


The amplitude of Wnt/β-catenin signaling is precisely controlled by the assembly of the cell surface–localized Wnt receptor signalosome and the cytosolic β-catenin destruction complex. How these two distinct complexes are coordinately controlled remains largely unknown. Here, we demonstrated that the signalosome scaffold protein Dishevelled 2 (Dvl2) undergoes liquid–liquid phase separation (LLPS). Dvl2 LLPS is mediated by an intrinsically disordered region and facilitated by components of the signalosome, such as the receptor Fzd5. Assembly of the signalosome is initiated by rapid recruitment of Dvl2 to the membrane, followed by slow and dynamic recruitment of Axin1. Axin LLPS mediates assembly of the β-catenin destruction complex, and Dvl2 attenuates LLPS of Axin. Compared with the destruction complex, Axin partitions into the signalosome at a lower concentration and exhibits a higher mobility. Together, our results revealed that Dvl2 LLPS is crucial for controlling the assembly of the Wnt receptor signalosome and disruption of the phase-separated β-catenin destruction complex.

Introduction
The Wnt/β-catenin signaling pathway governs cell proliferation and cell fate specification, which is essential for early embryogenesis and tissue homeostasis, and deregulation of Wnt/β-catenin signaling is associated with developmental disorders, multiple types of cancer, osteoporosis, and other diseases (Clevers and Nusse, 2012; MacDonald et al., 2009; Rim et al., 2022; Steinhart and Angers, 2018). Wnt/β-catenin signaling is precisely controlled by the β-catenin destruction complex and the receptor signalosome, which are assembled in the cytoplasm and on the plasma membrane (PM), respectively (Gammons and Bienz, 2018; Nusse and Clevers, 2017). In the absence of Wnt ligands, the soluble β-catenin protein is recruited to the Axin-scaffolded destruction complex, which contains adenomatous polyposis coli protein (APC), glycogen synthase kinase 3β (GSK3β), and casein kinase 1α (CK1α). Following recruitment to the destruction complex, β-catenin is phosphorylated, ubiquitylated, and then degraded by the proteasome. Recently, it has been shown that the destruction complex is organized via liquid–liquid phase separation (LLPS) of Axin (Li et al., 2020; Nong et al., 2021; Shi et al., 2021). Upon binding of Wnt ligands to the cell surface, Frizzled (Fzd) receptor and low-density-lipoprotein-related protein 5/6 (LRP5/6) coreceptor, cytosolic Dishevelled (Dvl) is recruited to the PM via its interaction with Fzd (Cliffe et al., 2003) and provides a docking site for Axin (Cliffe et al., 2003). Axin then promotes LRP6 phosphorylation by GSK3β and CK1, which in turn enhances the association of Axin with phosphorylated LRP6 to form the signalosome (Bienz, 2014; Bilic et al., 2007; MacDonald and He, 2012). Upon assembly of the signalosome, the destruction complex is disassembled via an unknown mechanism. Inhibition of Axin homopolymerization and the Axin-APC interaction by Axin-Dvl heteropolymerization has been suggested to contribute to the disruption of the destruction complex (Fiedler et al., 2011). Disruption of the destruction complex ultimately results in increased levels and nuclear accumulation of β-catenin and activation of β-catenin/T cell factor/lymphoid enhancer-binding factor (TCF/LEF) target genes (Bilic et al., 2007; Metcalfe et al., 2010; Nusse and Clevers, 2017).
Among the three Dvl homologs in humans and mice (Dvl1-3), Dvl2 is significantly more abundant (Gao and Chen, 2010; Sharma et al., 2018). Dvl proteins possess three conserved domains that can mediate multiprotein interactions: an N-terminal DIX domain responsible for Dvl polymerization and Dvl–Axin1 interaction, a central PDZ domain that directly interacts with Fzd, and a C-terminal DEP domain (Gao and Chen, 2010; Sharma et al., 2018; Shi and Chen, 2021). In addition to these structured domains, Dvl2 also contains intrinsically disordered regions (IDRs; Schaefer and Peifer, 2019; Shi et al., 2021). Dvl forms puncta in a manner that depends on its DIX domain (Axelrod et al., 1998; Miller et al., 1999; Schwarz-Romond et al., 2005; Yang-Snyder et al., 1996). The formation of Dvl puncta has been suggested to be crucial for signalosome formation (Bienz, 2014). Upon Wnt stimulation, endogenous Dvl forms oligomers containing <10 Dvl2 molecules on the PM, as revealed by TIRF imaging (Kan et al., 2020; Ma et al., 2020). Dvl oligomerization was suggested to be regulated by the binding affinity of Dvl2 for Fzd (Ma et al., 2020). The mechanism by which Dvl couples the assembly of the signalosome to the disassembly of the destruction complex remains largely unknown.
LLPS has recently been shown to drive the assembly of biomolecular condensates, facilitating biochemical reactions and signaling events (Alberti et al., 2019; Wang and Zhang, 2019). Dvl possesses characteristic features for LLPS, such as multivalent protein-interacting motifs and IDRs (DeBruine et al., 2017; Schaefer and Peifer, 2019), and has been predicted to undergo phase separation (Sear, 2007). In this study, we demonstrate that Dvl2 forms endogenous condensates through IDR-mediated LLPS. Dvl2 LLPS initiates the assembly of the membrane-associated signalosome, a process that is enhanced by Wnt stimulation. By single-molecule tracking in combination with mass action model prediction, we found that the recruitment of Dvl2 to the PM is rapid and stable and is followed by Axin1 recruitment. Accumulation of Axin1 in the signalosome is relatively slow and unstable compared with Dvl2. Phase-separated Dvl2 disrupts the destruction complex by attenuating Axin1 LLPS and also by promoting the recruitment of Axin into the signalosome. Our study reveals a critical role of Dvl LLPS in coordinating the assembly of the Wnt receptor signalosome and disruption of the destruction complex.
