


Tail-anchored (TA) membrane proteins have a potential risk to be mistargeted to the mitochondrial outer membrane (OM). Such mislocalized TA proteins can be extracted by the mitochondrial AAA-ATPase Msp1 from the OM and transferred to the ER for ER protein quality control involving ubiquitination by the ER-resident Doa10 complex. Yet it remains unclear how the extracted TA proteins can move to the ER crossing the aqueous cytosol and whether this transfer to the ER is essential for the clearance of mislocalized TA proteins. Here we show by time-lapse microscopy that mislocalized TA proteins, including an authentic ER-TA protein, indeed move from mitochondria to the ER in a manner strictly dependent on Msp1 expression. The Msp1-dependent mitochondria-to-ER transfer of TA proteins is blocked by defects in the GET system, and this block is not due to impaired Doa10 functions. Thus, the GET pathway facilitates the transfer of mislocalized TA proteins from mitochondria to the ER.
Introduction
Membrane proteins comprise about one third of the eukaryotic cellular proteome, and 3–5% of the membrane protein proteome belongs to a class of membrane proteins with a single transmembrane (TM) domain near the C-terminus, called tail-anchored (TA) proteins (Wattenberg and Lithgow, 2001; Chio et al., 2017; Wang and Walter, 2020). TA proteins are anchored to organelle membranes through their TM domain and perform diverse cellular functions, which rely on correct targeting to their destination membranes. Since the TM domain of TA proteins is sequestered within ribosomes during translation, TA proteins are posttranslationally targeted to different cellular membranes (Borgese and Fasana, 2011). Recent studies indicated the presence of multiple pathways for targeting of TA proteins to the ER membrane, mitochondrial outer membrane (OM), and peroxisomal membrane. Targeting of ER-destined TA proteins requires the guided entry of TA protein (GET) pathway in yeast and the TM recognition complex (TRC) pathway in mammals (Schuldiner et al., 2008; Chio et al., 2017). Briefly, newly synthesized TA proteins are initially recognized by a chaperone Sgt2 in the cytosol and then transferred to a cytosolic ATPase Get3, with the aid of the Get4-Get5 complex. The Get1-Get2 complex in the ER membrane functions as a receptor for Get3 to facilitate insertion of TA proteins into the ER membrane (Denic, 2012). In addition to the GET pathway, the signal recognition particle–independent (SND) pathway and ER membrane protein complex pathway were also identified as routes for the ER-targeting of classes of TA proteins (Aviram et al., 2016; Guna et al., 2018). Understanding of the targeting mechanism of TA proteins to mitochondria is much behind that of the ER targeting, but the mitochondrial import complex has been reported to promote insertion of TA proteins into the OM (Doan et al., 2020).
Gene deletion of the GET components results in mistargeting of a subset of TA proteins to mitochondria and/or formation of TA protein aggregates in the cytosol (Jonikas et al., 2009; Schuldiner et al., 2008). Mutations in the targeting signal of TA proteins also lead to mislocalization of the mutant proteins to mitochondria (Chen et al., 2014; Okreglak and Walter, 2014). Two independent studies reported that the mitochondrial AAA-ATPase Msp1 (Nakai et al., 1993) removes mislocalized TA proteins such as Pex15, Gos1, and Pex15Δ30 (a truncation variant of the C-terminal 30 residues of Pex15) from mitochondria (Chen et al., 2014; Okreglak and Walter, 2014). The recombinant Msp1 protein was shown by in vitro reconstitution to extract artificial TA proteins from lipid liposomes in an ATP-dependent manner (Wohlever et al., 2017). Cryo-EM structures of Msp1 showed that it forms a hexameric spiral assembly, engaging a substrate peptide within the conserved central pore loops (Wang et al., 2020). Consistently, Msp1 was found to act as a processive translocase capable of unfolding diverse protein substrates (Castanzo et al., 2020).
We (Matsumoto et al., 2019) and Dederer et al. (2019) independently found that mistargeted TA proteins such as Pex15Δ30 were ubiquitinated by Doa10, an E3 ubiquitin ligase that forms the Doa10 complex with Ubc6, Ubc7, and Cue1 in the ER membrane. Our microscopic observations revealed that Pex15Δ30 was mainly localized to mitochondria but partly to the ER when the Doa10 complex was defective, and that the ER localization of Pex15Δ30 from mitochondria was significantly enhanced by overexpression of Msp1 (Matsumoto et al., 2019). When the MSP1 gene was deleted in Doa10 complex–defective mutant cells, Pex15Δ30 was localized solely in mitochondria. We further found that ubiquitinated Pex15Δ30 in the ER was extracted from the ER membrane by the Cdc48 complex for cytosolic proteasomal degradation (Matsumoto et al., 2019). On the other hand, Dederer et al. (2019) failed to observe ER localization of Pex15Δ30 in Doa10-complex mutant cells and concluded that Doa10 ubiquitinates cytosolic Pex15Δ30, which was extracted from the mitochondrial OM by Msp1. Thus, both groups found that the Doa10 complex plays an important role in the ubiquitination followed by proteasomal degradation of Pex15Δ30, but the precise location of Pex15Δ30 for Doa10-mediated ubiquitination is still disputed. When mistargeted TA proteins escape the Doa10-mediated ubiquitination, as in the case of Golgi protein Gos1, the TA protein transferred to the ER membrane follows the secretory pathway to reach the original destination like the Golgi. Msp1 thus functions as a factor to achieve proofreading or correction of the mistargeting of TA proteins (Matsumoto et al., 2019; Wang and Walter, 2020).
In this study, we used Pex15Δ30 as a model mislocalized TA protein and showed by time-lapse fluorescent microscopy that Pex15Δ30 moved from mitochondria to the ER in an Msp1-dependent manner. We then asked whether the GET pathway is involved in this transfer of mislocalized TA proteins to the ER. We found that the Msp1-dependent transfer of Pex15Δ30 from mitochondria to the ER was impaired by the defects in the GET system and that this block of the mitochondria to the ER transfer of Pex15Δ30 was not due to impaired localization of the Doa10-complex subunit Ubc6 to the ER. We also revealed that an authentic ER protein mislocalized to mitochondria was also transferred to the ER with the aid of the GET system. Possible roles of the GET system in the clearance of mistargeted TA proteins in cooperation with Msp1 are discussed.
Results
Msp1-dependent transfer of Pex15Δ30 from mitochondria to the ER
To monitor the transfer of mistargeted TA proteins from mitochondria to the ER in real time, we took an approach of time-lapse imaging by fluorescence microscopy. A model TA protein, Pex15Δ30, which lacks the C-terminal 30 residues of peroxisomal protein Pex15, was previously found to be mistargeted to the mitochondrial OM and cleared by an Msp1-dependent mechanism (Okreglak and Walter, 2014). We used FLAG-tagged mNG-Pex15Δ30, a fusion protein containing the 3xFLAG tag and mNeonGreen (mNG) followed by Pex15Δ30, to visualize Pex15Δ30 localization. To coordinate the expression of mNG-Pex15Δ30 and Msp1 separately in the absence of Doa10-mediated ubiquitination, we used two drug-inducible expression systems for doa10Δ cells (Weir et al., 2017). The endogenous MSP1 promoter was replaced with the β-estradiol–inducible Z4EV promoter (McIsaac et al., 2013), and mNG-Pex15Δ30 was expressed under the control of the cyanamide-inducible DDI2 promoter (Wang et al., 2019) in doa10Δ cells, in which Doa10-mediated ubiquitination and following proteasomal degradation of Pex15Δ30 proteins would be blocked (Fig. 1 A). Upon expression of mNG-Pex15Δ30, but without expression of Msp1, mNG-Pex15Δ30 was localized mainly in mitochondria, but partially in peroxisomes in doa10Δ cells (Fig. S1 A). Subsequent expression of Msp1 made mNG-Pex15Δ30 degraded in a Doa10-dependent manner (Fig. 1 B), as previously observed for Pex15Δ30 (Matsumoto et al., 2019).

