an AKTA Purifier UPC10 FPLC protein purification system (GE Healthcare) as described previously (Wang et al., 2015). 15 μl of each sample at a concentration of 2 mg/ml was injected and separated using an Agilent BioSEC3 size-exclusion column (Agilent). All experiments were performed in 50 mM Tris-HCl, pH 7.6, 200 mM NaCl, 10% (vol/olv) glycerol, 0.2 mM TCEP, 2 mM Mg(OAc)2, and 1 mM EGTA at a flow rate of 0.3 ml/min. The protein concentrations of individual elution peaks in the SECs were derived from the observed refractive index as a function of elution volume by a refractive index detector (Wyatt Optilab T-rEX; Wyatt Technology Corp.). A standard refractive index increment (dn/dc) value of 0.185 ml/g was used, and the buffer viscosity η = 1.0257 cP at 25°C was estimated using SEDNTERP (http://philo.rasmb.org/software/sednterp/). The reference refractive index of SEC buffer (1.3458 RIU) was determined by passing buffer through the reference cell.
In vitro mUb assay
In vitro mUb was conducted in a total volume of 100 μl mixture containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM MgCl2, 2 mM DTT, 250 nM ubiquitin-activating enzyme UBE1, 50 μM ubiquitin/fluorescein-ubiquitin (UbWT or UbK0), 5 μM purified recombinant UBE2O, and 5 μM BAP1 substrate (unlabeled BAP1CTD, BAP1699–729, or FITC-labeled BAP1699–729). For unlabeled BAP1CTD and BAP1699–729, the reaction was performed with a mixture of fluorescein-labeled ubiquitin and unlabeled ubiquitin at a ratio of 1:4. For FITC-labeled BAP1699–729, unlabeled ubiquitin was used for the experiment.
For competition assay, FITC-labeled BAP1699–729 was preincubated with TNPO1 at the molar ratio of 1:1 and 1:2 for 1 h, and used as the substrate for the following reactions. All reactions were performed with constant shaking at 32°C for 0, 5, 10, 30, 60, and 90 min. The reaction at each time point was quenched by mixing with the SDS-loading dye and boiled at 95°C for 3 min. In-gel fluorescence was visualized with an excitation wavelength of 495 nm and an emission wavelength of 525 nm (iBright FL1000; Thermo Fisher Scientific). Samples were also confirmed by Coomassie blue staining.
Protein in-solution digestion
The sample was reduced with 10 mM dithioerythreitol and 25 mM ammonium bicarbonate (ABC), followed by the denaturation with 8 M urea and 25 mM ABC, pH 8.5, at 37°C for 1 h. After reduction and denaturation, the sample was alkylated with 25 mM iodoacetamide and 25 mM ABC, pH 8.5, at room temperature in the dark for 1 h. The reaction was quenched in 25 mM dithioerythreitol and 25 mM ABC.
Protein digestion was accomplished in two steps by using Lys-C (mass spectrometry grade; Wako) for 3 h in 25 mM ABC, pH 8.5, and 4 M urea, and using trypsin (sequencing grade; Promega) for 16 h in 25 mM ABC, pH 8.5, and 1 M urea. The enzyme-to-protein ratio for both digestion processes was 1:50. The reaction was quenched with 0.1% formic acid, and the sample was subsequently dried in Speedvac. The peptide mixture was aliquoted, desalted, and concentrated on a C18-ZipTip (Millipore) and eluted with 50% acetonitrile in 0.1% formic acid.
Shotgun proteomic identifications
NanoLC-nanoESI-MS/MS analysis was performed on an EASY-nLC 1200 system connected to a Thermo Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) equipped with a Nanospray Flex ion source (Thermo Fisher Scientific). Peptide mixtures were loaded onto a 75-μm internal diameter, 25-cm length PepMap C18 column (Thermo Fisher Scientific) packed with 2-μm particles with a pore size of 100 Å and separated using a segmented gradient in 120 min from 5 to 45% solvent B (80% acetonitrile with 0.1% formic acid) at a flow rate of 300 nl/min. Solvent A was 0.1% formic acid in water. The mass spectrometer was operated in the data-dependent mode. Briefly, survey scans of peptide precursors from 350 to 1,600 m/z were performed at 240K resolution with a 2 × 105 ion count target.