Results and discussion
Dvl2 undergoes phase separation
Dvl2 has been shown to form punctate structures driven by head-to-tail polymerization when overexpressed in cells (Axelrod et al., 1998; Miller et al., 1999; Yang-Snyder et al., 1996). To examine the dynamic distribution of endogenous Dvl2, we generated Dvl2-mEGFP knock-in (KI) HEK293T cells by fusing mEGFP to the C-terminus of the endogenous Dvl2 protein using CRISPR/Cas9-mediated genome editing. Similar to WT HEK293T cells, the clones with one or two knock-in alleles responded to Wnt3a stimulation in activating the downstream reporter (Fig. S1 A), which indicates that mEGFP insertion did not impair Dvl2 function. Confocal microscopy analysis revealed diffuse Dvl2–mEGFP in the cytoplasm with one to two large and stable puncta (Fig. S1 A), which may be associated with the MTOC/centrosome (Cervenka et al., 2016; Kan et al., 2020; Seto and Bellen, 2006). We then used grazing incidence structured illumination microscopy (GI-SIM) to examine the spatiotemporal expression of Dvl2-mEGFP (Guo et al., 2018). Compared with confocal microscopy, GI-SIM provides a higher spatiotemporal resolution of 95 nm and 266 frames per second, and also its axial illumination depth reaches ∼1 µm. GI-SIM analysis revealed that in addition to one to two large puncta (the ones detected by confocal microscopy), endogenous Dvl2-mEGFP formed many small punctate structures (Fig. 1 A). Their sizes ranged from 0.2 to 0.5 μm, and their abundance significantly increased under Wnt3a stimulation (Fig. 1 A). The high fluorescence background in the cytoplasm likely explains why the distinct punctate structures were not detected by confocal microscopy. The fluorescence signal of endogenous Dvl2-mEGFP quickly recovered to about 20% after photobleaching (FRAP), and the recovery rate was increased after Wnt3a stimulation (Fig. 1 B). When EGFP-tagged Dvl2 was expressed in Dvl1/2/3 knockout (KO) HEK293T cells at a level that was about two times higher than the endogenous protein, it also formed punctate structures with a larger size and higher mobility compared with endogenous Dvl2 (Fig. 1, C and D; and Fig. S1, B and C). The results suggest that Dvl2 forms dynamic condensates in living cells.
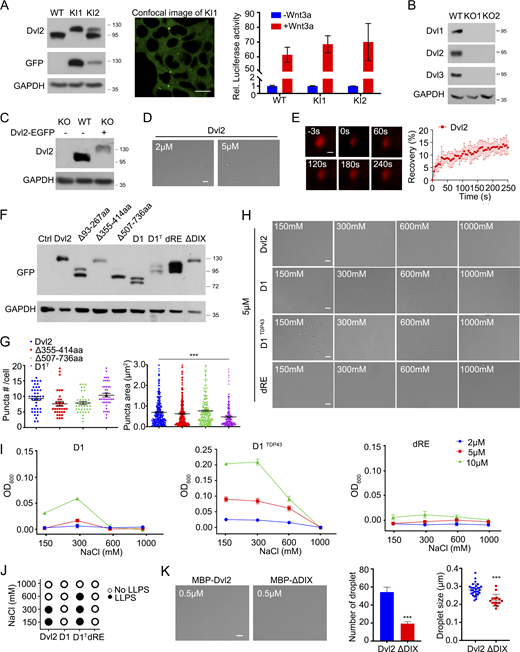
Dvl2 undergoes LLPS. (A) Verification of Dvl2-mEGFP knock-in (KI) HEK293T cells by immunoblotting (left) and confocal imaging (middle). Two KI lines are shown, KI1 (KI in both alleles) and KI2 (KI in one allele). The TopFlash-luciferase reporter were transfected into WT and Dvl2-mEGFP KI cells (KI1 and KI2) and then treated with or without Wnt3a (right). (B) Verification of Dvl1/2/3 knock out (KO) HEK293T cells examined by immunoblotting. Two KO lines are shown as KO1 and KO2. (C) Levels of endogenous Dvl2 in WT HEK293T cells and Dvl2-EGFP expressing Dvl1/2/3 KO HEK293T cells. (D) Droplet formation by 2 µM (left) and 5 µM (right) Dvl2 without PEG8000. (E) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl2 droplets (red) induced without crowder. (F) The expression level of Dvl2 WT and mutant proteins detected by immunoblotting. (G) Statistical analysis of puncta number and puncta area of Dvl2 WT and mutant proteins in Dvl1/2/3 KO cells. (H) DIC images of 5 µM Dvl2 and its mutants at different NaCl concentrations after induction by 2.5% PEG8000. (I) Turbidity assay of Dvl2 mutants after induction of LLPS by 2.5% PEG8000 in different protein and salt concentrations. (J) LLPS of Dvl2 and its mutants at different NaCl concentrations after induction by 2.5% PEG8000 as assessed by DIC imaging. (K) Droplet formation by 0.5 µM purified Dvl2 or ΔDIX with 10% PEG8000. Statistical analyses were performed with the two-tailed unpaired t test. Quantitative data in G are shown as mean ± SEM (n = 3). ***, P < 0.001. Quantitative data in K are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 10 µm; in D, H and K, 2 µm; in E, 0.2 µm. Source data are available for this figure: SourceData FS1.
Dvl2 undergoes LLPS. (A) Verification of Dvl2-mEGFP knock-in (KI) HEK293T cells by immunoblotting (left) and confocal imaging (middle). Two KI lines are shown, KI1 (KI in both alleles) and KI2 (KI in one allele). The TopFlash-luciferase reporter were transfected into WT and Dvl2-mEGFP KI cells (KI1 and KI2) and then treated with or without Wnt3a (right). (B) Verification of Dvl1/2/3 knock out (KO) HEK293T cells examined by immunoblotting. Two KO lines are shown as KO1 and KO2. (C) Levels of endogenous Dvl2 in WT HEK293T cells and Dvl2-EGFP expressing Dvl1/2/3 KO HEK293T cells. (D) Droplet formation by 2 µM (left) and 5 µM (right) Dvl2 without PEG8000. (E) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl2 droplets (red) induced without crowder. (F) The expression level of Dvl2 WT and mutant proteins detected by immunoblotting. (G) Statistical analysis of puncta number and puncta area of Dvl2 WT and mutant proteins in Dvl1/2/3 KO cells. (H) DIC images of 5 µM Dvl2 and its mutants at different NaCl concentrations after induction by 2.5% PEG8000. (I) Turbidity assay of Dvl2 mutants after induction of LLPS by 2.5% PEG8000 in different protein and salt concentrations. (J) LLPS of Dvl2 and its mutants at different NaCl concentrations after induction by 2.5% PEG8000 as assessed by DIC imaging. (K) Droplet formation by 0.5 µM purified Dvl2 or ΔDIX with 10% PEG8000. Statistical analyses were performed with the two-tailed unpaired t test. Quantitative data in G are shown as mean ± SEM (n = 3). ***, P < 0.001. Quantitative data in K are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 10 µm; in D, H and K, 2 µm; in E, 0.2 µm. Source data are available for this figure: SourceData FS1.