Msp1-dependent transfer of Pex15Δ30 from mitochondria to the ER. (A) Schematic representation of time-lapse imaging. 3xFLAG-mNG-Pex15Δ30 was expressed under the control of the cyanamide-inducible DDI2 promoter, and Msp1 was from the β-estradiol-inducible Z4EV promoter. DOA10 was deleted to block the ubiquitination and degradation of Pex15Δ30. Yeast cells were grown in SCD medium at 30°C, and 3xFLAG-mNG-Pex15Δ30 was induced by 5 mM cyanamide for 4 h at 30°C. The cells were washed with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. (B) Promoter shutoff chase of 3xFLAG-mNG-Pex15Δ30. doa10Δ and its isogenic WT (DOA10) cells in A were grown in SCD medium at 30°C. Cell extracts were prepared at the indicated times after addition of 1 μM β-estradiol, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in doa10Δ cells, right after the addition of β-estradiol (chase time = 0), relative to that in DOA10 cells, is shown (center). Values are mean ± SD from three independent experiments. (C) Yeast cells in A were grown in SCD medium at 30°C and treated with 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Pex15Δ30. Time-lapse images were then taken at 1-min intervals. Single-plane images are shown. The ER was labeled with BipN-mCherry-HDEL (ER-mCherry). Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (D) Colocalization of 3xFLAG-mNG-Pex15Δ30 with the ER in the presence (+β-estradiol, Fig. 1 C) and absence (−β-estradiol, Fig. S1 B) of Msp1 was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single cell was selected as a region of interest (ROI), and Pearson’s correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol, n = 45; +β-estradiol, n = 60) from three independent experiments; n represents the number of cells. ****, P < 0.0001 compared with DOA10 cells by two-tailed paired t test. Source data are available for this figure: SourceData F1.
Msp1-dependent transfer of Pex15Δ30 from mitochondria to the ER. (A) Schematic representation of time-lapse imaging. 3xFLAG-mNG-Pex15Δ30 was expressed under the control of the cyanamide-inducible DDI2 promoter, and Msp1 was from the β-estradiol-inducible Z4EV promoter. DOA10 was deleted to block the ubiquitination and degradation of Pex15Δ30. Yeast cells were grown in SCD medium at 30°C, and 3xFLAG-mNG-Pex15Δ30 was induced by 5 mM cyanamide for 4 h at 30°C. The cells were washed with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. (B) Promoter shutoff chase of 3xFLAG-mNG-Pex15Δ30. doa10Δ and its isogenic WT (DOA10) cells in A were grown in SCD medium at 30°C. Cell extracts were prepared at the indicated times after addition of 1 μM β-estradiol, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in doa10Δ cells, right after the addition of β-estradiol (chase time = 0), relative to that in DOA10 cells, is shown (center). Values are mean ± SD from three independent experiments. (C) Yeast cells in A were grown in SCD medium at 30°C and treated with 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Pex15Δ30. Time-lapse images were then taken at 1-min intervals. Single-plane images are shown. The ER was labeled with BipN-mCherry-HDEL (ER-mCherry). Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (D) Colocalization of 3xFLAG-mNG-Pex15Δ30 with the ER in the presence (+β-estradiol, Fig. 1 C) and absence (−β-estradiol, Fig. S1 B) of Msp1 was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single cell was selected as a region of interest (ROI), and Pearson’s correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol, n = 45; +β-estradiol, n = 60) from three independent experiments; n represents the number of cells. ****, P < 0.0001 compared with DOA10 cells by two-tailed paired t test. Source data are available for this figure: SourceData F1.
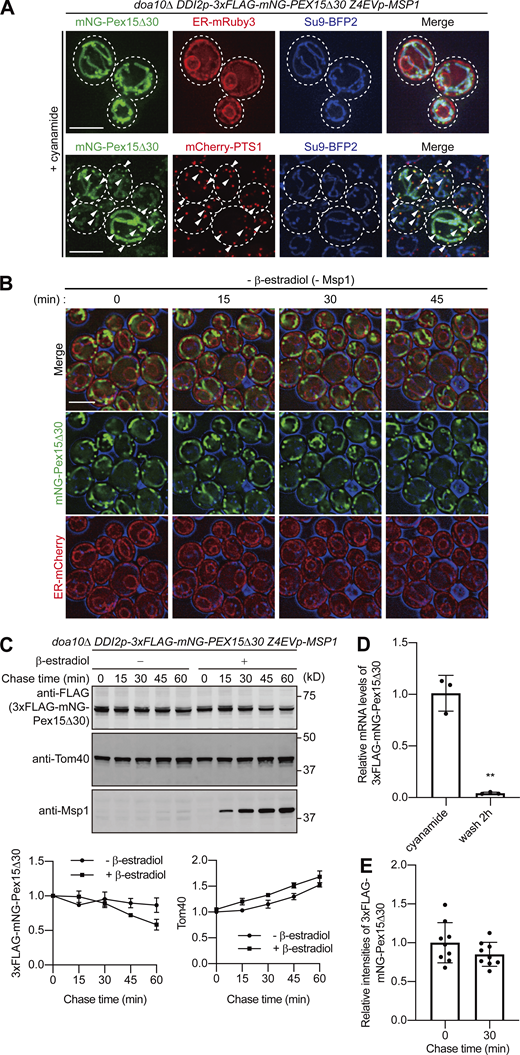
Msp1-mediated transfer of Pex15Δ30 from mitochondria to the ER. (A) Yeast cells in Fig. 1 A were grown in SCD at 30°C, treated with 5 mM cyanamide for 4 h, and imaged by fluorescence microscopy. The ER and mitochondria were labeled with BipN-mRuby3-HDEL (ER-mRuby3) and Su9-mTagBFP2 (Su9-BFP2), respectively (upper panel). Peroxisomes and mitochondria were labeled with mCherry-PTS1 and Su9-BFP2, respectively (lower panel). Cell shapes are outlined with broken lines, and arrowheads indicate mNG-Pex15Δ30 in peroxisomes. Maximum-projection images reconstituted from the z-stacks are shown (13 plane stacks, 0.4-μm spacing). Scale bar, 5 μm. (B) Time-lapse microscopy images were taken at the indicated times (min) for cells in Fig. 1 A without treatment with β-estradiol. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (C) Promoter shutoff chase of 3xFLAG-mNG-Pex15Δ30. Yeast cells in Fig. 1 A were grown in SCD at 30°C. Cell extracts were prepared at the indicated times in the presence (+β-estradiol) or absence (−β-estradiol) of Msp1 induction, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. Relative amounts of 3xFLAG-mNG-Pex15Δ30 (left) and Tom40 (right) were plotted against chase time. Values are mean ± SD from three independent experiments. (D) Yeast cells in Fig. 1 A were grown in SCD at 30°C. Total RNA was isolated from yeast cells before (cyanamide) and after (wash 2 h) washing off cyanamide, and mRNA levels of 3xFLAG-mNG-Pex15Δ30 (and control ACT1) were measured by quantitative real-time (RT)-PCR. mRNA levels were normalized to ACT1 mRNA levels, and values are mean ± SD from three independent experiments. The mean value before washing was set to 1.0. **, P = 0.01 compared with cyanamide-induced cells by two-tailed paired t test. (E) Quantification of 3xFLAG-mNG-Pex15Δ30 intensities in Fig. 1 C. A single cell was selected as a region of interest (ROI), and each mNG signal was quantified at chase time = 0 and 30. Values are mean ± SD (n = 9); n represents the number of cells. The mean value at chase time = 0 was set to 1.0. Source data are available for this figure: SourceData FS1.

Msp1-mediated transfer of Pex15Δ30 from mitochondria to the ER. (A) Yeast cells in Fig. 1 A were grown in SCD at 30°C, treated with 5 mM cyanamide for 4 h, and imaged by fluorescence microscopy. The ER and mitochondria were labeled with BipN-mRuby3-HDEL (ER-mRuby3) and Su9-mTagBFP2 (Su9-BFP2), respectively (upper panel). Peroxisomes and mitochondria were labeled with mCherry-PTS1 and Su9-BFP2, respectively (lower panel). Cell shapes are outlined with broken lines, and arrowheads indicate mNG-Pex15Δ30 in peroxisomes. Maximum-projection images reconstituted from the z-stacks are shown (13 plane stacks, 0.4-μm spacing). Scale bar, 5 μm. (B) Time-lapse microscopy images were taken at the indicated times (min) for cells in Fig. 1 A without treatment with β-estradiol. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (C) Promoter shutoff chase of 3xFLAG-mNG-Pex15Δ30. Yeast cells in Fig. 1 A were grown in SCD at 30°C. Cell extracts were prepared at the indicated times in the presence (+β-estradiol) or absence (−β-estradiol) of Msp1 induction, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. Relative amounts of 3xFLAG-mNG-Pex15Δ30 (left) and Tom40 (right) were plotted against chase time. Values are mean ± SD from three independent experiments. (D) Yeast cells in Fig. 1 A were grown in SCD at 30°C. Total RNA was isolated from yeast cells before (cyanamide) and after (wash 2 h) washing off cyanamide, and mRNA levels of 3xFLAG-mNG-Pex15Δ30 (and control ACT1) were measured by quantitative real-time (RT)-PCR. mRNA levels were normalized to ACT1 mRNA levels, and values are mean ± SD from three independent experiments. The mean value before washing was set to 1.0. **, P = 0.01 compared with cyanamide-induced cells by two-tailed paired t test. (E) Quantification of 3xFLAG-mNG-Pex15Δ30 intensities in Fig. 1 C. A single cell was selected as a region of interest (ROI), and each mNG signal was quantified at chase time = 0 and 30. Values are mean ± SD (n = 9); n represents the number of cells. The mean value at chase time = 0 was set to 1.0. Source data are available for this figure: SourceData FS1.
Next, we performed time-lapse fluorescent microscopy observation to monitor the transfer of mNG-Pex15Δ30 from mitochondria to the ER. In the absence of Msp1 expression, mNG-Pex15Δ30 stayed localized on mitochondria, not in the ER, irrespective of the presence of Doa10 (Figs. 1 D and S1 B, Video 1, and Video 3). Upon induction of Msp1 by β-estradiol, the mitochondrial signal of mNG-Pex15Δ30 was rapidly decayed in WT cells (Video 2), suggesting the efficient Doa10-dependent turnover of mNG-Pex15Δ30 as in Fig. 1 B. In contrast, expression of Msp1 made the fluorescent signal of mNG-Pex15Δ30 on mitochondria to be gradually shifted to the ER in doa10Δ cells (Fig. 1, C and D; and Video 4). The protein level of mNG-Pex15Δ30 remained constant or even slightly decreased after promoter shutoff of mNG-Pex15Δ30, while the protein level of Tom40 as a control even increased (Fig. S1 C), indicating that the observed ER localization of mNG-Pex15Δ30 in the presence of Msp1 did not reflect the ER-targeting of newly synthesized mNG-Pex15Δ30. Indeed, while the mRNA level of mNG-Pex15Δ30 was reduced to 4.3% after 2-h promoter shutoff (Fig. S1 D), as much as 86% of the mNG-Pex15Δ30 signal in mitochondria was found to be localized in the ER after 30-min chase with Msp1 expression following the promoter shutoff of Pex15Δ30 (Figs. 1 C and S1 E), supporting the above interpretation. Thus, the time-lapse fluorescent microscopy observations demonstrated the Msp1-dependent transfer of mitochondrially localized mNG-Pex15Δ30 from mitochondria to the ER.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, time-lapse imaging was recorded every minute. Single-plane image is shown. Mitochondria were labeled with Su9-RFP (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, time-lapse imaging was recorded every minute. Single-plane image is shown. Mitochondria were labeled with Su9-RFP (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the presence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every minute. Single-plane image is shown. Mitochondria were labeled with Su9-RFP (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the presence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every minute. Single-plane image is shown. Mitochondria were labeled with Su9-RFP (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10 and Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) in doa10Δ cells was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, time-lapse imaging was recorded every minute. Single-plane image is shown. The ER was labeled with BipN-mCherry-HDEL (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10 and Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) in doa10Δ cells was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, time-lapse imaging was recorded every minute. Single-plane image is shown. The ER was labeled with BipN-mCherry-HDEL (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10, but in the presence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) in doa10Δ cells was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every minute. Single-plane image is shown. The ER was labeled with BipN-mCherry-HDEL (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10, but in the presence of Msp1. Expression of 3xFLAG-mNG-Pex15Δ30 (green) in doa10Δ cells was induced by addition of cyanamide without expression of Msp1. After shutoff of 3xFLAG-mNG-Pex15Δ30 expression, the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every minute. Single-plane image is shown. The ER was labeled with BipN-mCherry-HDEL (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
The GET pathway is responsible for the ER localization of Pex15∆30
We confirmed the mitochondrial localization of mNG-Pex15Δ30 expressed from the galactose-inducible GAL1 promoter in WT cells (Fig. 2 A) and msp1Δ cells (Fig. 2 B). It may be noted that a minor fraction of mNG-Pex15Δ30 was localized to the ER (5%; Fig. 2 A), in addition to mitochondria and peroxisomes in WT cells (Fig. S2 A). However, mNG-Pex15Δ30 was not detected in the ER at all in msp1Δ cells (Fig. 2 B). In the absence of Doa10, mNG-Pex15Δ30 was localized in both mitochondria and the ER in doa10Δ cells (Fig. 2 C), but stayed only in mitochondria in doa10Δmsp1Δ cells (Fig. 2 D). These observations and their quantification (Fig. 2, I and J) indicate that the ER localization of Pex15Δ30 strongly depends on Msp1 expression, which is consistent with the above time-lapse observation of mNG-Pex15Δ30 (Fig. 1 C).