Tandem MS was performed by isolation window at 1.6 D with the quadrupole, HCD fragmentation with a normalized collision energy of 30, and rapid scan MS analysis in the ion trap. The MS2 ion count target was set to 1 × 104, and the max injection time was 35 ms. Only those precursors with charge states 2–6 were sampled for MS2. The instrument was run in top speed mode with 3-s cycles; the dynamic exclusion duration was set to 15 s with a 10-ppm tolerance around the selected precursor and its isotopes. Monoisotopic precursor selection was turned on.
The raw data obtained from LC-MS/MS acquisition was processed using Proteome Discoverer (v2.4; Thermo Fisher Scientific). The processed data were used to search against the Swissprot Homo sapiens database using Mascot search engine (v2.7.0; Matrix Science). All searches included carbamidomethyl (cysteine) and oxidation (methionine) as variable modifications. Two missed tryptic cleavages were allowed. The product mass tolerance of the parent ions and the fragment ions was set to 10 ppm and 0.6 D, respectively. A decoy database search was performed. Identified peptides were filtered with 1% false discovery rate cutoff.
FP
The residues 699–729 of BAP1 were chemically synthesized, and FITC was introduced to the N-terminus with an AHX linker (FITC-AHX-699RRRQGVSIGRLHKQRKPDRRKRSRPYKAKRQ729). FP measurements were performed in a filter-based microplate reader (Infinite M1000; TECAN) using a fluorescein filter set with an excitation wavelength (λex) of 485 nm and an emission wavelength (λem) of 535 nm. The total reaction volume was fixed to 100 μl containing a serial dilution of different nuclear import carriers (TNPO1, importin α1 and α5) and 50 nM of FITC-labeled BAP1699–729. The reactions were carried out at 25°C in triplicate. Kd values were determined by nonlinear regression using the one site, total binding model in Prism 9 (GraphPad).
Crystallization, data collection, and structure determination
To form the BAP1:TNPO1 complex, 7 mg/ml of untagged TNPO1 was preincubated with recombinant BAP1CTD and two chemically synthesized NLS peptides of BAP1699–729 and BAP1PY-NLS (residues 706–724) at a molar ratio of 1:5 in 50 mM Tris-HCl, pH 7.6, 200 mM NaCl, 10% (vol/vol) glycerol, 0.2 mM TCEP, 2 mM Mg(OAc)2, and 1 mM EGTA, respectively. The excess of free BAP1 peptides was removed by a 10-kD cutoff Amicon centrifuge tube. The initial trial of crystallization was accomplished by automatic robot screening (Phoenix RE; Rigaku). Further optimization was manually operated using the sitting-drop vapor diffusion method at 20°C overnight. 1 µl of protein sample was mixed with the same equivalent of crystallization reservoir buffer (0.49 M sodium phosphate monobasic monohydrate and 0.91 M potassium phosphate dibasic monohydrate, pH 7.4). Plate-like crystals of the BAP1:TNPO1 complex were grown overnight and harvested for x-ray diffraction.
The best crystal of the BAP1PY-NLS:TNPO1 complex was diffracted to 3.7 Å and indexed to the space group P22121. The initial structure was solved by the molecular replacement method with Phaser (McCoy et al., 2007), using the reported structure (TNPO1:JKTBPNLS complex, Protein Data Bank accession no. 2Z5O) as a searching template. The BAP1PY-NLS peptide ligand was manually modeled by Coot (Emsley et al., 2010), refined by ISOLDE (Croll, 2018) and Phenix (Adams et al., 2010), and validated by MolProbity (Chen et al., 2010). The final structure was refined to an Rwork value of 0.2293 and an Rfree value of 0.2584. The structural validation parameters are shown in Table 1. A missing fragment from residues 706–717 of BAP1 shows no electron densities in terms of structural flexibility and disorder. Structural visualization and rendering were accomplished by a combination of UCSF-ChimeraX (Goddard et al., 2018) and PyMOL (Schrödinger).