Dvl2 undergoes LLPS. (A) GI-SIM images of Dvl2-mEGFP-KI cells with or without Wnt3a stimulation. The puncta number was counted from three independent cells. (B) FRAP showing the recovery of an area of Dvl2-mEGFP in Dvl2-mEGFP KI cells. (C) Confocal image showing Dvl2-EGFP puncta in Dvl1/2/3 KO HEK293T cells. The nuclei were counterstained by DAPI (blue). (D) FRAP showing the recovery of the fluorescent signal in Dvl2-EGFP puncta in Dvl1/2/3 KO HEK293T cells. (E) Formation of droplets by 0.2 µM (left) and 0.5 µM (right) Dvl2 protein with 10% PEG8000. (F) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl2 droplets (red) induced by 10% PEG8000. (G) Differential interference contrast (DIC) images showing the fusion of Dvl2 droplets. (H and I) Dvl2 LLPS induced by 2.5% PEG8000 in different protein and salt concentrations. DIC image analysis (H) and turbidity measurement (I) were shown. (J) Schematic diagram of Dvl2 and its mutants. (K) Confocal images of EGFP-tagged proteins in Dvl1/2/3 KO cells. Statistical analyses were performed with the two-tailed unpaired t test. Data in A and B are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 25 µm and insets in A, 5 µm; in C and K, 10 µm; in E, 2 µm; in F and G, 0.2 µm.
Dvl2 undergoes LLPS. (A) GI-SIM images of Dvl2-mEGFP-KI cells with or without Wnt3a stimulation. The puncta number was counted from three independent cells. (B) FRAP showing the recovery of an area of Dvl2-mEGFP in Dvl2-mEGFP KI cells. (C) Confocal image showing Dvl2-EGFP puncta in Dvl1/2/3 KO HEK293T cells. The nuclei were counterstained by DAPI (blue). (D) FRAP showing the recovery of the fluorescent signal in Dvl2-EGFP puncta in Dvl1/2/3 KO HEK293T cells. (E) Formation of droplets by 0.2 µM (left) and 0.5 µM (right) Dvl2 protein with 10% PEG8000. (F) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl2 droplets (red) induced by 10% PEG8000. (G) Differential interference contrast (DIC) images showing the fusion of Dvl2 droplets. (H and I) Dvl2 LLPS induced by 2.5% PEG8000 in different protein and salt concentrations. DIC image analysis (H) and turbidity measurement (I) were shown. (J) Schematic diagram of Dvl2 and its mutants. (K) Confocal images of EGFP-tagged proteins in Dvl1/2/3 KO cells. Statistical analyses were performed with the two-tailed unpaired t test. Data in A and B are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 25 µm and insets in A, 5 µm; in C and K, 10 µm; in E, 2 µm; in F and G, 0.2 µm.
To explore the possibility of Dvl2 phase separation in vitro, we purified maltose-binding protein (MBP)-tagged Dvl2 protein and incubated it in a buffer containing 150 mM NaCl and 20 mM HEPES (pH 7.4). Dvl2 (0.5 μM) formed droplets after induction with 10% polyethylene glycol (PEG) 8000 (Fig. 1 E), which indicates that Dvl2 undergoes LLPS. Dvl droplets exhibited dynamic properties as shown by FRAP analysis with sulfo-cyanine3 maleimide-labeled Dvl2 proteins: the fluorescent signal recovered to 15% within 3 min after photobleaching (Fig. 1 F). Moreover, two encountering Dvl2 droplets fused and coalesced into a larger sphere (Fig. 1 G). Differential interference contrast (DIC) microscopy imaging revealed that Dvl2 LLPS induced by 2.5% PEG8000 was sensitive to protein concentration and salt concentration (Fig. 1 H), which was further confirmed by a turbidity assay at OD600 (Fig. 1 I). Moreover, after cleavage of the MBP tag by PreScission protease, Dvl2 at the higher concentration of 5 μM formed droplets in the absence of PEG8000 (Fig. S1 D), and the fluorescent signal was recovered to 15% within 4 min after photobleaching (Fig. S1 E). Nonetheless, these results together demonstrate that Dvl2 undergoes phase separation and the droplets display liquid-like properties in vitro.
The intrinsically disordered region (IDR) of Dvl2 mediates LLPS
To assess which region in Dvl2 is critical for LLPS, we constructed a series of deletion mutants (Fig. 1 J) and examined their expression and subcellular distribution in Dvl1/2/3 KO HEK293T cells (Fig. 1 K and Fig. S1 F). As shown in Fig. 1 K and Fig. S1 G, the Dvl2 mutant lacking IDR1 (Δ93–267aa) failed to form puncta, while the mutants with deletion of IDR2 (Δ355–414aa) or IDR3 (Δ507–736aa) exhibited no significant difference in their puncta formation compared with WT Dvl2 in cells. This indicates that IDR1 plays an essential role in Dvl2 punctum formation. The mutant D1, with a deletion of 141–259aa in IDR1, and the mutant dRE, with a deletion of 20 charged residues spanning two arginine-rich domains and one glutamate-rich domain (Δ160-167/210–215/235-240aa), exhibited diffuse distribution (Fig. 1 K). Replacement of the 141–259aa region with the LLPS-driving C-terminal domain (267–414aa) of TDP43 (D1TDP43) restored punctum formation, while the puncta size of D1TDP43 was smaller than WT Dvl2 (Fig. 1 K and Fig. S1 G). Thus, the 141–259aa region in IDR1, especially the charged residues, is essential for Dvl2 LLPS. Consistent with this, D1 and dRE failed to form droplets in in vitro LLPS assays, while WT Dvl2 and D1TDP43 did form droplets (Fig. S1 H). Turbidity measurements confirmed these results (Fig. S1, I and J).
The DIX domain of Dvl was shown to be responsible for the polymerization of Dvl and its interaction with the Axin DAX domain (Fiedler et al., 2011). Accordingly, the DIX-deleted Dvl2 mutant (ΔDIX) exhibited a diffuse distribution when overexpressed in Dvl1/2/3 KO HEK293T cells (Fig. 1 K). However, the purified Dvl2 ΔDIX mutant protein still formed droplets, albeit less efficiently than WT Dvl2 in vitro (Fig. S1 K). Taken together, these results show that IDR1, especially the charged residues in IDR1, mediates Dvl2 LLPS, and the DIX domain facilitates this process.