Localization of Pex15Δ30 to the ER is impaired by defects in Msp1 and the GET pathway. (A–H) WT (A), msp1Δ (B), doa10Δ (C), doa10Δmsp1Δ (D), get3Δ (E), get1Δget2Δ (F), doa10Δget3Δ (G), and doa10Δget1Δget2Δ (H) cells, expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, were grown in SCD medium at 30°C and then in SCGal medium for 4 h at 30°C and imaged by fluorescence microscopy. Mitochondria and the ER were labeled with Su9-RFP (upper panels) and ER-mCherry (lower panels), respectively. Single-plane images are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (I and J) Colocalization of 3xFLAG-mNG-Pex15Δ30 with mitochondria (I) and the ER (J) was measured using Pearson’s correlation coefficient between mNG and mitochondria (RFP)/ER(mCherry) signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ***, P < 0.01; ****, P < 0.0001 compared with WT cells by one-way ANOVA with Dunnett’s multiple comparison test.
Localization of Pex15Δ30 to the ER is impaired by defects in Msp1 and the GET pathway. (A–H) WT (A), msp1Δ (B), doa10Δ (C), doa10Δmsp1Δ (D), get3Δ (E), get1Δget2Δ (F), doa10Δget3Δ (G), and doa10Δget1Δget2Δ (H) cells, expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, were grown in SCD medium at 30°C and then in SCGal medium for 4 h at 30°C and imaged by fluorescence microscopy. Mitochondria and the ER were labeled with Su9-RFP (upper panels) and ER-mCherry (lower panels), respectively. Single-plane images are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (I and J) Colocalization of 3xFLAG-mNG-Pex15Δ30 with mitochondria (I) and the ER (J) was measured using Pearson’s correlation coefficient between mNG and mitochondria (RFP)/ER(mCherry) signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ***, P < 0.01; ****, P < 0.0001 compared with WT cells by one-way ANOVA with Dunnett’s multiple comparison test.

mNG-Pex15Δ30 localizes to peroxisomes in GET-deficient cells. (A–C) WT (A), get3Δ (B), and get1Δget2Δ (C) cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and imaged by fluorescence microscopy. Peroxisomes were labeled with mCherry-PTS1. Single-plane images (upper panel) and maximum-projection images (lower panel) reconstituted from the z-stacks (25 plane stacks, 0.2-μm spacing) are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy.

mNG-Pex15Δ30 localizes to peroxisomes in GET-deficient cells. (A–C) WT (A), get3Δ (B), and get1Δget2Δ (C) cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and imaged by fluorescence microscopy. Peroxisomes were labeled with mCherry-PTS1. Single-plane images (upper panel) and maximum-projection images (lower panel) reconstituted from the z-stacks (25 plane stacks, 0.2-μm spacing) are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy.
Next, we asked whether the GET system would be required for the transfer of Pex15Δ30 from mitochondria to the ER. We thus analyzed the localization of mNG-Pex15Δ30 in yeast GET-defective mutant cells. In get1Δget2Δ and get3Δ cells, mNG-Pex15Δ30 was observed only in mitochondria, not in the ER, even in the presence of Msp1 (Fig. 2, E, F, I, and J). A previous report suggested that Pex15 is first targeted to the ER by the GET system and then delivered to peroxisomes (Schuldiner et al., 2008). However, mNG-Pex15Δ30 was partly localized to peroxisomes even in get1Δget2Δ cells and get3Δ cells (Fig. S2, B and C) as in WT cells (Fig. S2 A) and doa10Δ cells (Fig. S1 A), suggesting that mNG-Pex15Δ30 is targeted to peroxisomes without using the GET-mediated ER targeting pathway. This suggests that the GET pathway is involved in the Msp1-dependent ER localization of mitochondrially mistargeted Pex15Δ30, not of newly synthesized Pex15Δ30.
Deletion of DOA10 simultaneously with the get1Δget2Δ or get3Δmutation moderately enhanced the mNG-Pex15Δ30 signals from mitochondria, not from the ER (Fig. 2, G–J). Therefore, the mitochondria-to-ER transfer of mNG-Pex15Δ30 was partly sensitive to the ubiquitination by Doa10, even in the absence of the normal GET system. This indicates that the GET system is important, but not essential, for the transfer of mNG-Pex15Δ30 to the ER.
Doa10 is functional in the GET mutant cells
Deletion of the GET3 gene was previously shown to have a negative genetic interaction with the MSP1 gene deletion (Costanzo et al., 2010; Chen et al., 2014; Okreglak and Walter, 2014). Dederer et al. (2019) further observed that mutants lacking the GET components Sgt2, Get2, Get3, Get4, or Get5 stabilized Pex15Δ30. These results were interpreted that the defective GET system caused secondary consequences of altered TA protein homeostasis, resulting in stabilized Pex15Δ30. Indeed, since Ubc6, an E2 enzyme for Doa10, is a TA protein in the ER membrane, mistargeting of Ubc6 could stabilize Pex15Δ30 when the GET system is defective. In relation to this, Ubc6 fused to GFP (GFP-Ubc6) was reported to be mistargeted to mitochondria upon its overexpression in get1Δget2Δ cells (Schuldiner et al., 2008); when expressed from the chromosomal gene with its own promoter, only a small fraction (<5%) of the Ubc6 population was mistargeted to mitochondria in get3Δ cells (Li et al., 2019). A Doa10 substrate Ste6*was reported to be partially stabilized in get3Δ cells (Dederer et al., 2019).
We thus expressed mNG followed by Ubc6 (mNG-Ubc6) from the chromosome with its own promoter in WT, get3Δ, and get1Δget2Δ cells, and monitored its localization. mNG-Ubc6 was exclusively localized in the ER in WT cells (Fig. 3, A, D, and E). Even in get1Δget2Δ and get3Δ cells, mNG-Ubc6 was mainly localized in the ER and mislocalized to mitochondria only partially (Fig. 3, B–E). We then prepared the total cell lysate from WT, get1Δget2Δ, and get3Δ cells and quantified the cellular levels of endogenous Ubc6 and mNG-Ubc6 by immunoblotting using anti-Ubc6 antibodies. Steady-state levels of Ubc6 and mNG-Ubc6 in get1Δget2Δ and get3Δ cells were similar to those in WT cells (Fig. 3 F). Importantly, Doa10 substrate Ste6*-3xHA was degraded in get3Δ cells as rapidly as in WT cells, as revealed by cycloheximide (CHX) chase experiments (Fig. 3 G). Collectively, our results indicate that the get1Δget2Δ and get3Δ mutations affected only partly the localization of Ubc6, so they did not have significant impact on the Doa10-facilitated degradation of its substrates.

Doa10 is active in the GET mutants. (A–C) WT (A), get3Δ (B), and get1Δget2Δ (C) cells, expressing mNG-Ubc6 from the own promoter of Ubc6, were grown in SCD and imaged by fluorescence microscopy. Mitochondria and the ER were labeled with Su9-RFP and ER-mCherry, respectively. Single-plane images are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (D and E) Colocalization of mNG-Ubc6 with the ER (D) and mitochondria (E) was analyzed using Pearson’s correlation coefficient between mNG and RFP/mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with WT cells and get3Δ or get1Δget2Δ cells by one-way ANOVA with Dunnett’s multiple comparison test. (F) WT, get3Δ, and get1Δget2Δ cells expressing Ubc6 or mNG-Ubc6 from their own promoters were grown in SCD. Cell extracts were prepared, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (upper panel). Protein amounts of endogenous Ubc6 and mNG-Ubc6 were normalized to that of Tom40 (lower panel). Values are mean ± SD from three independent experiments. One-way ANOVA with Dunnett’s multiple comparison test was used for comparison with WT-Ubc6 cells. (G) WT, get3Δ, and doa10Δ cells expressing Ste6*-3xHA from the GAL1 promoter were grown in SCD at 30°C and then in SCGal for 5 h at 30°C. Cell extracts were prepared at the indicated times at 30°C after addition of 100 µg/ml CHX, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of Ste6*-3xHA were plotted against chase time as in Fig. 1 B. Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData F3.
Doa10 is active in the GET mutants. (A–C) WT (A), get3Δ (B), and get1Δget2Δ (C) cells, expressing mNG-Ubc6 from the own promoter of Ubc6, were grown in SCD and imaged by fluorescence microscopy. Mitochondria and the ER were labeled with Su9-RFP and ER-mCherry, respectively. Single-plane images are shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (D and E) Colocalization of mNG-Ubc6 with the ER (D) and mitochondria (E) was analyzed using Pearson’s correlation coefficient between mNG and RFP/mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with WT cells and get3Δ or get1Δget2Δ cells by one-way ANOVA with Dunnett’s multiple comparison test. (F) WT, get3Δ, and get1Δget2Δ cells expressing Ubc6 or mNG-Ubc6 from their own promoters were grown in SCD. Cell extracts were prepared, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (upper panel). Protein amounts of endogenous Ubc6 and mNG-Ubc6 were normalized to that of Tom40 (lower panel). Values are mean ± SD from three independent experiments. One-way ANOVA with Dunnett’s multiple comparison test was used for comparison with WT-Ubc6 cells. (G) WT, get3Δ, and doa10Δ cells expressing Ste6*-3xHA from the GAL1 promoter were grown in SCD at 30°C and then in SCGal for 5 h at 30°C. Cell extracts were prepared at the indicated times at 30°C after addition of 100 µg/ml CHX, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of Ste6*-3xHA were plotted against chase time as in Fig. 1 B. Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData F3.
To complement the results of the CHX-chase experiments and to minimize the adaptive effects by the lack of Get3, we removed Get3 by rapid degradation by the auxin-inducible degradation (AID) system, which induces acute loss of a target protein specified by the AID tag (AID*-9xMYC) by the addition of auxin, indole-3-acetic acid (IAA; Nishimura et al., 2009). Get3 carrying the AID tag was functionally comparable to WT Get3 with respect to Pex15Δ30 degradation in the absence of IAA (Fig. 4, A and B). Acute degradation of Get3 by the addition of auxin stabilized Pex15Δ30, making the half-time of the Pex15Δ30 degradation twofold longer (Fig. 4 A). Taken together, our results showed that the defects in the GET pathway only partially affected the Ubc6 targeting and Doa10 function, but directly affected the ER localization of Pex15Δ30.

