


Fission yeast cells prevent mitotic entry until a threshold cell surface area is reached. The protein kinase Cdr2 contributes to this size control system by forming multiprotein nodes that inhibit Wee1 at the medial cell cortex. Cdr2 node anchoring at the cell cortex is not fully understood. Through a genomic screen, we identified the conserved GTPase Arf6 as a component of Cdr2 signaling. Cells lacking Arf6 failed to divide at a threshold surface area and instead shifted to volume-based divisions at increased overall size. Arf6 stably localized to Cdr2 nodes in its GTP-bound but not GDP-bound state, and its guanine nucleotide exchange factor (GEF), Syt22, was required for both Arf6 node localization and proper size at division. In arf6Δ mutants, Cdr2 nodes detached from the membrane and exhibited increased dynamics. These defects were enhanced when arf6Δ was combined with other node mutants. Our work identifies a regulated anchor for Cdr2 nodes that is required for cells to sense surface area.
Introduction
Many cell types maintain constant size during recurring cycles of growth and division. Such size control indicates the existence of mechanisms that delay cell cycle transitions until cells reach a critical size threshold (Amodeo and Skotheim, 2016; Rupes, 2002). The fission yeast Schizosaccharomyces pombe is a strong model system to study size-dependent control of entry into mitosis, also called the G2/M transition. These rod-shaped cells grow by linear extension and enter mitosis and divide at a reproducible size (Wood and Nurse, 2015). Although cell length, volume, and surface area scale together during linear extension growth, recent studies show that S. pombe cells divide at a specific surface area, as opposed to length or volume (Pan et al., 2014; Facchetti et al., 2019). Thus, the underlying size control network likely operates through mechanisms connected to the cell cortex.
The G2/M transition is controlled by regulated activation of the cyclin-dependent kinase Cdk1 (Harashima et al., 2013). Multiple mechanisms contribute to cell size–dependent Cdk1 activation in S. pombe. The protein kinase Wee1 phosphorylates and inhibits Cdk1 in small cells (Russell and Nurse, 1987a; Gould and Nurse, 1989), while a regulatory network progressively inhibits Wee1 as cell size increases (Lucena et al., 2017; Allard et al., 2018; Opalko et al., 2019). The concentrations of mitotic inducers including Cdc13 (mitotic cyclin) and Cdc25 (protein phosphatase that counteracts Wee1) increase as cells grow (Moreno et al., 1990; Keifenheim et al., 2017; Patterson et al., 2019). The Wee1 regulatory network draws particular interest because it functions at the cell cortex through the conserved protein kinases Cdr1 and Cdr2. Mutations in cdr1 and cdr2 cause cells to divide at a larger size due to uninhibited Wee1 (Russell and Nurse, 1987b; Young and Fantes, 1987; Breeding et al., 1998; Kanoh and Russell, 1998). In addition to increased cell size, cdr2Δ mutants fail to divide according to surface area and instead shift toward volume-based size control, meaning that Cdr1/2-Wee1 signaling links cell surface area with the G2/M transition (Facchetti et al., 2019).
Cdr1 and Cdr2, members of the conserved synapses of amphids defective (SAD) family of protein kinases, play distinct roles in Wee1 inhibition. Cdr1 directly phosphorylates the Wee1 kinase domain to inhibit its catalytic activity (Coleman et al., 1993; Parker et al., 1993; Wu and Russell, 1993; Opalko et al., 2019). Cdr2 does not regulate Wee1 kinase activity directly and instead provides spatial control to this pathway. Cdr2 forms oligomeric “nodes,” which are stable structures tethered to the plasma membrane and positioned in the cell middle (Morrell et al., 2004). Cdr2 recruits both Cdr1 and Wee1 to these sites, and Cdr2 kinase activity correlates with the dwell time of Wee1 at individual nodes (Moseley et al., 2009; Martin and Berthelot-Grosjean, 2009; Allard et al., 2018). Since Cdr2 is progressively activated as cells grow, Wee1 spends more time at nodes as cells grow larger, resulting in its cell size–dependent phosphorylation and inhibition (Deng et al., 2014; Allard et al., 2018). Importantly, this mechanism acts at the plasma membrane, consistent with its role connecting cell surface area with the G2/M transition. Cdr2 nodes also function after mitotic entry by serving as spatial landmarks for cytokinetic ring assembly (Almonacid et al., 2009).
Nodes are critical to Cdr1/2-Wee1 signaling, so it is important to identify mechanisms of node formation and anchoring at the cell cortex. Cdr2 contains a KA1 domain that is required for node formation in cells and binds to lipids, in part through an RKRKR motif (Rincon et al., 2014; Morrell et al., 2004). The Cdr2 KA1 domain also displays some clustering activity, while Cdr2-interacting factors, including anillin-like Mid1, contribute to Cdr2 clustering in a KA1-independent manner (Rincon et al., 2014). These node-forming activities are countered by the protein kinase Pom1, which directly phosphorylates Cdr2 to inhibit interactions with the membrane and Mid1 (Rincon et al., 2014). Pom1 is concentrated at cell tips to restrict Cdr2 node assembly to the medial cell cortex (Bähler and Pringle, 1998; Moseley et al., 2009; Martin and Berthelot-Grosjean, 2009). Understanding how Cdr2 oligomerizes into nodes and interacts with the plasma membrane is crucial to explaining how Wee1 is regulated by cell surface area.
In this study, we identify the conserved GTPase Arf6 as a novel component and regulator of Cdr1/2-Wee1 nodes. Loss of Arf6 impairs signaling to Wee1 and leads to Cdr2 clusters that are detached from the plasma membrane. Genetic experiments indicate that Arf6 anchors nodes at the cortex in parallel with other mechanisms including the Cdr2 KA1 domain and Cdr2–Mid1 interactions. The role of Arf6 in assembling these multicomponent signaling platforms at the plasma membrane has implications for kinase clusters in other systems.
Results and discussion
Arf6 regulates the cell cycle through the Cdr2 pathway
The Cdr2 pathway inhibits Wee1, and cdr1Δ and cdr2Δ mutants are synthetically lethal in combination with the temperature-sensitive cdc25-22 mutation (Russell and Nurse, 1987b; Young and Fantes, 1987; Breeding et al., 1998; Kanoh and Russell, 1998). To screen for new components of Cdr2 signaling (Fig. 1 A), we individually combined a library of 3,004 viable S. pombe deletion mutants (∼75% of the nonessential fission yeast genome) with cdc25-22 using synthetic genetic array (SGA) analysis (as in Roguev et al., 2007; Fig. 1, B and C). As controls, we combined each library deletion with either a cdr2Δ mutant or a cdc25+ wild-type allele. We reasoned that mutations in Cdr2 signaling should exhibit synthetic sick/synthetic lethal (SS/SL) interactions with cdc25-22, but should be nonadditive with cdr2Δ, while cdc25+ controlled for linkage effects. From the resulting double mutants, 66 were SS/SL with only cdc25-22, 31 were SS/SL with only cdr2Δ, and 28 were SS/SL with both cdc25-22 and cdr2Δ (Fig. 1 D and Table S1).