The endogenous assembly process of signalosomes visualized by TIRF
Wnt3a stimulation promotes the formation of the signalosome comprised of Fzd, LRP5/6, Dvl2, Axin, and other components (Bienz, 2014; Bilic et al., 2007; DeBruine et al., 2017; MacDonald and He, 2012). To visualize the recruitment of Dvl2 to the receptor complex on the PM, we imaged Dvl2-mEGFP KI HEK293T cells with the TIRF-SIM mode of a multi-SIM microscope (Guo et al., 2018). Endogenous Dvl2 puncta were observed on the membrane, and Wnt3a treatment significantly enhanced the number and intensity of the membrane Dvl2 puncta (Fig. 2 A). This indicates that Wnt stimulation enhances the membrane recruitment and condensate formation of Dvl2.

Dvl2 LLPS is crucial for signalosome organization. (A) TIRF-SIM images of Dvl2-mEGFP-KI cells with or without Wnt3a stimulation. The puncta number was counted from three independent cells. The relative intensity of membrane-associated Dvl2-mEGFP puncta was normalized to the group without Wnt3a treatment. (B and C) The average intensity of membrane-associated Dvl2-mEGFP (B) and Axin1-tdTomato (C) puncta detected by TIRF-SIM. (D) The dissociation constant of RD (Wnt receptor/Dvl2 complex) and Dvl versus dissociation constant of RD and Axin. Scatter plot comparing dissociation constant of solutions that fit the experimental dynamic averaged Dvl2 intensity data. (E) Average intensity of Dvl puncta from the 106 fitted modeling solutions at different concentration of Axin. The ODE modeling was solved at the time interval of 20 min after Wnt3a treatment; red dash line marked the absolute total Axin level of 590 nM in the cell. (F) Formation of 0.5 µM Dvl2 droplets with or without 0.5 µM purified Fzd5 C-terminal fragment (522–585aa). Droplet formation was induced with 10% PEG8000. (G and H) Formation of droplets by mixtures containing 0.2 μM of the indicated components with 10% PEG8000. D: Dvl2; A: Axin1; F: Fzd5 C-terminal fragment; L: LRP6 C-terminal fragment; dRE: Dvl2(dRE). (I) Labeled Axin1, Dvl2, Fzd-C-terminal fragment, and unlabeled LRP6-C-terminal fragment were mixed (0.5 µM each) for LLPS assay (top row); labeled Axin1, Dvl2, LRP6-C-terminal fragment and unlabeled Fzd5-C-terminal fragment were mixed (0.5 µM each) for LLPS assay (bottom row). (J)Dvl1/2/3 KO HEK293T cells expressing Dvl2-EGFP or Dvl2(dRE)-EGFP were treated with Wnt3a stimulation. Quantification data showing the size and intensity of membrane-associated puncta detected by TIRF-SIM. The data were normalized to that in 0 min. (K) Axin1-mCherry was expressed alone or with untagged Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO HEK293T cells with Wnt3a stimulation. Quantification data showing the size of membrane-associated puncta detected by TIRF-SIM. The data corresponding to other time points were normalized to that in 0 min. Statistical analyses were performed with the two-tailed unpaired t test. Data in A, F, J and K are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 10 µm; in F, G, H and I, 2 µm; in J and K, 5 µm; and insets in F, G, and H, 0.5 µm.
Dvl2 LLPS is crucial for signalosome organization. (A) TIRF-SIM images of Dvl2-mEGFP-KI cells with or without Wnt3a stimulation. The puncta number was counted from three independent cells. The relative intensity of membrane-associated Dvl2-mEGFP puncta was normalized to the group without Wnt3a treatment. (B and C) The average intensity of membrane-associated Dvl2-mEGFP (B) and Axin1-tdTomato (C) puncta detected by TIRF-SIM. (D) The dissociation constant of RD (Wnt receptor/Dvl2 complex) and Dvl versus dissociation constant of RD and Axin. Scatter plot comparing dissociation constant of solutions that fit the experimental dynamic averaged Dvl2 intensity data. (E) Average intensity of Dvl puncta from the 106 fitted modeling solutions at different concentration of Axin. The ODE modeling was solved at the time interval of 20 min after Wnt3a treatment; red dash line marked the absolute total Axin level of 590 nM in the cell. (F) Formation of 0.5 µM Dvl2 droplets with or without 0.5 µM purified Fzd5 C-terminal fragment (522–585aa). Droplet formation was induced with 10% PEG8000. (G and H) Formation of droplets by mixtures containing 0.2 μM of the indicated components with 10% PEG8000. D: Dvl2; A: Axin1; F: Fzd5 C-terminal fragment; L: LRP6 C-terminal fragment; dRE: Dvl2(dRE). (I) Labeled Axin1, Dvl2, Fzd-C-terminal fragment, and unlabeled LRP6-C-terminal fragment were mixed (0.5 µM each) for LLPS assay (top row); labeled Axin1, Dvl2, LRP6-C-terminal fragment and unlabeled Fzd5-C-terminal fragment were mixed (0.5 µM each) for LLPS assay (bottom row). (J)Dvl1/2/3 KO HEK293T cells expressing Dvl2-EGFP or Dvl2(dRE)-EGFP were treated with Wnt3a stimulation. Quantification data showing the size and intensity of membrane-associated puncta detected by TIRF-SIM. The data were normalized to that in 0 min. (K) Axin1-mCherry was expressed alone or with untagged Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO HEK293T cells with Wnt3a stimulation. Quantification data showing the size of membrane-associated puncta detected by TIRF-SIM. The data corresponding to other time points were normalized to that in 0 min. Statistical analyses were performed with the two-tailed unpaired t test. Data in A, F, J and K are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bars in A, 10 µm; in F, G, H and I, 2 µm; in J and K, 5 µm; and insets in F, G, and H, 0.5 µm.
To trace the signalosome assembly on the PM, both Dvl2-mEGFP KI cells and Axin1-tdTomato KI cells (Fig. S2 A) were imaged by TIRF after Wnt3a stimulation (Fig. S2, B and C). We analyzed the intensity distribution of the fluorescent spots during the monitoring period, followed by a Gaussian mixture modeling fitting to remove the noise signal and establish an average intensity value. The results showed that the fluorescence intensity of membrane Dvl2 increased rapidly and reached a stable state at 7 min of Wnt3a treatment (Fig. 2 B and Fig. S2 B). This is consistent with a previous report (Ma et al., 2020). Membrane Axin1 started to accumulate at 7 min of Wnt3a treatment, and the fluorescence intensity increased more slowly and reached a maximum at 15 min of Wnt3a treatment (Fig. 2 C and Fig. S2 C). Axin1 began to appear at the membrane when the level of Dvl2 was stable (about 7 min after Wnt3a treatment), which indicates delayed recruitment of Axin1 to the membrane compared with Dvl2. Moreover, within 26–40 min of Wnt3a treatment, the fluorescence intensity of Dvl2 was still stable on the PM, while Axin1 showed a gradual decline starting at 30 min.