Overexpression of Msp1 restores the degradation inhibition of Pex15Δ30 in the GET mutants. (A) Cells expressing Get3-AID*-9xMyc from its own promoter and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter were grown in SCD at 30°C, in SCGal for 2 h at 30°C, and further incubated for 30 min after addition of 1 mM IAA. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (bottom). Values are mean ± SD from three independent experiments. (B)get3Δ cells expressing Get3-HA or its ATPase-inactive mutant Get3(D57N)-HA under the control of its own promoter were grown in SCD at 30°C and then in SCGal for 3 h at 30°C to induce 3xFLAG-mNG-Pex15Δ30, with or without Msp1 overexpression (Msp1 O/E) from the ADH1 promoter. Cell extracts were prepared at the indicated times at 30°C after addition of 100 µg/ml CHX, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in get3Δ cells, right after the addition of CHX (chase time = 0), relative to that in get3Δ cells with Get3-HA, but without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. (C)doa10Δ cells used in Fig. 1 were grown in SCD at 30°C, and then 3xFLAG-mNG-Pex15Δ30 was induced by addition of 5 mM cyanamide for 3 h at 30°C. The cells were washed with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression and incubated for 2 h at 30°C. The cells were treated with or without 1 μM β-estradiol for 30 min at 30°C and were harvested. Total cell lysates (T) were separated into the supernatant/cytosol fraction (S) and pellet/membrane fraction (P) by centrifugation (1 h, 100,000 g), and 3xFLAG-mNG-Pex15Δ30 was immunoprecipitated from the supernatant fractions with anti-FLAG magnetic beads (IP: FLAG; see Coimmunoprecipitation in Materials and methods). Amounts of 3xFLAG-mNG-Pex15Δ30 in fraction S relative to the total amounts in fractions S and P are indicated below the uppermost gel. Proteins in the flow-through (F), washed (W), and eluted (E) fractions were analyzed by SDS-PAGE and immunoblotted with the indicated antibodies. The arrowheads indicate the proteins of interest. The asterisk in immunoblotting using anti-Pgk1 antibodies indicates an IgG heavy chain of anti-FLAG antibodies. (D)get3Δ cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, with or without overexpression of Msp1 (Msp1O/E) under the control of the ADH1 promoter, were grown in SCD at 30°C, and then in SCGal for 3 h at 30°C. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 are plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30, right after the addition of CHX (chase time = 0), relative to that in WT cells without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData F4.
Overexpression of Msp1 restores the degradation inhibition of Pex15Δ30 in the GET mutants. (A) Cells expressing Get3-AID*-9xMyc from its own promoter and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter were grown in SCD at 30°C, in SCGal for 2 h at 30°C, and further incubated for 30 min after addition of 1 mM IAA. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (bottom). Values are mean ± SD from three independent experiments. (B)get3Δ cells expressing Get3-HA or its ATPase-inactive mutant Get3(D57N)-HA under the control of its own promoter were grown in SCD at 30°C and then in SCGal for 3 h at 30°C to induce 3xFLAG-mNG-Pex15Δ30, with or without Msp1 overexpression (Msp1 O/E) from the ADH1 promoter. Cell extracts were prepared at the indicated times at 30°C after addition of 100 µg/ml CHX, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in get3Δ cells, right after the addition of CHX (chase time = 0), relative to that in get3Δ cells with Get3-HA, but without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. (C)doa10Δ cells used in Fig. 1 were grown in SCD at 30°C, and then 3xFLAG-mNG-Pex15Δ30 was induced by addition of 5 mM cyanamide for 3 h at 30°C. The cells were washed with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression and incubated for 2 h at 30°C. The cells were treated with or without 1 μM β-estradiol for 30 min at 30°C and were harvested. Total cell lysates (T) were separated into the supernatant/cytosol fraction (S) and pellet/membrane fraction (P) by centrifugation (1 h, 100,000 g), and 3xFLAG-mNG-Pex15Δ30 was immunoprecipitated from the supernatant fractions with anti-FLAG magnetic beads (IP: FLAG; see Coimmunoprecipitation in Materials and methods). Amounts of 3xFLAG-mNG-Pex15Δ30 in fraction S relative to the total amounts in fractions S and P are indicated below the uppermost gel. Proteins in the flow-through (F), washed (W), and eluted (E) fractions were analyzed by SDS-PAGE and immunoblotted with the indicated antibodies. The arrowheads indicate the proteins of interest. The asterisk in immunoblotting using anti-Pgk1 antibodies indicates an IgG heavy chain of anti-FLAG antibodies. (D)get3Δ cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, with or without overexpression of Msp1 (Msp1O/E) under the control of the ADH1 promoter, were grown in SCD at 30°C, and then in SCGal for 3 h at 30°C. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 are plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30, right after the addition of CHX (chase time = 0), relative to that in WT cells without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData F4.
Pex15∆30 extracted by Msp1 physically interacts with Get3
To detect a direct physical interaction between Get3 and Pex15Δ30, we expressed an HA-tagged version of WT Get3 (Get3-HA) or its ATPase-deficient mutant (Get3D57N-HA), which would slowly release substrate TA proteins (Mariappan et al., 2011), in get3Δ cells and monitored the degradation of mNG-Pex15Δ30 by CHX-chase experiments (Fig. 4 B). When WT Get3-HA was present in the get3Δ cells, mNG-Pex15Δ30 was degraded rapidly, which was not affected by co-overexpression of Msp1 (Fig. 4 B). On the other hand, when Get3-HA carried the D57N mutation (Get3D57N-HA), mNG-Pex15Δ30 was highly stabilized (Fig. 4 B), suggesting that Get3D57N-HA trapped mNG-Pex15Δ30, thereby preventing it from ubiquitination by Doa10 on the ER membrane. Interestingly, when Msp1 was overexpressed, mNG-Pex15Δ30 was efficiently degraded even with Get3-HA carrying the D57N mutation (Fig. 4 B). This suggests that the Msp1-mediated extraction of mNG-Pex15Δ30 from the OM controls the flux of mNG-Pex15Δ30 for degradation.
We adopted the expression switch system in doa10Δ cells as shown in Fig. 1 A to follow the interaction between Get3 and Pex15Δ30 with and without Msp1. We first expressed mNG-Pex15Δ30 in the absence of Msp1 and then turned off the mNG-Pex15Δ30 expression; we found that Pex15Δ30 partly shifted from the membrane (P) to cytosolic (S) fraction upon expression of Msp1 (Fig. 4 C, lanes 2, 3, 8, and 9). Then, by immunoprecipitating mNG-Pex15Δ30 through its attached FLAG tag from the cytosolic fraction, we found that Get3 was coimmunoprecipitated only in the presence of Msp1 (Fig. 4 C, lanes 6 and 12), indicating that mNG-Pex15Δ30 extracted from membranes was indeed bound to Get3.
ER-targeting of Pex15∆30 bypasses the GET pathway upon overexpression of Msp1
The present results suggest that the GET-mediated transfer of Pex15Δ30 from mitochondria to the ER represents a major pathway for Pex15Δ30 degradation. However, this does not rule out operation of a parallel GET-independent pathway for the transfer of Pex15Δ30 from the mitochondrial OM to the ER, as suggested by the effect of overexpressed Msp1 (Fig. 4 B). To address this point, we enhanced the Msp1-mediated extraction of mNG-Pex15Δ30 from the OM by constitutive overexpression of Msp1 from the ADH1 promoter in get1Δget2Δ or get3Δ cells and evaluated the mNG-Pex15Δ30 degradation by CHX-chase experiments. While degradation of mNG-Pex15Δ30 was suppressed in get1Δget2Δ and get3Δ cells with the endogenous level of Msp1, overexpression of Msp1 led to efficient degradation of mNG-Pex15Δ30 in get3Δ and get1Δget2Δ cells (Figs. 4 D and S3), which is similar to the case of Msp1 overexpression in Get3D57N-expressing cells (Fig. 4 B). Therefore, a minor GET-independent pathway can work for the transfer of extracted Pex15Δ30 to the ER in GET-deficient cells such as get1Δget2Δ, get3Δ, and Get3D57N-expressing cells when the Msp1-mediated extraction from the OM is enhanced by Msp1 overexpression.
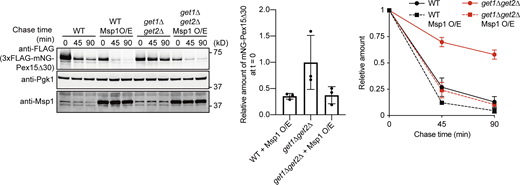
Overexpression of Msp1 leads to efficient degradation of mNG-Pex15Δ30 in get1Δget2Δ cells.get1Δget2Δ cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, with or without overexpression of Msp1 (Msp1 O/E) under the control of the ADH1 promoter, were grown in SCD at 30°C, and then in SCGal for 3 h at 30°C. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in get3Δ cells, right after the addition of CHX (chase time = 0), relative to that in WT cells without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData FS3.