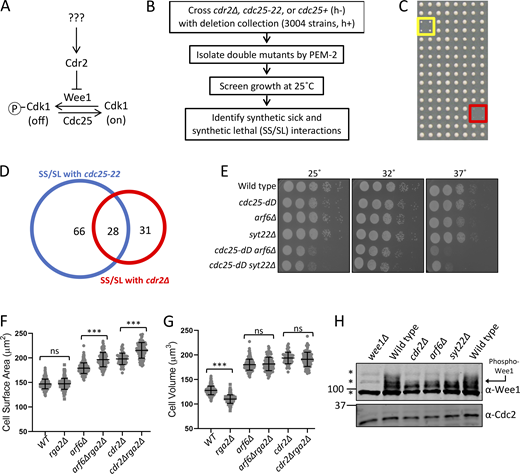
Arf6 regulates cell size through the Cdr2-Wee1 pathway.(A) Schematic of the Cdr2-Wee1 pathway. (B) SGA workflow. (C) Example of SGA growth screening. Strains were spotted in quadruplicate. Yellow box shows slow growth; red box shows no growth. (D) Venn diagram of SGA hits. (E) Serial dilution assay. (F and G) Surface area (F) and volume (G) of dividing cells from the indicated strains. n ≥ 70 cells per strain; ns, P ≥ 0.05; ***, P ≤ 0.001 by one-way ANOVA followed by Tukey’s multiple comparison test. Graphs show mean ± SD. (H) Western blot of whole-cell extracts for endogenous Wee1. Asterisks indicate background bands; Cdc2 is loading control. Source data are available for this figure: SourceData F1.
Arf6 regulates cell size through the Cdr2-Wee1 pathway.(A) Schematic of the Cdr2-Wee1 pathway. (B) SGA workflow. (C) Example of SGA growth screening. Strains were spotted in quadruplicate. Yellow box shows slow growth; red box shows no growth. (D) Venn diagram of SGA hits. (E) Serial dilution assay. (F and G) Surface area (F) and volume (G) of dividing cells from the indicated strains. n ≥ 70 cells per strain; ns, P ≥ 0.05; ***, P ≤ 0.001 by one-way ANOVA followed by Tukey’s multiple comparison test. Graphs show mean ± SD. (H) Western blot of whole-cell extracts for endogenous Wee1. Asterisks indicate background bands; Cdc2 is loading control. Source data are available for this figure: SourceData F1.
Mutants in the Cdr2 pathway exhibit altered cell length at division, and this phenotype is synthetic with mutations in cdc25 (Breeding et al., 1998; Kanoh and Russell, 1998). We were interested to find that arf6Δ was SS/SL with cdc25-22 and increased cell length at division when combined with cdc25-22 (Fig. S1 A). Arf6 is a conserved GTPase linked in animal cells to formation of multivalent protein assemblies at the plasma membrane (Cherfils, 2014), the site of Cdr2 nodes in fission yeast. In growth assays and analysis of cell length at division, arf6Δ was nonadditive with cdr2Δ but showed synthetic defects with the hypomorphic allele cdc25-dD (dD stands for degron-DAmP; Fig. 1 E and Fig. S1, B–D). GTPase signaling is activated by guanine nucleotide exchange factors (GEFs) and inhibited by GTPase-activating proteins (GAPs). Deletion of the Arf6 GEF (syt22Δ) phenocopied arf6Δ genetic interactions with cdr2Δ and cdc25-dD (Fig. S1, B–D). Previous studies identified a role for fission yeast Arf6 and its regulatory GEF (Syt22) and GAP (Ucp3) in bipolar cell growth (Fujita, 2008; Fujita and Misumi, 2009, 2011), but a role in cell cycle progression has not been reported. Our data suggest a novel function for active Arf6 in the Cdr2 cell cycle pathway.

Arf6 regulates cell size at division through Cdr2-Wee1 . (A) Cell length at division measurements, reporting mean ± SD for n > 50 cells each. (B) Serial dilution growth assays. (C and D) Cell length at division for the indicated strains. n > 50 cells each. Graphs show mean ± SD. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison. (E) Cell width measurement for the indicated strains. ****, P ≤ 0.0001 by Welch’s unpaired t test (n > 100 cells for each strain).
Arf6 regulates cell size at division through Cdr2-Wee1 . (A) Cell length at division measurements, reporting mean ± SD for n > 50 cells each. (B) Serial dilution growth assays. (C and D) Cell length at division for the indicated strains. n > 50 cells each. Graphs show mean ± SD. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison. (E) Cell width measurement for the indicated strains. ****, P ≤ 0.0001 by Welch’s unpaired t test (n > 100 cells for each strain).
The cell length at division phenotype for arf6Δ was minor, but these cells were wider than wild type (Fig. S1 E). With the increased width, the enlarged cell size phenotype of arf6Δ was nearly as severe as that of cdr2Δ when comparing cell surface area (Fig. 1 F). Recent work using cell width mutants showed that fission yeast cells divide at a specific surface area, as opposed to length or volume (Facchetti et al., 2019; Pan et al., 2014). In addition, cdr2Δ mutants shift to dividing at a set volume that is larger than dividing wild-type cells (Facchetti et al., 2019). By comparing wild type and rga2Δ, which has reduced cell width (Villar-Tajadura et al., 2008), we confirmed that fission yeast cells divide at a specific surface area, not volume (Fig. 1, F and G). Perhaps more importantly, arf6Δ cells and arf6Δ rga2Δ cells divided at the same volume but at distinct surface areas, similar to cdr2Δ and cdr2Δ rga2Δ (Fig. 1, F and G). Thus, arf6Δ cells (like cdr2Δ) divide at a larger size and shift from surface area–based to volume-based divisions.
The molecular output of the Cdr2 pathway is inhibitory phosphorylation of Wee1, which can be monitored by SDS-PAGE mobility shifts (Allard et al., 2018, 2019; Opalko et al., 2019). The slower-migrating, hyperphosphorylated form of Wee1 was lost in arf6Δ and syt22Δ, similar to cdr2Δ (Fig. 1 H). We conclude that activated Arf6 functions in the Cdr2 pathway to control cell size at division through inhibition of Wee1.