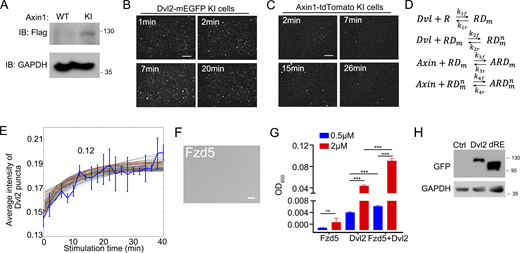
Visualizing the endogenous signalosome assembly process at the single-molecule level by TIRF. (A) Validation of Axin1-Flag-tdTomato KI HEK293T cells (Axin1-tdTomato KI cells) by immunoblotting. (B) Time-lapse TIRF-SIM images showing the membrane localization of Dvl2 puncta in Dvl2-mEGFP KI cells under Wnt3a stimulation. (C) Time-lapse TIRF-SIM images showing membrane localization of Axin1 puncta in Axin1-tdTomate KI cells under Wnt3a stimulation. (D) The binding interactions between the Wnt receptor (R), Dvl, and Axin for signalosome formation. RD, receptor-Dvl complex; ARD, Axin-receptor-Dvl complex. m represents the Dvl on the membrane, and n represents multiple Dvl in the system. (E) Average intensity of Dvl2-mEGFP puncta compared to the constrained and fitted well modeling solutions. (F) DIC images of 0.5 μM purified intracellular domain of Fzd5 (522–585aa) that were induced with 10% PEG8000. (G) Turbidity of purified Dvl2 (0.5 μM, 2 μM) with or without the same concentrations of intracellular domain of Fzd5 (0.5 μM, 2 μM) induced be 10% PEG8000. (H) Dvl2 WT and dRE have the similar expression in cells. The expression level of Dvl2 WT and dRE was detected with immunoblotting. Statistical analyses were performed with the two-tailed unpaired t test. Quantification data are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bar in F, 2 µm. Source data are available for this figure: SourceData FS2.
Visualizing the endogenous signalosome assembly process at the single-molecule level by TIRF. (A) Validation of Axin1-Flag-tdTomato KI HEK293T cells (Axin1-tdTomato KI cells) by immunoblotting. (B) Time-lapse TIRF-SIM images showing the membrane localization of Dvl2 puncta in Dvl2-mEGFP KI cells under Wnt3a stimulation. (C) Time-lapse TIRF-SIM images showing membrane localization of Axin1 puncta in Axin1-tdTomate KI cells under Wnt3a stimulation. (D) The binding interactions between the Wnt receptor (R), Dvl, and Axin for signalosome formation. RD, receptor-Dvl complex; ARD, Axin-receptor-Dvl complex. m represents the Dvl on the membrane, and n represents multiple Dvl in the system. (E) Average intensity of Dvl2-mEGFP puncta compared to the constrained and fitted well modeling solutions. (F) DIC images of 0.5 μM purified intracellular domain of Fzd5 (522–585aa) that were induced with 10% PEG8000. (G) Turbidity of purified Dvl2 (0.5 μM, 2 μM) with or without the same concentrations of intracellular domain of Fzd5 (0.5 μM, 2 μM) induced be 10% PEG8000. (H) Dvl2 WT and dRE have the similar expression in cells. The expression level of Dvl2 WT and dRE was detected with immunoblotting. Statistical analyses were performed with the two-tailed unpaired t test. Quantification data are shown as mean ± SD (n = 3). ***, P < 0.001. Scale bar in F, 2 µm. Source data are available for this figure: SourceData FS2.
Our TIRF microscopy analysis quantified the intensity of both Dvl2 and Axin1 on the PM during Wnt stimulation and revealed that there was a lag time between the recruitment of Dvl2 and Axin1 on the PM (Fig. 2, B and C). This suggests that Axin1 recruitment may require polymerization of Dvl2 on the membrane, and the membrane association of Dvl2 may be more stable than that of Axin1. A recent study showed that in response to Wnt3a, the Fzd-Dvl binding affinity is enhanced, which promotes the oligomerization of Dvl on the membrane (Ma et al., 2020). With mathematical modeling, we attempted to explain the changes and interactions of both membrane-associated Dvl2 and Axin1 after Wnt stimulation. The ordinary differential equations (ODE) were formulated to describe the interaction of the proteins by a simple application of the law of mass action. Theoretically, the complex formed by Wnt receptors and Dvl (RD) in an initial response to Wnt3a can subsequently interact with more Dvl or with Axin. Thus, there are four essential binding reactions in the model regulating the membrane-associated concentration of Dvl and Axin (Fig. S2 D). To simulate the above system, we need the following model parameters: (1) dissociation constant of cytosolic Dvl binding with the receptor (k1r/k1f); (2) dissociation constant of cytosolic Dvl binding with the Dvl on the membrane (k2r/k2f); (3) dissociation constant of Axin recruited to the membrane when one Dvl binding to the receptor (k3r/k3f); and (4) dissociation constant of Axin recruited to the membrane when more Dvl binding on the membrane (k4r/k4f). We selected the dissociation constant randomly within the physiological ranges at log space (10−2 to 101). Then we performed 100,000 computational screens to simulate the above binding process with random searched dissociation constant for different time intervals. We compared the membrane-associated Dvl concentration in the model for each simulation with our experimental measurements. We calculated the sum of squared errors (SSE) to measure the variation of modeling errors. Our screen generated 106 model solutions (SSE < 0.02) that fit well with the average dynamic intensity of Dvl puncta by TIRF (Fig. S2 E). The model predicts that the RD-Dvl dissociation constant is smaller than the RD-Axin dissociation constant in most of the solutions (76.86%; SSE < 0.08; Fig. 2 D). This suggests that the binding affinity of RD to Dvl is higher and RD is inclined to further associate with Dvl instead of Axin, which is consistent with the rapid accumulation of membrane Dvl revealed by TIRF imaging.
We further investigated the effect of Axin on the intensity of Dvl puncta. We calculated the average Dvl punctum intensity after Wnt3a stimulation for 20 min from the 106 model solutions, which fitted well with the experimental data, at different total Axin concentrations. Interestingly, increasing the Axin concentration between 100 and 1,000 nM attenuates the intensity of Dvl2 puncta (Fig. 2 E). The endogenous Axin concentration was quantified as 590 nM through mass spectrometry analysis (red dash line in Fig. 2 E). Thus, a fluctuation of Axin level in the physiological conditions may impact the Dvl puncta intensity. The TIRF images combined with the mass action model prediction indicate that Dvl2 accumulation on the membrane is an initial and important step for signalosome assembly followed by Axin1 recruitment.