Statistical analysis
Data analyses were performed using GraphPad Prism 9. Unpaired, two-tailed t test was used to compare two groups, and P < 0.05 was considered statistically significant. Two-way ANOVA followed by the Bonferroni posttest was used for multiple comparisons. Kd values reported in Fig. 3 were derived from nonlinear regression fitting to a one-site binding model with error bars indicating 95% confidence intervals based on technical triplicates (n = 3).
Online supplemental material
Fig. S1 shows the CRISPR/Cas9-based genome editing strategy for and the results of generating three TNPO1-, KPNA1-, and KPNA2-KO cells. Fig. S2 shows the sequence alignment between human BAP1 and its Drosophila ortholog Calypso, which share a conserved C-terminal region. Fig. S3 illustrates expanded views of the crystal structure of TNPO1:BAP1706–724 crystal structure. Fig. S4 shows the results of the in vitro UBE2O-mediated mUb with the BAP1 NLS peptide. Related to Fig. 6, Fig. S5 shows how TNPO1 impairs UBE2O-mediated mUb of BAP1 NLS peptide. Table S1 lists the BAP1-associated proteins identified by AP-MS.
Data availability
The atomic coordinates of TNPO1 in complex with the PY-NLS motif (residues 706–724) of BAP1 are deposited in Protein Data Bank under accession no. 7VPW. In addition, the AP-MS raw dataset of BAP1-associated proteins identification has been deposited to Proteomics IDEntifications Database (PRIDE, https://www.ebi.ac.uk/pride/) under accession code PXD030044.
Acknowledgments
Mass spectrometry data were acquired at the Academia Sinica Common Mass Spectrometry Facilities for Proteomics and Protein Modification Analysis located at the Institute of Biological Chemistry, Academia Sinica, supported by Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-107). The x-ray crystallography data were collected at the Taiwan Photon Source (TPS) beamline 05A, National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan. We thank the supports from the biophysics core facility managed by Dr. Meng-Ru Ho and the mammalian cell culture facility of Institute of Biological Chemistry, Academia Sinica.
This research was supported by intramural funding Academia Sinica to S.T.D. Hsu, K.P. Wu, and S. Lin, Academia Sinica Career Development Awards to S.T.D. Hsu (AS-CDA-109-L08) and to K.P. Wu (AS-CDA-110-L03), research grants from the Ministry of Science and Technology, Taiwan, to S.T.D. Hsu (110-2113-M-001-050-MY3), K.P. Wu (109-2311-B-001-022), and L.H.C. Wang (110-2320-B-007-004-MY3), funding from National Health Research Institutes to L.H.C. Wang (NHRI-EX111-111-1124BI), and funding from National Tsing Hua University to L.H.C. Wang (NTHU 110-Q27-13E1).
The authors declare no competing financial interests.
Author contributions: T.J. Yang: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing - Original Draft, Writing - Review & Editing; T.N. Li: Data curation, Formal analysis, Investigation, Methodology; R.S. Huang: Data curation, Formal analysis, Investigation, Methodology; Y.C. Pan: Data curation, Formal analysis, Investigation, Methodology; S.Y. Lin: Methodology, Writing - original draft; S. Lin: Funding acquisition, Methodology, Resources, Supervision, Writing - original draft; K.P. Wu: Funding acquisition, Methodology, Resources, Validation; L.H.C. Wang: Formal analysis, Funding acquisition, Methodology, Resources, Supervision, Validation, Writing - original draft; S.T.D. Hsu: Conceptualization, Methodology, Formal analysis, Investigation, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing - Original Draft, Writing - Review & Editing.