Arf6 localizes to nodes
We tested if Arf6 functions at nodes by examining its localization. Arf6-mNeonGreen (mNG) localized to the plasma membrane as previously shown (Fujita, 2008), but was strongly enriched at cortical nodes in the cell middle (Fig. 2 A). Arf6 and Cdr2 colocalized at nodes (Figs. 2 B and S2 A), identifying Arf6 as a new node component. The Arf6-mNG signal at individual nodes was stable by time-lapse microscopy (Fig. 2 C), similar to Cdr2 and Cdr1 but distinct from Wee1 (Pan et al., 2014; Allard et al., 2018). To test the timing of Arf6 node localization, we tracked mitosis with the spindle pole body marker Sad1 (Hagan and Yanagida, 1995) and tracked cytokinesis with the myosin-II regulatory light chain Rlc1 (Naqvi et al., 2000; Le Goff et al., 2000). Arf6 localized to nodes throughout interphase but left nodes after cells entered mitosis (Fig. 2 D), similar to Cdr2 (Morrell et al., 2004). Arf6 returned to the cell middle during cytokinesis but did not constrict with the ring. Following cell separation, Arf6 reappeared at nodes. Arf6 node localization required Cdr2 but not other node proteins (Figs. 2 E and S2 B). These data show that Arf6 is a component of Cdr2 nodes in addition to functioning in the Cdr2 pathway.

Arf6 is a novel node component. (A and B) Maximum-intensity projection of Airyscan images. Insets show zoom of boxed area. (C) Maximum-intensity projections of deconvolved z series from images taken every 5 min. Scale bar, 3 µm. (D) Localization of Arf6 at different cell cycle stages. Sad1 and Rlc1 mark mitosis and cytokinesis, respectively. (E) Localization of Arf6-mNG in the indicated mutants. Images are sum intensity projections of deconvolved z series. (F and G) Sum intensity projections of deconvolved z series. Scale bars in A, B, and D–G are 5 µm; inset in B is 1 µm.
Arf6 is a novel node component. (A and B) Maximum-intensity projection of Airyscan images. Insets show zoom of boxed area. (C) Maximum-intensity projections of deconvolved z series from images taken every 5 min. Scale bar, 3 µm. (D) Localization of Arf6 at different cell cycle stages. Sad1 and Rlc1 mark mitosis and cytokinesis, respectively. (E) Localization of Arf6-mNG in the indicated mutants. Images are sum intensity projections of deconvolved z series. (F and G) Sum intensity projections of deconvolved z series. Scale bars in A, B, and D–G are 5 µm; inset in B is 1 µm.

Localization, regulation, and function of Arf6 at cortical nodes . (A) Arf6 is a component of most Cdr2 nodes. Each point indicates the percentage of Cdr2 nodes with Arf6 present from a single cell. Bars represent mean ± SD; n = 25 cells. (B) Localization of Arf6 in the indicated mutants. Images are sum intensity projections of deconvolved z series. (C) Maximum-intensity projection of a deconvolved z series. (D) Maximum-intensity projection of a deconvolved z series. Insets show zooms of boxed areas. (E) Maximum-intensity projection of a deconvolved z series. (F) Middle single focal plane images. Insets show zooms of boxed areas. (G) Schematic showing relative conservation of the Arf6 Nter from fission yeast, budding yeast, and humans. (H) Single middle focal plane and maximum-intensity projections (Max Project) from z series. (I) Cytoplasmic fluorescence intensity of the indicated strains. n > 50 cells each. Graphs show mean ± SD. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison. (J) Cdr2 cytoplasmic puncta counted from five middle focal planes. n > 50 cells each. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison (left) or Welch’s unpaired t test (right). (K) Single focal plane images. Red arrows point to Cdr2 at cell tips. (L) Quantification of Cdr2 nodes/spots at cell sides and cell tips. Note the increase in Cdr2 spots at cell tips in arf6Δ cells. Statistical analysis was evaluated by Welch’s unpaired t test, n = 25 cells. (M) Quantification of Cdr2-mEGFP signal per node from the indicated strains. Each point represents a single node. Statistical analysis was evaluated by Welch’s unpaired t test. n = 25 cells. Bars represent mean ± SD; ****, P ≤ 0.0001; *, P ≤ 0.05. Scale bars of insets are 2 µm; all other scale bars are 5 µm.
Localization, regulation, and function of Arf6 at cortical nodes . (A) Arf6 is a component of most Cdr2 nodes. Each point indicates the percentage of Cdr2 nodes with Arf6 present from a single cell. Bars represent mean ± SD; n = 25 cells. (B) Localization of Arf6 in the indicated mutants. Images are sum intensity projections of deconvolved z series. (C) Maximum-intensity projection of a deconvolved z series. (D) Maximum-intensity projection of a deconvolved z series. Insets show zooms of boxed areas. (E) Maximum-intensity projection of a deconvolved z series. (F) Middle single focal plane images. Insets show zooms of boxed areas. (G) Schematic showing relative conservation of the Arf6 Nter from fission yeast, budding yeast, and humans. (H) Single middle focal plane and maximum-intensity projections (Max Project) from z series. (I) Cytoplasmic fluorescence intensity of the indicated strains. n > 50 cells each. Graphs show mean ± SD. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison. (J) Cdr2 cytoplasmic puncta counted from five middle focal planes. n > 50 cells each. Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison (left) or Welch’s unpaired t test (right). (K) Single focal plane images. Red arrows point to Cdr2 at cell tips. (L) Quantification of Cdr2 nodes/spots at cell sides and cell tips. Note the increase in Cdr2 spots at cell tips in arf6Δ cells. Statistical analysis was evaluated by Welch’s unpaired t test, n = 25 cells. (M) Quantification of Cdr2-mEGFP signal per node from the indicated strains. Each point represents a single node. Statistical analysis was evaluated by Welch’s unpaired t test. n = 25 cells. Bars represent mean ± SD; ****, P ≤ 0.0001; *, P ≤ 0.05. Scale bars of insets are 2 µm; all other scale bars are 5 µm.
We next asked how GTP binding and hydrolysis regulate Arf6 localization to nodes. A GDP-locked mutant arf6(T52N)-mNG lost node localization, but the GTP-locked allele arf6(Q75L)-mNG remained at nodes (Fig. 2 F). Further, Arf6 localization to nodes was lost upon deletion of its GEF Syt22 (Fig. 2 G). The Arf6 GAP Ucp3 is essential (Fujita and Misumi, 2011), so we could not test its role in Arf6 localization. If the Arf6 localization defect in syt22Δ is due to loss of the GTP-bound state, then it should be suppressed by arf6(Q75L). Indeed, arf6(Q75L)-mNG localized to nodes even in syt22Δ cells (Fig. 2 G). These data show that GTP binding is required for Arf6 localization to nodes and suggest that Arf6 localizes to nodes in its GTP-bound state. Loss of Arf6 from Cdr2 nodes explains syt22Δ defects in cell size and Wee1 phosphorylation.