Dvl2 LLPS promotes signalosome assembly
The above data indicate that Dvl plays a critical role in signalosome assembly. Next, we attempted to address if LLPS is important for the function of Dvl in this process. As Wnt stimulation induces the interaction of Dvl with Fzd (Tauriello et al., 2012), we first examined if Fzd5 could promote Dvl2 droplet formation. Purified Dvl2 was incubated with the intracellular domain (522–585aa) of Fzd5, and then phase separation was induced. Although the intracellular domain of Fzd5 did not form droplets (Fig. S2 F), we found that Dvl2 droplets became larger and more abundant and the turbidity was increased in the presence of Fzd5 (Fig. 2 F and Fig. S2 G), which indicates that Fzd5 promotes Dvl2 LLPS. As the membrane-associated Dvl2 provides a platform for the interaction of LRP and Axin to assemble the signalosome (Nusse and Clevers, 2017), we then added purified Axin1 and the intracellular domain of LRP6 to the droplets formed by Dvl2 and the intracellular domain of Fzd5. The endogenous protein concentration of Axin1 is 0.6 μM in HEK293T cells (Nong et al., 2021). By synthesizing three peptides of Dvl2 as standards for mass-spectrometry quantification, the endogenous Dvl2 protein concentration was assessed as 0.2 μM. Dvl2 failed to form droplets at 0.2 μM in the presence of 10% PEG8000 as the inducer, and the addition of 0.2 μM of the intracellular domains of Fzd5 and LRP6 had no significant effect (Fig. 2 G). In contrast, the addition of 0.2 μM WT Axin1 protein to the mixture triggered droplet formation. Similarly, Axin1 failed to form droplets at 0.2 μM, but the droplet formation was greatly facilitated by the presence of the intracellular domains of Fzd5, LRP6, and Dvl2, but not with Dvl2(dRE) (Fig. 2 H). The addition of labeled Dvl2 and Axin1 together with the intracellular domains of Fzd5 or LRP6 showed that these proteins were recruited to the droplets (Fig. 2 I). These results suggest that Dvl2 LLPS is critical to organize the droplet formation, which reflects the simplified assembly of the signalosome in vitro. We then examined whether Dvl2 LLPS also contributes to the recruitment of Dvl2 and Axin1 on the PM, which is the critical step for signalosome assembly in vivo. We expressed Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO cells with a comparable expression level (Fig. S2 H), and the membrane-associated puncta were analyzed by TIRF. Under Wnt3a stimulation, Dvl2-EGFP formed more and larger PM-associated puncta, while Dvl2(dRE)-EGFP failed to be recruited to the PM (Fig. 2 J). We then coexpressed Axin1-mCherry with Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO cells, and the TIRF images were used to measure Axin1 recruitment to the PM under Wnt stimulation. Dvl2 enhanced the Axin1 puncta number and size under Wnt stimulation (Fig. 2 K), while Dvl2(dRE)-EGFP showed no effect on Axin1 recruitment. These results reveal that Dvl2 LLPS contributes to the assembly of the signalosome.
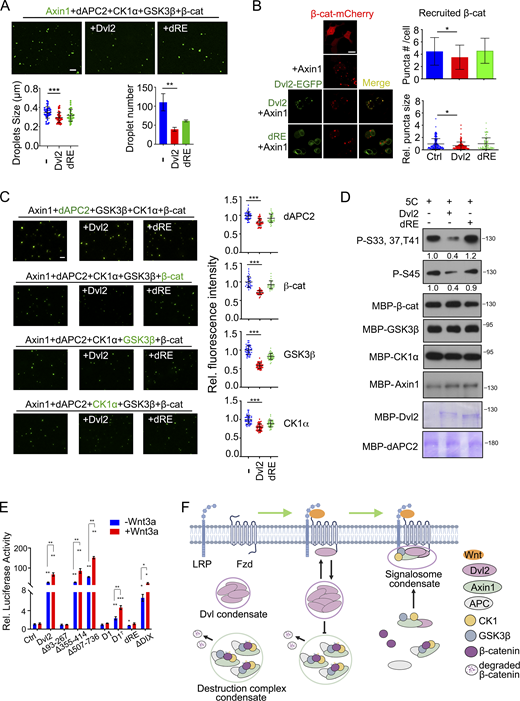
Dvl2 modulates the biophysical properties of Axin1
In addition to its important role in mediating signalosome formation, Dvl has been reported to disrupt the β-catenin destruction complex (Fiedler et al., 2011; Sharma et al., 2018; Shi and Chen, 2021). Axin scaffolds the assembly of the destruction complex via its LLPS property, which is promoted by APC (Nong et al., 2021). We attempted to address whether Dvl2 regulates Axin1 LLPS and whether the LLPS property of Dvl2 is important for this regulation. Dvl2-EGFP and Axin1-mCherry both formed puncta in Dvl1/2/3 KO HEK293T cells, and they were colocalized when coexpressed (Fig. 3 A). Dvl2(dRE)-EGFP was still diffusely distributed even when coexpressed with Axin1. WT Dvl2, but not Dvl2(dRE), reduced the number and the internal mobility of Axin1-mCherry puncta (Fig. 3 A and Fig. S3 B). Moreover, under Wnt3a stimulation, WT Dvl2 reduced the size and relative fluorescence intensity of Axin1-mCherry puncta in the cytoplasm (Fig. S3 A). The internal mobility and puncta number of Axin1-mcherry were reduced by Wnt3a stimulation in WT HEK293T but not Dvl1/2/3 KO cells (Fig. S3, C and D). These results indicate that Dvl2 suppresses Axin1 condensate formation via LLPS, and this suppressive function is potentiated by Wnt stimulation. To confirm the inhibitory effect of Dvl on Axin condensate formation, we mixed purified Axin1 protein (0.5 μM, close to its endogenous level) with 0.5 μM Drosophila APC2 (dAPC2) or 0.5 μM Dvl2 in in vitro LLPS assays. dAPC2 was used as it is difficult to express human APC due to its huge size, and dAPC has been shown to be functional in suppressing Wnt/β-catenin signaling (Roberts et al., 2012). As shown in Fig. 3 B, dAPC2 promoted Axin1 LLPS, while Dvl2 attenuated both the size and number of Axin1 droplets. Moreover, Dvl2 further abolished dAPC2-enhanced Axin1 LLPS. Dvl2 also decreased the internal mobility of Axin1 droplets (Fig. S3 E). Importantly, Dvl2(dRE), which exhibited an impaired droplet-forming ability (Fig. S3 F), had no significant impact on the formation and dynamics of Axin1 droplets (Fig. 3 B and Fig. S3 E), indicating that the LLPS property of Dvl2 is critical for attenuating the droplet-forming ability of Axin1. The turbidity assay also supported that Dvl2 attenuated the Axin1 LLPS (Fig. S3 G).