Overexpression of Msp1 leads to efficient degradation of mNG-Pex15Δ30 in get1Δget2Δ cells.get1Δget2Δ cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter, with or without overexpression of Msp1 (Msp1 O/E) under the control of the ADH1 promoter, were grown in SCD at 30°C, and then in SCGal for 3 h at 30°C. Normalized relative amounts of 3xFLAG-mNG-Pex15Δ30 were plotted against chase time (right), and the amount of 3xFLAG-mNG-Pex15Δ30 in get3Δ cells, right after the addition of CHX (chase time = 0), relative to that in WT cells without Msp1 overexpression, is shown (center). Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData FS3.
To further address this point, we analyzed the localization of mNG-Pex15Δ30 in get1Δget2Δ and get3Δ cells under Msp1-overexpressing conditions in the absence of Doa10. In doa10Δ cells, mNG-Pex15Δ30 significantly shifted from mitochondria to the ER when Msp1 was overexpressed (Fig. 5, A, D, and E). In doa10Δget3Δ cells with overexpressed Msp1, mNG-Pex15Δ30 was partly localized to the ER as well as mitochondria (Fig. 5, B, D, and E). However, in doa10Δget1Δget2Δ cells with overexpressed Msp1, localization of mNG-Pex15Δ30 to the ER or mitochondria was not obvious anymore (Fig. 5, C–E), but mNG-Pex15Δ30 tended to show discrete foci in the cytosol (Fig. 5 F). This is consistent with the previous report that mislocalized TA proteins made foci in the cytosol in get1Δget2Δ cells (Schuldiner et al., 2008; Powis et al., 2013). We observed that mNG-Pex15Δ30 foci were colocalized with Get3 and Hsp104 (Fig. 5 F), but not with other cytosolic chaperones such as Ssa1, Ydj1, and Hsp42 (Fig. S4), in doa10Δget1Δget2Δ cells with overexpressed Msp1. Taking these results together, we conclude that overexpression of Msp1 promotes transfer of mistargeted Pex15Δ30 to the ER at least partly independently of Get3, yet in the absence of Get1/2, Pex15Δ30 tended to stay in the cytosol and formed aggregates involving Get3 and Hsp104.

Overexpression of Msp1 leads to accumulation of Pex15Δ30 in the cytosol and ER in the GET mutants. (A–C) doa10Δ (A), doa10Δget3Δ (B), and doa10Δget1Δget2Δ (C) cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpression of Msp1 (Msp1O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and were imaged by fluorescence microscopy. Single-plane images are shown. The ER and mitochondria were labeled with ER-mCherry (lower panels in A, B, and C) and Su9-RFP (upper panels in A, B, and C), respectively. Arrowheads indicate mNG-Pex15Δ30 localized in mitochondria (upper panels in B and C) and the ER (lower panels in B and C). Scale bars, 5 μm. DIC, differential interference contrast microscopy. (D and E) Colocalization of 3xFLAG-mNG-Pex15Δ30 with mitochondria (D) or the ER (E) was measured by using Pearson’s correlation coefficient between mNG and RFP/mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with doa10ΔMsp1O/E cells and doa10Δget3ΔMsp1O/E or doa10Δget1Δget2ΔMsp1O/E cells by one-way ANOVA with Dunnett’s multiple comparison test. (F)doa10Δget1Δget2Δ cells expressing Get3-mCherry (upper panel) or Hsp104-mCherry (lower panel) from their own promoters and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpressed Msp1 (Msp1 O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and were imaged by fluorescence microscopy. Single-plane images are shown. Arrowheads indicate 3xFLAG-mNG-Pex15Δ30 foci that were merged with Get3-mCherry or Hsp104-mCherry. Scale bar, 5 μm. The percentages and numbers of cells showing 3xFLAG-mNG-Pex15Δ30 foci colocalized with Get3 (n = 75) and Hsp104 (n = 119) are indicated on the right. DIC, differential interference contrast microscopy.
Overexpression of Msp1 leads to accumulation of Pex15Δ30 in the cytosol and ER in the GET mutants. (A–C) doa10Δ (A), doa10Δget3Δ (B), and doa10Δget1Δget2Δ (C) cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpression of Msp1 (Msp1O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and were imaged by fluorescence microscopy. Single-plane images are shown. The ER and mitochondria were labeled with ER-mCherry (lower panels in A, B, and C) and Su9-RFP (upper panels in A, B, and C), respectively. Arrowheads indicate mNG-Pex15Δ30 localized in mitochondria (upper panels in B and C) and the ER (lower panels in B and C). Scale bars, 5 μm. DIC, differential interference contrast microscopy. (D and E) Colocalization of 3xFLAG-mNG-Pex15Δ30 with mitochondria (D) or the ER (E) was measured by using Pearson’s correlation coefficient between mNG and RFP/mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with doa10ΔMsp1O/E cells and doa10Δget3ΔMsp1O/E or doa10Δget1Δget2ΔMsp1O/E cells by one-way ANOVA with Dunnett’s multiple comparison test. (F)doa10Δget1Δget2Δ cells expressing Get3-mCherry (upper panel) or Hsp104-mCherry (lower panel) from their own promoters and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpressed Msp1 (Msp1 O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and were imaged by fluorescence microscopy. Single-plane images are shown. Arrowheads indicate 3xFLAG-mNG-Pex15Δ30 foci that were merged with Get3-mCherry or Hsp104-mCherry. Scale bar, 5 μm. The percentages and numbers of cells showing 3xFLAG-mNG-Pex15Δ30 foci colocalized with Get3 (n = 75) and Hsp104 (n = 119) are indicated on the right. DIC, differential interference contrast microscopy.

Ssa1, Ydj1, or Hsp42 does not colocalize with the mNG-Pex15Δ30 foci formed by overexpression of Msp1 in doa10Δget1Δget2Δ cells. doa10Δget1Δget2Δ cells expressing Ssa1-mCherry (upper panel), mCherry-Ydj1 (middle panel), and Hsp42-mCherry (lower panel) from their own promoters and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpression of Msp1 (Msp1O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and imaged by fluorescence microscopy. Single-plane images were shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy.

Ssa1, Ydj1, or Hsp42 does not colocalize with the mNG-Pex15Δ30 foci formed by overexpression of Msp1 in doa10Δget1Δget2Δ cells. doa10Δget1Δget2Δ cells expressing Ssa1-mCherry (upper panel), mCherry-Ydj1 (middle panel), and Hsp42-mCherry (lower panel) from their own promoters and 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter with overexpression of Msp1 (Msp1O/E) from the ADH1 promoter were grown in SCD at 30°C and in SCGal for 4 h at 30°C and imaged by fluorescence microscopy. Single-plane images were shown. Scale bar, 5 μm. DIC, differential interference contrast microscopy.
Mistargeted authentic ER-TA proteins were transferred to the ER via the Msp1-GET pathway
We next asked if authentic ER-TA proteins can be, when mistargeted to the OM, transferred to the ER with the aid of Msp1 via the GET pathway. We chose Frt1, whose ER targeting is Get3 dependent (Li et al., 2019), as an authentic ER-TA protein and used the AID system to trigger the acute loss of Get3 for its efficient mitochondrial mistargeting (Fig. 6 A). The expression of FLAG-tagged mNG-Frt1, a fusion protein containing the 3xFLAG tag and mNG followed by Frt1, and Msp1 was controlled by the DDI2 promoter and Z4EV promoter, respectively, like the scheme shown in Fig. 1 A.