Next, we tested if Arf6 localized to nodes with its GEF (Syt22) or GAP (Ucp3). Syt22-mNG localized to cortical puncta as previously shown (Fig. S2 C; Fujita and Misumi, 2009). Syt22 puncta were spread throughout the cell periphery and did not colocalize with Cdr2 (Fig. S2, C and D). Ucp3-mNG localized to spots at the cell tips, which likely represent endocytic actin patches due to colocalization with actin patch component Pan1 (Fig. S2, E and F). We conclude that Syt22 and Ucp3 do not localize to nodes with Arf6, but they regulate Arf6 node localization through its nucleotide-bound state.
Arf6 GTPases have a conserved myristoylated glycine and an α helix that work together to promote membrane binding (Gillingham and Munro, 2007; Fig. S2 G). To test if membrane binding contributes to Arf6 node localization, we made a nonmyristoylated arf6 mutant (G2A) and two stepwise helix deletions (N3-S10Δ and K11-F17Δ), because complete helix deletion abrogated expression. The resulting arf6 alleles reduced node localization and instead enriched at the cytoplasm (Fig. S2, H and I). Therefore, reducing Arf6 membrane binding also reduces Arf6 node localization. Together, these results show that Arf6 localizes stably to Cdr2 nodes during interphase in a manner that depends on nucleotide binding, membrane binding, and Cdr2 itself.
Arf6 promotes cortical tethering of Cdr2 nodes
How does Arf6 promote Cdr2 node function? We imaged Cdr2-mEGFP in arf6Δ cells and observed several node defects. First, arf6Δ cells had cytoplasmic Cdr2 clusters that were absent in wild-type cells (Figs. 3 A and S2 J), indicating defects in cortical anchoring. Second, Cdr2 was present at cell tips in addition to nodes in arf6Δ cells (Fig. S2, K and L). Third, Cdr2 node intensity was reduced in arf6Δ cells (Fig. S2 M). The brightest nodes, which are lacking in arf6Δ cells, are likely diffraction-limited clusters of smaller/unitary nodes (Akamatsu et al., 2017). Fourth, time-lapse imaging revealed reduced stability of Cdr2 signal at some but not all nodes in arf6Δ cells (Figs. 3 B and S3 A). In addition, FRAP experiments showed increased Cdr2 dynamics at nodes of arf6Δ cells consistent with loss of anchoring (Figs. 3 C and S3 B), but slower Cdr2 dynamics at nodes in the GTP-locked mutant arf6(Q75L) consistent with hyperstable anchoring (Fig. S3 C). These defects indicate that Arf6 anchors Cdr2 stably at nodes, meaning that Arf6 and Cdr2 reciprocally promote each other’s node localization.

Cdr2 nodes are disrupted in arf6Δ. (A) Maximum-intensity projections (Max Project) of z series (top) or single middle focal planes (bottom). Red arrows indicate cytoplasmic Cdr2 puncta. (B) Time-lapse imaging of Cdr2-mEGFP from middle focal plane. Montages show 5-min time points from boxed regions. (C) FRAP analysis of Cdr2-mEGFP in wild-type and arf6Δ cells. n = 10 cells each. Points are mean ± SD; *, P ≤ 0.05 by Welch’s unpaired t test. (D) Middle focal plane images of Wee1-mNG. Insets are zooms of yellow boxes. (E) Middle focal plane images. Scale bars for A, B, D, and E are 5 µm; for B inset, 2 µm; and for D inset, 1 µm.
Cdr2 nodes are disrupted in arf6Δ. (A) Maximum-intensity projections (Max Project) of z series (top) or single middle focal planes (bottom). Red arrows indicate cytoplasmic Cdr2 puncta. (B) Time-lapse imaging of Cdr2-mEGFP from middle focal plane. Montages show 5-min time points from boxed regions. (C) FRAP analysis of Cdr2-mEGFP in wild-type and arf6Δ cells. n = 10 cells each. Points are mean ± SD; *, P ≤ 0.05 by Welch’s unpaired t test. (D) Middle focal plane images of Wee1-mNG. Insets are zooms of yellow boxes. (E) Middle focal plane images. Scale bars for A, B, D, and E are 5 µm; for B inset, 2 µm; and for D inset, 1 µm.

Node defects in arf6∆ cells . (A) Quantification of Cdr2-mEGFP dynamics from time-lapse imaging. For single cells, the Pearson correlation coefficient was measured for each time point compared with the initial image (time = 0); n = 10 cells for each strain. A faster rate of decay for the correlation coefficient indicates loss of stability for Cdr2-mEGFP localization. **, P ≤ 0.01; *, P ≤ 0.05 by Welch’s unpaired t test performed for each time point. (B) Examples of FRAP experiment where red boxed region was photobleached and analyzed for recovery. Scale bar is 2 µm. (C) FRAP analysis of Cdr2-mEGFP at nodes in the indicated strains. n = 10 cells each. Points are mean ± SD; *, P ≤ 0.05 by Welch’s unpaired t test performed for each time point. (D) Percentage of cells with Wee1 localization at cortical nodes for wild-type versus arf6Δ strains. Bars indicate mean ± SD; n = 100 cells each. NS, not significant by Welch’s unpaired t test. (E) Middle focal plane images. Insets are zooms of yellow boxes. (F) Fluorescence intensity of line scans along cortex of cells as in E (dashed yellow lines). Asterisks mark node peaks for Cdr2 (red) and Cdr1 (blue). Note overlapping peaks in wild-type but not in arf6Δ cells (two cells each). (G) Middle focal plane images. (H) Middle focal plane images; insets are zooms of yellow boxes. (I–K) Quantification of cell morphology (I), Mid1-Nter localization (J), and Cdr2 localization (K) for the indicated strains. Bars represent mean ± SD from biological triplicate experiments with n > 50 cells each. ****, P ≤ 0.0001 and ***, P ≤ 0.001 by Welch's unpaired t test. (L) Timing of cytokinesis measured with Sad1 and Rlc1. n = 20 cells each, bars represent mean ± SD. (M) Serial dilution growth assay at 37°C (from Fig. 5 C). Main panel scale bars are 5 µm; inset scale bars are 2 µm.