Dvl2 modulates the biophysical properties of Axin1 condensates. (A) Confocal images of Axin1-mCherry puncta when EGFP-tagged Dvl2 or Dvl2(dRE) were coexpressed in Dvl1/2/3 KO cells. The puncta size was normalized to that in the control group. (B) 0.5 µM Axin1 droplet formation induced by 10% PEG8000 after incubation with 0.5 µM dAPC2 and 0.5 µM Dvl2 or dRE. Quantification of droplet size and number are shown below. (C) TIRF images showing the membrane-associated Axin1-EGFP puncta in Axin1 KO cells with or without Wnt3a stimulation. (D) FRAP analysis of the membrane-associated Axin1-EGFP puncta detected by TIRF. (E) FRAP analysis of Alexa Fluor 488 NHS ester labeled Axin1 droplets incubated with or without the indicated components. The recovery of the fluorescent signal in Axin1 droplets was shown in the right. Statistical analyses were performed with the two-tailed unpaired t test. Data are shown as mean ± SD (n = 3). *, P < 0.05, **, P < 0.01, ***, P < 0.001. Scale bars in A and C, 10 µm; B, 2 µm; E, 0.2 µm.
Dvl2 modulates the biophysical properties of Axin1 condensates. (A) Confocal images of Axin1-mCherry puncta when EGFP-tagged Dvl2 or Dvl2(dRE) were coexpressed in Dvl1/2/3 KO cells. The puncta size was normalized to that in the control group. (B) 0.5 µM Axin1 droplet formation induced by 10% PEG8000 after incubation with 0.5 µM dAPC2 and 0.5 µM Dvl2 or dRE. Quantification of droplet size and number are shown below. (C) TIRF images showing the membrane-associated Axin1-EGFP puncta in Axin1 KO cells with or without Wnt3a stimulation. (D) FRAP analysis of the membrane-associated Axin1-EGFP puncta detected by TIRF. (E) FRAP analysis of Alexa Fluor 488 NHS ester labeled Axin1 droplets incubated with or without the indicated components. The recovery of the fluorescent signal in Axin1 droplets was shown in the right. Statistical analyses were performed with the two-tailed unpaired t test. Data are shown as mean ± SD (n = 3). *, P < 0.05, **, P < 0.01, ***, P < 0.001. Scale bars in A and C, 10 µm; B, 2 µm; E, 0.2 µm.

Dvl2 LLPS regulates the mobility of Axin1 and β-catenin. (A) Statistical analysis of the size and intensity of Axin1-mCherry puncta when Dvl2-EGFP or Dvl2(dRE)-EGFP was co-expressed in Dvl1/2/3 KO cells with Wnt stimulation. (B) FRAP analysis of Axin1-mCherry puncta in the cytoplasm when Dvl2-EGFP or Dvl2(dRE)-EGFP was co-expressed in Dvl1/2/3 KO cells. (C) Statistical analysis of overexpressed Axin1-mcherry puncta size in WT or Dvl1/2/3 KO HEK293T cells with or without Wnt3a stimulation for 30 min. Quantitative data are shown as mean ± SEM (n = 3). *, P < 0.05. (D) FRAP showing the recovery of the fluorescent signal in Axin1-mcherry puncta in WT or Dvl1/2/3 KO HEK293T cells with or without Wnt3a stimulation for 30 min. (E) FRAP analysis of Axin1 droplets labeled with Alexa Fluor 488 NHS ester with or without Dvl2 or the Dvl2(dRE) mutant. (F) Droplet formation of 0.5 μM Dvl2 and dRE induced by 10% PEG8000. (G) Turbidity assay of 2 μM Axin1 after incubation with indicated proteins. A: Axin1, P: dAPC2, D: Dvl2, dRE: Dvl2(dRE). (H) FRAP analysis of Axin1 puncta in Axin1-tdTomato KI cells localized in the cytoplasm or near the PM with or without Wnt3a stimulation. (I) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl droplets (red) with or without the indicated components. (J) FRAP analysis of β-cat-mCherry puncta when Axin1 and Dvl2-EGFP or Dvl2(dRE)-EGFP were co-expressed in Dvl1/2/3 KO cells. (K) FRAP analysis of Alexa Fluor 488 NHS ester labeled β-cat (green) recruited to destruction complex with or without Dvl2 or the Dvl2(dRE). Statistical analyses were performed with the two-tailed unpaired t test. Quantitative data are shown as mean ± SD (n = 3). *, P < 0.05, **, P < 0.01; ***, P < 0.001. Scale bars in B, F, and J, 2 µm; in E, I and K, 0.2 µm.
Dvl2 LLPS regulates the mobility of Axin1 and β-catenin. (A) Statistical analysis of the size and intensity of Axin1-mCherry puncta when Dvl2-EGFP or Dvl2(dRE)-EGFP was co-expressed in Dvl1/2/3 KO cells with Wnt stimulation. (B) FRAP analysis of Axin1-mCherry puncta in the cytoplasm when Dvl2-EGFP or Dvl2(dRE)-EGFP was co-expressed in Dvl1/2/3 KO cells. (C) Statistical analysis of overexpressed Axin1-mcherry puncta size in WT or Dvl1/2/3 KO HEK293T cells with or without Wnt3a stimulation for 30 min. Quantitative data are shown as mean ± SEM (n = 3). *, P < 0.05. (D) FRAP showing the recovery of the fluorescent signal in Axin1-mcherry puncta in WT or Dvl1/2/3 KO HEK293T cells with or without Wnt3a stimulation for 30 min. (E) FRAP analysis of Axin1 droplets labeled with Alexa Fluor 488 NHS ester with or without Dvl2 or the Dvl2(dRE) mutant. (F) Droplet formation of 0.5 μM Dvl2 and dRE induced by 10% PEG8000. (G) Turbidity assay of 2 μM Axin1 after incubation with indicated proteins. A: Axin1, P: dAPC2, D: Dvl2, dRE: Dvl2(dRE). (H) FRAP analysis of Axin1 puncta in Axin1-tdTomato KI cells localized in the cytoplasm or near the PM with or without Wnt3a stimulation. (I) FRAP analysis of Sulfo-Cyanine3 maleimide labeled Dvl droplets (red) with or without the indicated components. (J) FRAP analysis of β-cat-mCherry puncta when Axin1 and Dvl2-EGFP or Dvl2(dRE)-EGFP were co-expressed in Dvl1/2/3 KO cells. (K) FRAP analysis of Alexa Fluor 488 NHS ester labeled β-cat (green) recruited to destruction complex with or without Dvl2 or the Dvl2(dRE). Statistical analyses were performed with the two-tailed unpaired t test. Quantitative data are shown as mean ± SD (n = 3). *, P < 0.05, **, P < 0.01; ***, P < 0.001. Scale bars in B, F, and J, 2 µm; in E, I and K, 0.2 µm.