Authentic ER-TA proteins mistargeted to mitochondria return to the ER via the Msp1-GET pathway. (A) Schematic representation of time-lapse imaging. 3xFLAG-mNG-Frt1 was expressed under the control of the cyanamide-inducible DDI2 promoter and Msp1 from the β-estradiol–inducible Z4EV promoter. Get3-AID*-9xMyc, instead of endogenous Get3, was expressed from the chromosome under the control of its own promoter. Yeast cells were grown in SCD medium at 30°C, and Get3-AID*-9xMYC was depleted by 1 mM IAA for 30 min at 30°C. Then, 3xFLAG-mNG-ER-TA proteins were induced by 5 mM cyanamide for 3 h at 30°C. The cells were washed with fresh SCD medium to shut off the 3xFLAG-mNG-ER-TA protein expression and re-express Get3-AID*-9xMYC for 2 h at 30°C. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. (B) Localization of 3xFLAG-mNG-Frt1 was imaged by fluorescence microscopy in the presence (−IAA, upper panel) or absence (+IAA, lower panel) of Get3-AID*-9xMYC. Single-plane images are shown. Mitochondria were labeled with Tom70-mCherry. Arrowheads indicate the signal from mNG-Frt1 mislocalized to mitochondria. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (C) Colocalization of mNG-Frt1 with mitochondria was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with IAA untreated (−IAA) cells by two-tailed paired t test. (D) Yeast cells in A were grown in SCD at 30°C and treated with 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Frt1. Time-lapse images were then taken at 2-min intervals. Single-plane images are shown. The ER was labeled with Snd3-mCherry. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (E) Colocalization of 3xFLAG-mNG-Frt1 with the ER was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single yeast cell was selected as a region of interest (ROI), and Pearson correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol; n = 30, +β-estradiol; n = 38, +IAA, β-estradiol; n = 30) from three technical replicates; n represents the number of cells. ****, P < 0.0001, *, P = 0.0475 compared with the result of time-lapse imaging without β-estradiol treatment by one-way ANOVA with Dunnett’s multiple comparison test. (F) Yeast cells in B were grown in SCD at 30°C and treated with 1 mM IAA and 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Frt1. Time-lapse images were then taken at 2-min intervals. Single-plane images are shown. Mitochondria was labeled with Tom70-mCherry. Scale bar, 5 μm. (G) Colocalization of 3xFLAG-mNG-Frt1 with mitochondria was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single yeast cell was selected as a region of interest (ROI), and Pearson correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol; n = 30, +β-estradiol; n = 30, +IAA, β-estradiol; n = 30) from three independent experiments; n represents the number of cells. ****, P < 0.0001 compared with the result of time-lapse imaging without β-estradiol treatment by one-way ANOVA with Dunnett’s multiple comparison test.
Authentic ER-TA proteins mistargeted to mitochondria return to the ER via the Msp1-GET pathway. (A) Schematic representation of time-lapse imaging. 3xFLAG-mNG-Frt1 was expressed under the control of the cyanamide-inducible DDI2 promoter and Msp1 from the β-estradiol–inducible Z4EV promoter. Get3-AID*-9xMyc, instead of endogenous Get3, was expressed from the chromosome under the control of its own promoter. Yeast cells were grown in SCD medium at 30°C, and Get3-AID*-9xMYC was depleted by 1 mM IAA for 30 min at 30°C. Then, 3xFLAG-mNG-ER-TA proteins were induced by 5 mM cyanamide for 3 h at 30°C. The cells were washed with fresh SCD medium to shut off the 3xFLAG-mNG-ER-TA protein expression and re-express Get3-AID*-9xMYC for 2 h at 30°C. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. (B) Localization of 3xFLAG-mNG-Frt1 was imaged by fluorescence microscopy in the presence (−IAA, upper panel) or absence (+IAA, lower panel) of Get3-AID*-9xMYC. Single-plane images are shown. Mitochondria were labeled with Tom70-mCherry. Arrowheads indicate the signal from mNG-Frt1 mislocalized to mitochondria. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (C) Colocalization of mNG-Frt1 with mitochondria was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with IAA untreated (−IAA) cells by two-tailed paired t test. (D) Yeast cells in A were grown in SCD at 30°C and treated with 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Frt1. Time-lapse images were then taken at 2-min intervals. Single-plane images are shown. The ER was labeled with Snd3-mCherry. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (E) Colocalization of 3xFLAG-mNG-Frt1 with the ER was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single yeast cell was selected as a region of interest (ROI), and Pearson correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol; n = 30, +β-estradiol; n = 38, +IAA, β-estradiol; n = 30) from three technical replicates; n represents the number of cells. ****, P < 0.0001, *, P = 0.0475 compared with the result of time-lapse imaging without β-estradiol treatment by one-way ANOVA with Dunnett’s multiple comparison test. (F) Yeast cells in B were grown in SCD at 30°C and treated with 1 mM IAA and 1 μM β-estradiol after expression shutoff of 3xFLAG-mNG-Frt1. Time-lapse images were then taken at 2-min intervals. Single-plane images are shown. Mitochondria was labeled with Tom70-mCherry. Scale bar, 5 μm. (G) Colocalization of 3xFLAG-mNG-Frt1 with mitochondria was analyzed using Pearson’s correlation coefficient between mNG and mCherry signals. A single yeast cell was selected as a region of interest (ROI), and Pearson correlation coefficients were analyzed at each time point. Values are mean ± SD (−β-estradiol; n = 30, +β-estradiol; n = 30, +IAA, β-estradiol; n = 30) from three independent experiments; n represents the number of cells. ****, P < 0.0001 compared with the result of time-lapse imaging without β-estradiol treatment by one-way ANOVA with Dunnett’s multiple comparison test.
We confirmed that cyanamide-induced mNG-Frt1 colocalized with the ER and mitochondria in the presence (−IAA) and absence (+IAA) of Get3, respectively (Fig. 6, B and C; and Fig. S5, A and B). Then, auxin and cyanamide were washed out to promote Get3 re-expression and shut off mNG-Frt1 expression, respectively (Fig. 6 A). After 2-h wash, re-expressed Get3 reached ∼50% of the Get3 level of IAA-untreated cells (Fig. S5 C). We followed the signals from mNG-Frt1 by time-lapse fluorescent microscopy observations under different conditions. mNG-Frt1 signals (green) on mitochondria gradually shifted to the ER (red) with Msp1 expression (+β-estradiol; Fig. 6, D, E, and G; and Video 5), while mNG-Frt1 signals stayed on mitochondria without Msp1 expression (−β-estradiol; Fig. 6, E and G; Fig. S5 D; and Video 6). mNG-Frt1 was stable with and without expressed Msp1 (Fig. S5 E). Thus, Msp1 expression shifted mNG-Frt1 from mitochondria to the ER, where it escaped ER quality control.
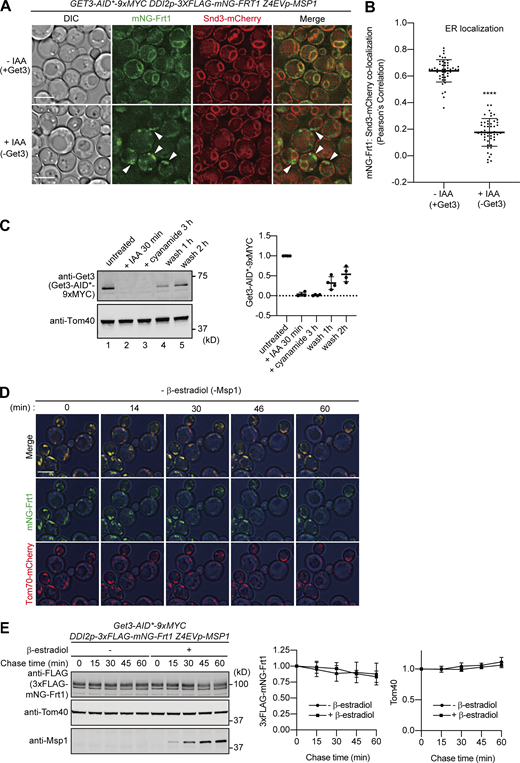
Frt1 mislocalized to the mitochondrial OM is transferred to the ER in a manner dependent on Msp1 and Get3. (A) Localization of 3xFLAG-mNG-Frt1 was imaged by fluorescence microscopy in the presence (−IAA, upper panel) or absence (+IAA, lower panel) of Get3-AID-9xMyc. Single-plane images are shown. The ER were labeled with Snd3-mCherry. Arrowheads indicate mNG-Frt1 mislocalized to mitochondria. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (B) Colocalization of mNG-Frt1 with the ER was analyzed by using Pearson’s correlation coefficient between mNG and mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with IAA untreated (−IAA) cells by two-tailed paired t test. (C) Yeast cells expressing Get3-AID*-9xMYC as in Fig. 6 A were grown in SCD medium. Cell extracts were prepared, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Lane 1: untreated cells, lane 2: IAA treatment for 30 min at 30°C, lane 3: cyanamide induction for 3 h at 30°C in the presence of IAA, lane 4: 1 h after washing off of IAA and cyanamide, and lane 5: 2 h after washing off of IAA and cyanamide. Protein amounts of Get3-AID*-9xMYC were normalized by that of Tom40 (right). Values are mean ± SD from four independent experiments. (D) Time-lapse microscopy images were taken at the indicated times (min) for the cells in Fig. 6 A without IAA and β-estradiol treatment. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (E) Promoter shutoff chase of 3xFLAG-mNG-Frt1. Yeast cells in Fig. 6 A were grown in SCD at 30°C. Cell extracts were prepared at the indicated times in the presence or absence of 1 μM β-estradiol, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Relative amounts of 3xFLAG-mNG-Frt1 (central) and Tom40 (right) were plotted against the protein levels at t = 0, respectively. Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData FS5.