Node defects in arf6∆ cells . (A) Quantification of Cdr2-mEGFP dynamics from time-lapse imaging. For single cells, the Pearson correlation coefficient was measured for each time point compared with the initial image (time = 0); n = 10 cells for each strain. A faster rate of decay for the correlation coefficient indicates loss of stability for Cdr2-mEGFP localization. **, P ≤ 0.01; *, P ≤ 0.05 by Welch’s unpaired t test performed for each time point. (B) Examples of FRAP experiment where red boxed region was photobleached and analyzed for recovery. Scale bar is 2 µm. (C) FRAP analysis of Cdr2-mEGFP at nodes in the indicated strains. n = 10 cells each. Points are mean ± SD; *, P ≤ 0.05 by Welch’s unpaired t test performed for each time point. (D) Percentage of cells with Wee1 localization at cortical nodes for wild-type versus arf6Δ strains. Bars indicate mean ± SD; n = 100 cells each. NS, not significant by Welch’s unpaired t test. (E) Middle focal plane images. Insets are zooms of yellow boxes. (F) Fluorescence intensity of line scans along cortex of cells as in E (dashed yellow lines). Asterisks mark node peaks for Cdr2 (red) and Cdr1 (blue). Note overlapping peaks in wild-type but not in arf6Δ cells (two cells each). (G) Middle focal plane images. (H) Middle focal plane images; insets are zooms of yellow boxes. (I–K) Quantification of cell morphology (I), Mid1-Nter localization (J), and Cdr2 localization (K) for the indicated strains. Bars represent mean ± SD from biological triplicate experiments with n > 50 cells each. ****, P ≤ 0.0001 and ***, P ≤ 0.001 by Welch's unpaired t test. (L) Timing of cytokinesis measured with Sad1 and Rlc1. n = 20 cells each, bars represent mean ± SD. (M) Serial dilution growth assay at 37°C (from Fig. 5 C). Main panel scale bars are 5 µm; inset scale bars are 2 µm.
Node defects in arf6Δ correlate with loss of Wee1 phosphorylation and altered cell size. To explain this connection, we examined the localization of Wee1 and Cdr1 at cortical nodes in arf6Δ mutants. Wee1 localized to nodes in arf6Δ, but Cdr1 did not (Fig. 3, D and E; and Fig. S3 D). Loss of Cdr1 from Cdr2 nodes was most obvious in line traces along the cortex of arf6Δ cdr1-3xGFP cdr2-RFP cells (Fig. S3, E and F). Because the 3xGFP tag on Cdr1 reduces cell size, we also tested Cdr1 localization in arf6Δ cells elongated with the cdc25-dD mutation and observed similar defects (Fig. 3 E). We conclude that loss of node integrity in arf6Δ prevents recruitment of Cdr1, leading to loss of Wee1 inhibitory phosphorylation and to cell size defects.
We next tested how Arf6 connects with three other mechanisms that contribute to Cdr2 node anchoring. First, Cdr2 has a membrane-binding KA1 domain required for cortical localization. Mutation of an RKRKR motif in the KA1 domain reduced Cdr2 node localization (Rincon et al., 2014), and this defect was enhanced by arf6Δ (Fig. S3 G). Second, the node protein Mid1 interacts with Cdr2 to promote clustering into nodes (Rincon et al., 2014). We combined arf6Δ with the mid1(400–450Δ) mutant that cannot bind Cdr2. In the resulting cells, Cdr2 was absent from the cell cortex and formed large cytoplasmic puncta (Figs. 4 A and S2 J). Thus, Arf6 and Mid1 are partially overlapping anchors for Cdr2 nodes. We note that the Cdr2 KA1 domain was still present in these cells but was not sufficient for cortical localization. Third, we combined arf6Δ with mild overexpression of Pom1, which phosphorylates Cdr2 to reduce its membrane binding, catalytic activity, and clustering (Deng et al., 2014; Bhatia et al., 2014; Rincon et al., 2014). P3nmt1-pom1 alone causes mild defects in Cdr2 localization (Moseley et al., 2009), but arf6Δ P3nmt1-pom1 largely eliminated Cdr2 from the cortex and caused localization to cytoplasmic puncta (Figs. 4 B and S2 J). We conclude that Arf6 contributes to Cdr2 cortical anchoring together with parallel mechanisms, most notably Mid1 and Pom1.

Cdr2 node mutants exacerbate arf6Δ phenotypes. (A and B) Cdr2-mEGFP localization in the indicated strains. Single focal planes and maximum-intensity projection (Max Project) images of z series. (C) Montage of no-delay time lapse for single focal plane. Arrow follows a moving Cdr2 punctum. (D) Mobile Cdr2 puncta colocalize with exocytic vesicle marker Syb1. Circles mark colocalizing puncta, and arrows mark moving colocalized punctum. Cells were imaged every 30 s. Scale bar in D is 2 µm; all other scale bars are 5 µm.
Cdr2 node mutants exacerbate arf6Δ phenotypes. (A and B) Cdr2-mEGFP localization in the indicated strains. Single focal planes and maximum-intensity projection (Max Project) images of z series. (C) Montage of no-delay time lapse for single focal plane. Arrow follows a moving Cdr2 punctum. (D) Mobile Cdr2 puncta colocalize with exocytic vesicle marker Syb1. Circles mark colocalizing puncta, and arrows mark moving colocalized punctum. Cells were imaged every 30 s. Scale bar in D is 2 µm; all other scale bars are 5 µm.
By time-lapse microscopy, we observed motile cytoplasmic Cdr2 puncta in 14 of 20 arf6Δ mid1(400–450Δ) cells analyzed, with most movement directed toward cell tips (Fig. 4 C). These Cdr2 puncta colocalized with Syb1, the v-SNARE synaptobrevin that marks exocytic vesicles, as puncta moved toward cell tips in arf6Δ mid1(400–450Δ) (Fig. 4 D). Cdr2 did not colocalize with Syb1 in wild-type cells (Fig. S3 H). These data suggest that Cdr2 becomes enriched on exocytic vesicles when it cannot be anchored at cortical nodes by Arf6 and Mid1.
Arf6 contributes to cytokinesis
Cdr2 contributes to cytokinesis by recruiting anillin-like protein Mid1 to nodes during interphase (Almonacid et al., 2009). The Mid1 N-terminus (Mid1-Nter) is necessary and sufficient for its cytokinesis function, and the localization and function of Mid1-Nter requires Cdr2 (Celton-Morizur et al., 2006; Almonacid et al., 2009). Unlike the mid1(400–450Δ) mutant that cannot interact with Cdr2, Mid1-Nter interacts with Cdr2 and depends on Cdr2 nodes for cytokinesis, meaning that Mid1-Nter provides a system to examine the function of Cdr2 nodes in cytokinesis. Given the Cdr2 node anchoring defects in arf6Δ, we tested the localization and function of Mid1-Nter in arf6Δ. In arf6+ cells, GFP-Mid1-Nter localized to the cortex to support proper cell morphology and division plane positioning (Fig. 5 A). In contrast, GFP-Mid1-Nter mislocalized in arf6Δ cells, which were multinucleated with grossly aberrant septa (Fig. 5 A and Fig. S3, I and J). In addition, Cdr2-mEGFP did not concentrate in medial nodes in arf6Δ mid1-Nter cells (Figs. 5 B and S3 K). These data indicate that Arf6 is required for the localization and function of Mid1-Nter in cytokinesis.

Arf6 promotes robust cytokinesis. (A) Middle focal plane images of GFP-Nter-Mid1. Insets are zooms of black boxes. (B) Cdr2-mEGFP localization in mid1-Nter arf6Δ cells stained with Blankophor. (C) Serial dilution growth assays. (D) Blankophor images of the indicated strains. (E) Quantification of septation defects. Values are means ± SD from three biological replicate experiments. n > 80 cells. (F) Localization of Rlc1 and Sad1 in representative arf6Δ pom1Δ cells. Maximum-intensity projection of a z series. DIC, differential interference contrast. Inset scale bar for A is 2 µm; all other scale bars are 5 µm.
Arf6 promotes robust cytokinesis. (A) Middle focal plane images of GFP-Nter-Mid1. Insets are zooms of black boxes. (B) Cdr2-mEGFP localization in mid1-Nter arf6Δ cells stained with Blankophor. (C) Serial dilution growth assays. (D) Blankophor images of the indicated strains. (E) Quantification of septation defects. Values are means ± SD from three biological replicate experiments. n > 80 cells. (F) Localization of Rlc1 and Sad1 in representative arf6Δ pom1Δ cells. Maximum-intensity projection of a z series. DIC, differential interference contrast. Inset scale bar for A is 2 µm; all other scale bars are 5 µm.
We wondered if this result reflected a broad role for Arf6 in promoting the robustness of cytokinesis. We did not observe defects in the timing of cytokinetic ring formation, maturation, or constriction in arf6Δ cells (Fig. S3 L), so we tested genetic interactions between arf6Δ and several cytokinesis mutants. We combined arf6Δ with cdc4-31, a temperature-sensitive mutant in the myosin-II essential light chain (McCollum et al., 1995), with rng2-D5, a temperature-sensitive mutant in IQGAP (Eng et al., 1998; Chang et al., 1996), or with pom1Δ, which positions the division plane asymmetrically (Bähler and Pringle, 1998). All three double mutants exhibited synthetic defects in cell growth and cytokinesis as judged by tilted and disorganized septa (Fig. 5, C–E; and Fig. S3 M). We attempted to visualize cytokinesis in arf6Δ pom1Δ cells using Rlc1-mNG and Sad1-mEGFP to mark the actomyosin ring and spindle pole bodies, respectively (Fig. 5 F). However, strong disorganization of Rlc1 in these cells prevented meaningful time-lapse microscopy experiments. We conclude that Arf6, like other node proteins, contributes to robust cytokinesis.
Conclusions
Our study identifies the conserved GTPase Arf6 as a new functional component of Cdr2 nodes. Human ARF6 functions in several membrane-localized processes such as endocytosis, lipid homeostasis, and cytokinesis (Schweitzer et al., 2011; Gillingham and Munro, 2007) and has been linked to the proliferation, invasion, and metastasis of multiple cancers (Hashimoto et al., 2004; Li et al., 2017). A general theme of Arf GTPases is the assembly and stabilization of multivalent membrane-bound platforms (Cherfils, 2014). We demonstrated a clear role for Arf6 in organizing nodes for cell size control. We found that Cdr2 nodes lose stability in arf6Δ cells, and unstable nodes fail to recruit Cdr1, leading to reduced Wee1 phosphorylation and cell size defects. These findings suggest that Arf6 promotes stable node coalescence by promoting multivalent protein–protein and protein–lipid interactions. In the absence of this clustered node anchoring, cells lose surface area sensing and shift to volume-based divisions. Arf6 localization to nodes depends on the state of its bound nucleotide under control of the GEF Syt22. It will be interesting to learn how cell growth and expansion control the balanced activities of both Syt22 and the Arf6 GAP Ucp3. Because arf6Δ mutants fail to divide according to cell surface area and instead shift to volume-based division, it seems likely that plasma membrane expansion regulates Arf6 through Syt22 and/or Ucp3 as part of a mechanism to link cell cycle progression with overall cell size.
Materials and methods
Strain construction and media
Standard S. pombe media and methods were used (Moreno et al., 1991). Strains and plasmids used in this study are listed in Table S1. The cdc25-dD mutant reduces Cdc25 levels by addition of a degradation tag (degron) in combination with truncation of the 3′ UTR (Breslow et al., 2008; Deng et al., 2014). Homologous recombination was used for C-terminal tagging and gene deletions as described previously (Bähler et al., 1998). To construct GTP binding and helix mutants of Arf6, the sequence pArf6-Arf6-mNG-Tadh1 was PCR amplified from genomic DNA of strain JM6125. This PCR product was ligated into pDC99 vector using KpnI/SacII sites. Each mutation was introduced using QuikChange II site-directed mutagenesis kit (StrataGene). All constructs were Sanger sequenced for verification. The pDC99 vector was then linearized using NotI and transformed into arf6Δ::natR leu1-32 ura4-D18 (JM6565). Colonies were selected for growth on EMM-Uri medium and no growth on EMM-Leu medium. For serial dilution assays, cells were spotted in 10-fold dilutions on YE4S medium and grown at the indicated temperature for 3–5 d.
SGA screening
We used the PEM-2 approach to combine cdc25-22 (strain JM942), cdr2Δ (strain JM945), and cdc25+ (strain JM943) with a collection of 3,004 fission yeast gene deletions (∼75% of the nonessential S. pombe genome; note that this collection did not include syt22Δ; Kim et al., 2010). For cdc25-22 and cdc25+, the natR cassette was integrated 507 bp downstream of the cdc25 stop codon. These strains contained cycloheximide-sensitive cyhS at the h locus, along with a cycloheximide-resistance allele (cyhR) inserted at the rpl42 locus, for antidiploid selection and mating type selection following mating and sporulation (Roguev et al., 2007, 2008). Mating and haploid selection were performed on a Singer ROTOR pinning station. The resulting double-mutant strains were grown in 1,536 array format on YE5S solid media plates. Colony size was used as a quantitative readout to derive scores covering each genetic interaction (e.g., SS/SL; Kim et al., 2009). Colony size was scored between 0 (no growth) and 3 (wild-type growth). For each deletion mutant, synthetic growth defects with cdc25-22 were assessed by subtracting the double-mutant colony size score when combined with cdc25-22 from that with cdc25+. Similar analysis was done for cdr2Δ. Mutants were considered SS/SL if the synthetic growth defect was −2 or −3. All scores are provided in Table S1.