Frt1 mislocalized to the mitochondrial OM is transferred to the ER in a manner dependent on Msp1 and Get3. (A) Localization of 3xFLAG-mNG-Frt1 was imaged by fluorescence microscopy in the presence (−IAA, upper panel) or absence (+IAA, lower panel) of Get3-AID-9xMyc. Single-plane images are shown. The ER were labeled with Snd3-mCherry. Arrowheads indicate mNG-Frt1 mislocalized to mitochondria. Scale bar, 5 μm. DIC, differential interference contrast microscopy. (B) Colocalization of mNG-Frt1 with the ER was analyzed by using Pearson’s correlation coefficient between mNG and mCherry signals. Values are mean ± SD (n = 50) from three technical replicates; n represents the number of cells. ****, P < 0.0001 compared with IAA untreated (−IAA) cells by two-tailed paired t test. (C) Yeast cells expressing Get3-AID*-9xMYC as in Fig. 6 A were grown in SCD medium. Cell extracts were prepared, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Lane 1: untreated cells, lane 2: IAA treatment for 30 min at 30°C, lane 3: cyanamide induction for 3 h at 30°C in the presence of IAA, lane 4: 1 h after washing off of IAA and cyanamide, and lane 5: 2 h after washing off of IAA and cyanamide. Protein amounts of Get3-AID*-9xMYC were normalized by that of Tom40 (right). Values are mean ± SD from four independent experiments. (D) Time-lapse microscopy images were taken at the indicated times (min) for the cells in Fig. 6 A without IAA and β-estradiol treatment. Cell outlines (blue) are shown by a DIC image. Scale bar, 5 μm. (E) Promoter shutoff chase of 3xFLAG-mNG-Frt1. Yeast cells in Fig. 6 A were grown in SCD at 30°C. Cell extracts were prepared at the indicated times in the presence or absence of 1 μM β-estradiol, and proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies (left). Relative amounts of 3xFLAG-mNG-Frt1 (central) and Tom40 (right) were plotted against the protein levels at t = 0, respectively. Values are mean ± SD from three independent experiments. Source data are available for this figure: SourceData FS5.
Fate of mislocalized Frt1 in the presence of Msp1 and Get3-AID*-9xMyc. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was re-expressed for 2 h, then the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every 2 min. Single-plane image is shown. The ER was labeled with Snd3-mCherry (red). Left, 3xFLAG-mNG-Frt1 (green); central, Snd3-mCherry (red); right, merge. Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of mislocalized Frt1 in the presence of Msp1 and Get3-AID*-9xMyc. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was re-expressed for 2 h, then the Msp1 expression was induced by addition of β-estradiol and time-lapse imaging was recorded every 2 min. Single-plane image is shown. The ER was labeled with Snd3-mCherry (red). Left, 3xFLAG-mNG-Frt1 (green); central, Snd3-mCherry (red); right, merge. Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of mislocalized Frt1 in the presence of Get3-AID*-9xMYC, but in the absence of Msp1. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was re-expressed for 2 h, then time-lapse imaging was recorded every 2 min. Single-plane image is shown. Mitochondria were labeled with Tom70-mCherry (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
Fate of mislocalized Frt1 in the presence of Get3-AID*-9xMYC, but in the absence of Msp1. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was re-expressed for 2 h, then time-lapse imaging was recorded every 2 min. Single-plane image is shown. Mitochondria were labeled with Tom70-mCherry (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm. Frame rate is 7 fps.
We next tested the role of Get3 in the Msp1-dependent shift of mNG-Frt1 from mitochondria to the ER. With Msp1 expression (+β-estradiol) and Get3 depletion (+IAA), mNG-Frt1 signals decreased from mitochondria but did not move to the ER, suggesting that mNG-Frt1 was extracted from the OM by Msp1, but did not reach the ER due to the lack of Get3 (Fig. 6, E, F, and G; and Video 7). Thus, our time-lapse observations showed that authentic ER-TA proteins mislocalized to mitochondria could also be retargeted to the ER by Msp1 and via the GET pathway.
Fate of mislocalized Frt1 in the presence of Msp1, but in the absence of Get3-AID*-9xMyc. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was again depleted by addition of IAA for 30 min before addition of β-estradiol for Msp1 expression and then time-lapse imaging was recorded every 2 min. Single-plane image is shown. Mitochondria was labeled with Tom70-mCherry (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm.
Fate of mislocalized Frt1 in the presence of Msp1, but in the absence of Get3-AID*-9xMyc. Expression of 3xFLAG-mNG-Frt1 (green) was induced by addition of cyanamide in the presence of IAA. After shutoff of 3xFLAG-mNG-Frt1 expression, Get3-AID*-9xMyc was again depleted by addition of IAA for 30 min before addition of β-estradiol for Msp1 expression and then time-lapse imaging was recorded every 2 min. Single-plane image is shown. Mitochondria was labeled with Tom70-mCherry (red). Cell outlines (blue) are shown by a single DIC image. Scale bar, 5 μm.
Discussion
In the present study, we identified the GET pathway as a major route for the transfer of mistargeted TA proteins from mitochondria to the ER (Fig. 7). The GET pathway is known as a targeting route for newly synthesized TA proteins to the ER. The GET pathway has also been shown to promote the ER localization of mitochondrial OM proteins when their targeting signals are masked or their import into mitochondria is impaired (Shakya et al., 2021; Vitali et al., 2018; Xiao et al., 2021). Such nonimported OM proteins can relocate to mitochondria through the ER-surface–mediated protein targeting (ER-SURF) pathway (Hansen et al., 2018; Xiao et al., 2021) and/or ER-localized P5A-ATPase (Spf1 in yeast; ATP13A1 in humans; McKenna et al., 2020; Qin et al., 2020) as safeguard mechanisms. However, the GET-dependent shift of mitochondrially mistargeted TA proteins such as mNG-Pex15Δ30 and mNG-Frt1 to the ER observed here is unlikely to reflect the ER targeting of newly synthesized TA proteins (Fig. S1, D and E), but instead indicates direct ER retransfer of the mistargeted TA proteins extracted from the mitochondrial OM by Msp1. Thus, the GET system is involved in the process other than targeting of newly synthesized ER-TA proteins, and our present results further expand the role of the GET system to the one in retransfer of mistargeted TA proteins to the ER.