Microscopy for cell geometry measurements
Fission yeast cells were grown at 25°C in YE4S medium to logarithmic phase for imaging. Cells were collected at 4,000 rpm for 15 s, placed on a coverglass-bottom dish (P35G-1.5-20C; MatTek), and covered with a piece of YE4S agar prewarmed to 25°C. Images were collected using a spinning disk confocal microscope: Yokogawa CSU-WI (Nikon Software) equipped with a 60× 1.4-NA CFI60 Apochromat Lambda S objective lens (Nikon); 405-, 488-, and 561-nm laser lines; and a photometrics Prime BSI camera on an inverted microscope (Eclipse Ti2; Nikon). Multiple fields per cell type were imaged within 1 h at room temperature, and images were acquired with 27 z-stacks and 0.2-µm steps.
Cell segmentation
Bright-field images were processed for cell size analysis using a partly automated pipeline. First using ImageJ, a smoothing filter and Gaussian blur was applied to each optical section of an image to reduce image noise. We performed global thresholding by selecting a gray value cutoff to produce a binary image for each z-section. An optical section outside of the focal plane with intact boundary bands around cells was selected for further processing. The flood fill tool was used (by hand) to generate a black background so that image pixel values in the background were set to 0 and cells in white were 1. Binary images were further processed by morphological erosion and subsequent dilation operators to remove white regions between cell clumps for better single-cell segmentation. The paintbrush tool was used to further separate clumped cells by hand, and the flood fill tool was used to delete any abnormal cells or unresolved clumps of cells. Images were also processed to remove cells along the edge of the image. The resulting binary image (“cell mask”) was compared with the original bright-field image and confirmed to be an accurate representation of cell size. Next, cell masks were compared with corresponding fluorescent mtagBFP2-NLS images to identify cells containing two nuclei, indicating active division. Cells containing only a single nucleus were deleted using the flood fill tool, thus generating a binary mask of only dividing cells.
Cell geometry measurements
For a given cell segmentation, the cell width was determined by hand measurements in ImageJ using the straight-line tool. The average cell width divided by 2 (cell radius) was determined for a population of cells (n > 100). We assumed that each cell in a given strain had the same cell radius (average of population) for calculation of cell surface area and volume (Table 1). In Matlab, the cell length or cell symmetry axis of individual segmented cells (from mask of dividing cells) was identified by principal component analysis of the cloud points internal to the cell (Facchetti et al., 2019). Cell surface area or volume of individual cells was calculated in Matlab using the equation for surface area and volume of a cylinder with hemispherical ends, because of the rod-like shape of fission yeast cells. ANOVA was performed to determine statistical differences between sets of data for cell geometry analyses.
Calculation of cell surface area and volume
| Cell radiusa | R |
| Cell length | L |
| Cell surface area | 2πRL |
| Cell volume | πR2L − 2/3πR3 |
Average over cell population of radius of each single cell measured by hand, with each strain considered separately.
Western blot
For Western blots, 2 ODs of logarithmic phase S. pombe cells were flash frozen in liquid nitrogen. Whole-cell extracts were made by lysing cells in 100 µl sample buffer (65 mM Tris, pH 6.8, 3% SDS, 10% glycerol, 10% 2-mercaptoethanol, 50 mM sodium fluoride, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, and complete EDTA-free protease inhibitor tablet [Pierce]) in a Mini-beadbeater-16 for 2 min at 4°C, and then incubated at 99°C for 5 min. Lysates were briefly centrifuged to pellet insoluble material, and the supernatant was isolated as whole-cell extract. Samples were resolved by reducing SDS-PAGE (10% acrylamide/bis-acrylamide for Cdc2 gels; 6% acrylamide/bis-acrylamide for all others) run at constant 20 mAmps until the 75-kDa marker approached the bottom of the gel. Gels were transferred to nitrocellulose using Trans-blot Turbo Transfer System (Bio-Rad). Wee1 was probed using a rabbit polyclonal antibody directed against S. pombe Wee1 (Allard et al., 2018). Cdc2 was used as a loading control using a mouse monoclonal antibody directed against S. pombe Cdc2 (SC-53217; Santa Cruz Biotechnologies).
Wide-field microscopy
For Fig. 2, E–G and Figs. 5 D, S2 B, and S2 F, cells were imaged at room temperature on a DeltaVision Imaging System (Applied Precision), with an Olympus IX-71 inverted wide-field microscope, a 100× UplanSApo 1.4-NA oil objective, a Photometrics CoolSNAP HQ2 camera, and an Insight solid-state illumination unit. For images with z-stacks, focal planes were imaged with 0.3-µm step size and deconvolved using 10 iterations in SoftWoRx software (Applied Precision) Blankophor was used to mark septating cells for cell length measurements and to highlight septum irregularities (Fig. 5 D). Cell length measurements, along with maximum and sum intensity projection images, were created using ImageJ.
Spinning disc confocal imaging
Two spinning disc confocal imaging systems were used, and cells were imaged at room temperature. The first system was a Nikon Eclipse Ti-E microscope with a Yokogawa, CSU-WI spinning disc system, a Nikon LU-N4 laser launch, and two Photometrics Prime BSI sCMOS cameras. The second system was a Yokogawa CSU-WI with a 100× 1.45-NA CFI Plan Apochromat Lambda objective on an Eclipse Ti2 Nikon base with a Photometrics Prime BSI sCMOS camera (as described above). For Fig. 2 C, cells were imaged every 5 min using 0.4-µm z-stack step size. Images in Fig. 2 C and Fig. S2, C–E, were deconvolved using 3D automatic deconvolution in Nikon Elements software. For time-lapse imaging in Fig. 3 B, cells were imaged on EMM4s agar pads, and middle-focal plane images were acquired every 5 min. For Fig. 4 C, cells were mounted on glass slides, and single focal planes were imaged every 200 ms using continuous acquisition. The delay between frames was 657 ms.
Image analysis and quantification
To count cytoplasmic nodes, z-series at 0.2-µm intervals were taken through the entire cell. Cytoplasmic clusters were counted if present within the middle five focal planes to avoid any cortical clusters. The number of clusters was compared with wild type by one-way ANOVA with Tukey’s multiple comparison test (Fig. S2 J, Mid1 graph) or Welch’s t test (Fig. S2 J, Pom1 graph). Cytoplasmic fluorescence intensity was measured for Fig. S2 I by measuring the mean intensity in a set region of interest (ROI) after background subtracting. Fluorescence intensity of each strain was then compared with wild type by one-way ANOVA with Tukey’s multiple comparison analysis. Timing of cytokinesis in Fig. S3 L was monitored using Sad1-mEGFP and Rlc1-mNG as respective markers of the spindle pole body and cytokinetic ring. Cells were imaged at room temperature on a Mat Tek dish with YE4S agar. 0.6-µm slices through the whole cell were imaged every 3 min.
To quantify Cdr2 node intensity (Fig. S2 M), cells were grown in EMM4S at 25°C and imaged under glass coverslips. 0.3-µm z-sections were taken through the whole cell, and sum projections for the top half of the cell were used for quantification. The ImageJ plugin comdetv5.5 was used to identify spots. An ROI was used, and spots were set to a pixel size of 4 with no intensity threshold. Large particles were set to be included and segmented. Integrated density per node was graphed for 25 cells. To quantify spots on the sides and tips of the cell (Fig. S2 L), single-focal-plane images were taken. As above, comdetv5.5 was used to identify spots. Pixel size was set at 4, intensity was set to 10, and 25 cells were analyzed. Welch’s unpaired t test was used to analyze significance of both node intensity and node number for these experiments.
For colocalization (Fig. S2 A), the comdetv5.5 was used as above to identify spots from a maximum-intensity projection of the top half of the cell (n = 25). The distance between spots was set to 3 pixels, pixel size was set to 3, and intensity threshold was 3 for both channels. Large particles were set to be included and segmented. To quantify Wee1 localization at nodes (Fig. S3 D), middle-focal-plane images were taken of cells grown in EMM4S at 25°C. Spots were counted as nodes when associated with the membrane flanking the nucleus of interphase cells. n = 100 cells for each strain were analyzed in biological triplicate experiments. Welch’s unpaired t test was performed to compare wild-type and arf6Δ cells.
For quantification of cytokinesis defects (Fig. 5 E), cells were stained with Blankophor to highlight the division septum. Phenotypes were marked as normal, multiseptated, asymmetric, and “other,” which included tilted septa, cell wall deposits, and cells that failed to separate but initiated growth at the division site. n > 80 cells for each strain were analyzed in biological triplicate experiments. To quantify Mid1 localization (Fig. S3 J), cells were marked to have nodes if spots were localized to the membrane flanking the nucleus in interphase cells; n > 50 cells were analyzed in biological triplicate experiments. Cells were marked as abnormal (Fig. S3 I) if septa appeared mispositioned or contained more than the normal number of nuclei. For Cdr2 localization (Fig. S3 K), medial Cdr2 nodes were counted if they were found at the plasma membrane around the nucleus. n > 50 cells were analyzed in biological triplicate experiments. Unpaired Welch’s t test was used for both analyses.
Cdr2 duration was quantified using a time course of single middle-focal-plane images in wild-type or arf6Δ cells. Multistackreg was used to correct for cell drift. An ROI was drawn around the entire cell, and Pearson’s correlation coefficient (PCC; Colocalization plugin, Fiji) was used to compare Cdr2 localization in each frame to time point 0. A decrease in PCC over time indicates changes in the localization pattern of nodes. 10 cells were analyzed for each strain. Mean PCC and SD were plotted, and Welch’s unpaired t test was used to compare wild type and arf6Δ within each individual time point. Similar data were obtained with or without background subtraction.
Airyscan imaging and analysis
An LSM880 laser scanning confocal microscope (Zeiss) with a 100× Plan Apochromat 1.46-NA oil objective, an Airyscan superresolution module, GaAsP detectors, and Zen Blue acquisition software (Zeiss) was used to image Arf6 nodes (Fig. 2 A) and colocalization of Cdr2 and Arf6 (Fig. 2 B). For best resolution, 0.17-µm stacks were taken through the entire cell in resolution versus sensitivity mode. Airyscan images were then processed using Zen Blue software.
FRAP
FRAP analysis was performed using the LSM880 laser scanning confocal microscope described above and following previously described methods (Miller et al., 2021). Cells were imaged on a single focal plane in EMM4S (Figs. 3 C and S3 B) or YE4S (Fig. S3 C) at room temperature on lectin-coated μ-slide 18-well chambers (81811; Ibidi). Cells were bleached by drawing an ROI within the node-containing region on one side of the cell as seen in a single middle focal plane. Two prebleach images were taken, and ROIs were bleached to <50% of the original fluorescence intensity. Unbleached cells were used to correct for photobleaching, and background was subtracted from each ROI. The intensity data were fitted using the exponential equation y = m1 + m2 × exp(−m3 × X), where m3 is the off-rate, using Prism 8 (GraphPad Software). The half-time of recovery was calculated using the equation t1/2 = (ln2)/m3. For experiments in Fig. S3 C, both arf6+ and arf6(Q75L) were integrated into the leu1+ locus of arf6Δ cells and expressed by the endogenous promoter.
Statistical analysis
One-way ANOVA followed by Tukey’s multiple comparison test was used to assess differences for Fig. 1, F and G, and Figs. S1 C, S2 I, and S2 J (left panel). This test was selected to compare each mean within an experiment to each other. Unpaired t tests with Welch’s correction were performed for Fig. S1 E; Fig. S2, L and M; Fig. S3 D; and Fig. S3, I–K and for each time point in Fig. 3 C and Fig. S3, A and C. This test was selected to compare two datasets that are not assumed to have equal variance. Data distribution was assumed to be normal, but this was not formally tested. All statistics and graphs were made using Prism8 (GraphPad Software).
Online supplemental material
Fig. S1 relates to Fig. 1 and shows data for Arf6 controlling cell size through the Cdr2 pathway. Fig. S2 shows data for interdependent node localization of Arf6 and Cdr2. Fig. S3 contains characterization and quantification of node defects in arf6Δ cells. Table S1 contains yeast strains used in our study and scores from SGA screens.
Acknowledgments
We thank members of the Moseley laboratory for discussions and comments on the manuscript and A. Paoletti for sharing strains. This project was initiated in the laboratory of Paul Nurse, whom we thank for support and advice.
We thank the Biomolecular Targeting Core (National Institute of General Medical Sciences P20-GM113132) and the Imaging Facility at Dartmouth for use of equipment. This work was supported by grants from the National Institute of General Medical Sciences (R01GM099774 and R01GM133856) to J.B. Moseley.
The authors declare no competing financial interests.
Author contributions: Conceptualization: H.E. Opalko and J.B. Moseley; data acquisition: H.E. Opalko, K.E. Miller, H.-S. Kim, and J.B. Moseley; data analysis: H.E. Opalko, K.E. Miller, H.-S. Kim, C. Vargas-Garcia, A. Singh, M.-C. Keogh, and J.B. Moseley; writing – original draft: H.E. Opalko and J.B. Moseley; writing – review and editing: all authors.