The role of the GET pathway in the transfer of mistargeted TA proteins from the mitochondrial OM to ER membrane. Msp1 extracts mislocalized TA proteins from the mitochondrial OM in an ATP-dependent manner. Get3 then binds to the extracted TA proteins in the cytosol and escorts them to the Get1-Get2 complex in the ER membrane. While TA proteins that failed to be captured by Get3 tend to return to the mitochondrial OM again, a fraction of them can move to the ER membrane via an uncharacterized Get-independent pathway upon Msp1 overexpression. See Discussion for details.
The role of the GET pathway in the transfer of mistargeted TA proteins from the mitochondrial OM to ER membrane. Msp1 extracts mislocalized TA proteins from the mitochondrial OM in an ATP-dependent manner. Get3 then binds to the extracted TA proteins in the cytosol and escorts them to the Get1-Get2 complex in the ER membrane. While TA proteins that failed to be captured by Get3 tend to return to the mitochondrial OM again, a fraction of them can move to the ER membrane via an uncharacterized Get-independent pathway upon Msp1 overexpression. See Discussion for details.
Because Msp1 facilitates ATP-dependent extraction of mistargeted TA proteins from the mitochondrial OM, the GET pathway likely mediates the subsequent retargeting of extracted TA proteins through the aqueous cytosol to the ER, minimizing aggregate formation. Indeed, mNG-Pex15Δ30 extracted from membranes by Msp1 was indeed bound to Get3 (Fig. 4 C), and when Get1/2 and Doa10 were absent, Pex15Δ30 tended to stay in the cytosol and formed aggregates involving Get3 and Hsp104 (Fig. 5 F).
Nevertheless, it is still not clear why the GET system is required for the retargeting of mislocalized Pex15Δ30 to the ER while it does not facilitate targeting of newly synthesized Pex15Δ30 to the ER. It is also a mystery how mistargeted TA proteins can travel to the ER, although inefficiently, even in the absence of the functional GET system (Fig. 4, B and D; and Fig. 7). This alternative GET-independent route to the ER could be a spontaneous insertion of extracted Pex15Δ30 to any membrane in the proximity. It is open to future studies, as well, if this route is facilitated by the close apposition of the ER and mitochondrial OM like ERMES in yeast cells.
Materials and methods
Strains and plasmids
Yeast strains, plasmids, and primers used in this study are described in Tables S1, S2, and S3, respectively. Yeast strains were constructed using standard homologous recombination methods (Knop et al., 1999).
For time-lapse imaging in Fig. 1, Fig. S1, Video 1, Video 2, Video 3, and Video 4, 3xFLAG-mNG-Pex15Δ30 was expressed from the DDI2 promoter by integrating the linearized pSM555 into the TRP1 locus in doa10Δ or WT cells. Subsequently, a transcriptional factor Z4EV was expressed from the ADH1 promoter by integrating the linearized pMT287 into the LEU2 locus, and the endogenous promoter of MSP1 was replaced with the Z4EV promoter (pMT289; McIsaac et al., 2013). BipN-mCherry-HDEL (ER-mCherry) and Su9-mTagBFP2 (Su9-BFP2) were expressed from the GPD (ER-mCherry) and TEF1 (Su9-BFP2) promoters by integrating the linearized pMT328 and pMT357 into the URA3 and HIS3 locus, respectively. The expression cassette for ER-mRuby3 (pMT341), Su9-RFP (pMT367), or mCherry-PTS1 (pMT375) was integrated into the URA3 locus in the same manner as ER-mCherry.
For fluorescence microscopy imaging in Figs. 2, 5, S2, and S4 and the CHX chase experiment in Fig. 4, B and D and Fig. S3, 3xFLAG-mNG-Pex15Δ30 was expressed from the GAL1 promoter by integrating the linearized pSM552 into the TRP1 locus. For fluorescence microscopy imaging in Figs. 3 and S4, mNG or mCherry tag was integrated into the position following the start codon of the UBC6 or YDJ1 ORF, respectively, by using CRISPR/Cas9 genome editing according to the published protocol (Okada et al., 2021).
For fluorescence microscopy imaging in Figs. 5 and S4, and the CHX chase experiment in Fig. 4, B and D and Fig. S3, Msp1 was expressed from the ADH1 promoter by integrating the linearized pSM574 into the LEU2 locus for overexpression. Get3-mCherry, Hsp104-mCherry, Ssa1-mCherry, or Hsp42-mCherry were expressed from their own promoters by integrating the mCherry tag (pMT294) into the position preceding the stop codon of each ORF in doa10Δget1Δ get2Δ cells with overexpression of Msp1.
For the CHX chase experiment in Fig. 4 A and time-lapse imaging in Figs. 6 and S5, OsTIR1-9xMYC was expressed from the ADH1 promoter by integrating the linearized pNHK53 into the URA3 locus (Nishimura et al., 2009). Subsequently, Get3-AID*-9xMYC was expressed from its own promoter by integrating AID*-9xMYC tag (pKan-AID*-9myc) into the position preceding the stop codon of the GET3 ORF (Morawska and Ulrich, 2013). 3xFLAG-mNG-Frt1 was expressed from the DDI2 promoter by integrating the linearized pSM558 into the TRP1 locus in Get3-AID*-9xMYC cells. Subsequently, the expression cassette for Z4EV (pMT287) was integrated into the LEU2 locus, and the endogenous promoter of MSP1 was replaced with the Z4EV promoter (pMT289) as described above. Tom70-mCherry or Snd3-mCherry was expressed from its own promoter by integrating mCherry tag (pMT294) into the position preceding the stop codon of the TOM70 or SND3 ORF. All plasmid constructs were verified by DNA sequencing.
Yeast growth conditions
Yeast strains were cultured in YP (1% yeast extract and 2% polypeptone) or SC (0.67% yeast nitrogen base without amino acids and 0.5% casamino acid) medium with appropriate carbon sources (2% glucose [D], 2% galactose [Gal]) and supplements.
CHX chase
Yeast cells for expression of 3xFLAG-mNG-Pex15Δ30 or Ste6*-3xHA from the GAL1 promoter were shifted from SCD medium to SCGal medium and cultured for 2–5 h at 30°C. CHX was added at 100 μg/ml to start a chase at 30°C. One OD600 unit cells were collected periodically and frozen at −80°C until use. The frozen cells were thawed in ice-cold 0.1 M NaOH and incubated on ice for 5 min. The cell pellets were precipitated by centrifugation at 20,000 g for 5 min. The resulting pellet was resuspended in SDS-PAGE sample buffer and subjected to SDS-PAGE and immunoblotting.
Immunoblotting
Proteins were transferred from polyacrylamide gels to PVDF membranes (Bio-Rad) in blotting buffer for SDS-PAGE (25 mM Tris, 192 mM glycine, and 5% MeOH) at a constant voltage of 25 V. Membranes were washed in TBST buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween-20) and blocked with 5% skim milk in TBST buffer for 1 h. The membranes were incubated with primary antibodies in TBST buffer overnight at 4°C and washed three times with TBST (5 min each). The membranes were then incubated with secondary antibodies for 30 min and washed three times with TBST. The PVDF membranes were scanned with an Amersham Typhoon scanner (Cytiva), and signal intensities were quantified using ImageQuant software (Cytiva).
The following antibodies were used: monoclonal anti-FLAG M2 mouse IgG (F3165; 1:2,000 dilution; Millipore-Sigma), monoclonal anti-Pgk1 mouse IgG (ab113687; 1:2,000 dilution; Abcam), monoclonal anti-HA mouse IgG (M180-3; 1:2,000 dilution; Medical & Biological Laboratories), polyclonal anti-Tom40 rabbit IgG (1:2,000 dilution), polyclonal anti-Msp1 rabbit IgG (1:2,000 dilution), goat anti-mouse IgG (H + L) secondary antibody, DyLight 800 4X PEG (SA5-35521; 1:2,000 dilution; Thermo Fisher Scientific), goat anti-rabbit IgG (H + L) secondary antibody, DyLight 800 4X PEG (SA5-35571; 1:2,000 dilution; Thermo Fisher Scientific), goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, cyanine5 (A10524; 1:2,000 dilution; Thermo Fisher Scientific), and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, cyanine5 (A10524; 1:2,000 dilution; Thermo Fisher Scientific).
Fluorescence microscopy
Yeast cells expressing 3xFLAG-mNG-Pex15Δ30 from the DDI2 promoter were grown in SCD medium, and those expressing the GAL1 promoter were grown in SCD medium at 30°C. Yeast cells expressing 3xFLAG-mNG-Pex15Δ30 from the DDI2 promoter were grown in SCD medium, and 3xFLAG-mNG-Pex15Δ30 was induced by addition of 5 mM cyanamide for 4 h at 30°C. Yeast cells expressing 3xFLAG-mNG-Pex15Δ30 from the GAL1 promoter were grown in SCD medium and then shifted to SCGal medium for 4 h at 30°C. Yeast cells expressing chromosomally tagged Get3-AID*-9xMYC were treated with or without 1 mM IAA for 30 min at 30°C in SCD and further incubated in the presence of 5 mM cyanamide to induce 3xFLAG-mNG-Frt1 from the DDI2 promoter for 3 h at 30°C. Cells were mounted on a 1% agarose pad containing synthetic dextrose growth medium and placed on a 15-mm glass-bottom dish (Matsunami Glass). Cells were observed under a DeltaVision Elite system (Cytiva) equipped with a 100× objective lens (UPLSAPO, NA/1.40; Olympus) and a sCMOS camera (Edge5.5; PCO) at room temperature. Deconvolution was performed using SoftWoRx software (Cytiva). Z-sections were collected every 0.2 or 0.4 μm from the upper to bottom surface of yeast cells. Acquired images were processed and analyzed using Fiji software.
Time-lapse fluorescence microscopy
For time-lapse imaging in Fig. 1 C, yeast cells were grown in SCD medium at 30°C, and 3xFLAG-mNG-Pex15Δ30 was induced by 5 mM cyanamide from the DDI2 promoter for 4 h at 30°C. Then the cells were washed twice with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression and incubated for 2 h at 30°C. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. Single-plane images were collected at 1-min intervals for 60 min. For time-lapse imaging in Fig. S1 B, time-lapse imaging was recorded without the β-estradiol treatment.
For time-lapse imaging in Fig. 6 D, yeast cells expressing chromosomally tagged Get3-AID*-9xMYC were treated with 1 mM IAA for 30 min at 30°C in SCD medium and further incubated in the presence of 5 mM cyanamide to induce 3xFLAG-mNG-Frt1 for 3 h at 30°C. Then, cells were washed twice with fresh SCD medium and incubated for 2 h at 30°C. Time-lapse imaging was recorded after addition of 1 μM β-estradiol to start expression of Msp1. Single-plane images were collected at 2-min intervals for 60 min.
Analysis of mRNA levels by quantitative RT-PCR
To determine the mRNA level of 3xFLAG-mNG-Pex15Δ30 in the time-lapse experiment, we performed quantitative RT-PCR. doa10Δ cells were grown in SCD medium at 30°C, and 3xFLAG-mNG-Pex15Δ30 was induced by 5 mM cyanamide from the DDI2 promoter for 4 h at 30°C. Then the cells were washed twice with fresh SCD medium to shut off 3xFLAG-mNG-Pex15Δ30 expression and incubated in SCD medium without cyanamide for 2 h at 30°C. One OD600 unit of yeast cells was collected 2 h before and after washing off cyanamide, and total RNA was isolated with a MAXwell RSC miRNA Tissue kit (Promega). cDNA was synthesized with the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa). Quantitative RT-PCR was performed with TB Green Premix ExTaq II (TaKaRa) and the primers listed in Table S3, using Mic real-time PCR cycler (Bio Molecular Systems). Data were analyzed by the 2−ΔΔCT method, normalized to the ACT1 gene.
Coimmunoprecipitation
Coimmunoprecipitation was performed as follows. doa10Δ cells expressing 3xFLAG-mNG-Pex15Δ30 from the DDI2 promoter were grown in SCD medium, and 3xFLAG-mNG-Pex15Δ30 was induced by addition of 5 mM cyanamide for 3 h at 30°C. The cells were washed twice with fresh SCD medium and incubated for 2 h at 30°C. Expression of Msp1 was initiated by addition of 1 mM β-estradiol, and cells were incubated for 30 min at 30°C. 100 OD600 units of yeast cells were collected and frozen at −80°C. The frozen cells were thawed in 1 ml of KMH buffer (20 mM Hepes-KOH, pH 7.4, 100 mM KCl, 2 mM MgCl2, and protease inhibitor cocktail [03969-34; Nacalai tesque]) and disrupted with glass beads using a vortex mixer. After removal of glass beads and insoluble debris by centrifugation at 1,000 g for 5 min at 4°C, the total cell lysates were centrifugated at 100,000 g for 1 h at 4°C to collect the cytosolic fraction as a supernatant and membrane fractions. Anti-FLAG beads (MBL) were added to the S100 cytosolic fraction, and the mixture was gently rotated for 1.5 h at 4°C. After collecting the flow-through fraction, the beads were washed three times with KMH buffer, and the anti-FLAG beads were boiled in SDS-PAGE sample buffer. Eluted proteins were analyzed by SDS-PAGE followed by immunoblotting.
Image representation
Microscopy images shown in Fig. 1 C; Fig. 2, A–H; Fig. 3, A–C; Fig. 5, A–C and F; Fig. 6, B, D, and F; Fig. S1 B; Fig. S2, A–C; Fig. S4; and Fig. S5, A and D are single z slices. Images in Fig. S1 A and Fig. S2, A–C are maximum projections of all z slices.
Statistics
Statistical tests used for data analysis are defined in the figure legends. These include one-way ANOVA with Dunnett’s multiple comparison test in Fig. 2, I and J; Fig. 3, D–F; Fig. 5, D and E; and Fig. 6, E and G and two-tailed paired t test in Fig. 1 D, S1 D, 6 C, and S5 B. Data distribution was assumed to be normal but was not formally tested.
Online supplemental material
Fig. S1 shows the Msp1-mediated transfer of mNG-Pex15Δ30 from mitochondria to the ER. Fig. S2 shows the peroxisomal localization of mNG-Pex15Δ30 in GET-deficient cells. Fig. S3 shows the CHX chase experiment of mNG-Pex15Δ30 in get1Δget2Δ cells with and without overexpression of Msp1. Fig. S4 shows that Ssa1, Ydj1, or Hsp42 does not colocalize with the mNG-Pex15Δ30 foci formed by overexpression of Msp1 in doa10Δget1Δget2Δ cells. Fig. S5 shows that Frt1 mislocalized to the mitochondrial OM is transferred to the ER in a manner dependent on Msp1 and Get3. Video 1 shows the fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Msp1. Video 2 shows the fate of 3xFLAG-mNG-Pex15Δ30 in the presence of Msp1. Video 3 shows the fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10 and Msp1. Video 4 shows the fate of 3xFLAG-mNG-Pex15Δ30 in the absence of Doa10, but in the presence of Msp1. Video 5 shows the fate of 3xFLAG-mNG-Frt1 in the presence of Msp1 and Get3-AID*-9xMYC. Video 6 shows the fate of 3xFLAG-mNG-Frt1 in the presence of Get3-AID*-9xMYC, but in the absence of Msp1. Video 7 shows the fate of 3xFLAG-mNG-Frt1 in the presence of Msp1, but in the absence of Get3-AID*-9xMYC. Table S1 lists the yeast strains used in this study. Table S2 lists the plasmids used in this study. Table S3 lists the primers used in this study.
Acknowledgments
We thank the members of the Endo lab for discussions and critical comments on the manuscript. We are grateful to K. Nakatsukasa for the ste6-166-3xHA expression plasmid and S. Rospert and E. Jarosch for the Get3 and Ubc6 polyclonal antibodies, respectively.
This work was supported by JSPS KAKENHI to T. Endo (15H05705, 20H04912, 20H00458, 20H05689, and 20H05929), to S. Matsumoto (20K15794), and to S. Okada (26891019 and 18K06062), AMED-CREST to T. Endo (21gm1410002), JST-CREST to T. Ito (JPMJCR19S1), and a grant from Takeda Science Foundation. S. Shinoda is supported by a Research Fellowship for Young Scientists from the Japan Society of the Promotion of Science (19J00899).
The authors declare no competing financial interests.
Author contributions: Conceptualization, methodology, project administration, supervision, and writing the original draft were accomplished by S. Matsumoto and T. Endo, and partly by S. Ono. T. Numata contributed to the project administration and supervision, and S. Okada and T. Ito partly to the methodology. The investigation was carried out by S. Matsumoto and partly by S. Ono and S. Shinoda, with the support for preparing materials by C. Kakuta. S. Matsumoto and S. Ono in part were responsible for data curation, formal analysis, and visualization.