Although Dvl2 decreased the Axin1 dynamics, the internal mobility of Axin1 puncta in the cytoplasm was not further affected by Wnt3a stimulation (Nong et al., 2021). Therefore, we next attempted to investigate whether the mobility of membrane-associated Axin1 differs from that of cytoplasm-localized Axin1. We expressed Axin1-EGFP in Axin1 KO cells with or without Wnt3a stimulation and then measured the mobility of membrane-associated puncta by TIRF. The FRAP assays revealed that membrane-associated Axin1-EGFP puncta exhibited higher dynamic after Wnt3a stimulation (Fig. 3, C and D). Moreover, the FRAP assay in Axin1-tdTomato KI cells also showed that the fluorescence near the PM recovered more quickly than the fluorescence in the cytoplasm after Wnt3a stimulation (Fig. S3 H). We also measured the mobility of Axin1 when mixed with Dvl2 and the intracellular domains of Fzd5 and LRP6. The results revealed that the mobility of Axin1 was reduced in the presence of Dvl2 (Fig. 3 E), which is consistent with the above data (Fig. S3 E). However, Axin1 exhibited increased mobility after adding the intracellular domains of Fzd5 and LRP6 to the mixture of Axin1 and Dvl2 in vitro (Fig. 3 E), which is consistent with the membrane-associated Axin1 that displays high dynamics in vivo (Fig. 3, C and D). Furthermore, we measured the dynamics of Dvl2 droplets after mixing with Axin1 and the intracellular domains of Fzd5 and LRP6. The results showed that Axin1 failed to modulate the mobility of Dvl2 (Fig. S3 I). Thus, the mobility of Axin1 is higher in the signalosome than in itself phase-separated condensates in the cytoplasm.
Dvl2 LLPS disrupts the organization and function of the destruction complex
Phase-separated Axin1/APC condensates concentrate β-catenin, CK1α, and GSK3β into droplets in vitro, recapitulating the formation of the destruction complex (Nong et al., 2021). To determine whether Dvl2 disrupts the organization of the destruction complex in vitro, we premixed Alexa Fluor 488 NHS ester -labeled Axin1, dAPC2, GSK3β, CK1α, and β-catenin with or without WT Dvl2 or Dvl2(dRE) and subsequently induced phase separation. Droplet formation was significantly impaired after Dvl2 addition, with fewer and smaller droplets formed (Fig. 4 A). Dvl2(dRE) still reduced the formation of droplets but less effectively, presumably, because it retains its DIX domain, which is critical for Dvl2 polymerization.
Dvl2 LLPS disrupts the organization and function of the β-catenin destruction complex. (A) Alexa Fluor 488 NHS ester labeled 1 µM Axin1 protein (green) and other destruction complex components (1 µM) were mixed with or without Dvl2 or the dRE deletion and subjected to in vitro LLPS assay. The number and size of droplets were counted from three independent fields. (B) Confocal images of β-catenin-mCherry puncta when Axin1 was expressed with or without Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO cells. The number and size of puncta were counted from three independent cells. The puncta size was normalized to that in the control group. (C) Destruction complex components were mixed with or without Dvl2 and the Dvl2(dRE) for in vitro LLPS assay. In each assay, only one protein was labeled with Alexa Fluor 488 NHS ester. Fluorescence images (left) and the fluorescence intensity of the labeled protein recruited into the Axin1 droplets (right) are shown. 5C: Axin1, dAPC, β-catenin, CK1α, and GSK3β. (D) Dvl2 protein or the Dvl2(dRE) mutant (1 µM) were incubated with 1 µM Axin1, 50 nM GSK3β and 2.5 µM CK1α for in vitro phosphorylation. The band intensity of phosphorylated β-catenin was normalized to total β-catenin protein. 5C: Axin1, dAPC, β-catenin, CK1α and GSK3β. (E)Dvl1/2/3 KO cells were transfected with TopFlash-luciferase reporter and WT Dvl2-EGFP or its mutants, then treated with or without Wnt3a conditional medium for 12 h and harvested for determination of luciferase activity. The expression level of Dvl2 WT and mutant proteins in the reporter assay was detected with immunoblotting (bottom panel). (F) Working model of Dvl2 LLPS in assembly of the Wnt receptor signalosome and disruption of the β-catenin destruction complex. Statistical analyses were performed with the two-tailed unpaired t test. Data are shown in A, B, C, and E as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars in B, 10 µm; A and C, 2 µm. Source data are available for this figure: SourceData F4.
Dvl2 LLPS disrupts the organization and function of the β-catenin destruction complex. (A) Alexa Fluor 488 NHS ester labeled 1 µM Axin1 protein (green) and other destruction complex components (1 µM) were mixed with or without Dvl2 or the dRE deletion and subjected to in vitro LLPS assay. The number and size of droplets were counted from three independent fields. (B) Confocal images of β-catenin-mCherry puncta when Axin1 was expressed with or without Dvl2-EGFP or Dvl2(dRE)-EGFP in Dvl1/2/3 KO cells. The number and size of puncta were counted from three independent cells. The puncta size was normalized to that in the control group. (C) Destruction complex components were mixed with or without Dvl2 and the Dvl2(dRE) for in vitro LLPS assay. In each assay, only one protein was labeled with Alexa Fluor 488 NHS ester. Fluorescence images (left) and the fluorescence intensity of the labeled protein recruited into the Axin1 droplets (right) are shown. 5C: Axin1, dAPC, β-catenin, CK1α, and GSK3β. (D) Dvl2 protein or the Dvl2(dRE) mutant (1 µM) were incubated with 1 µM Axin1, 50 nM GSK3β and 2.5 µM CK1α for in vitro phosphorylation. The band intensity of phosphorylated β-catenin was normalized to total β-catenin protein. 5C: Axin1, dAPC, β-catenin, CK1α and GSK3β. (E)Dvl1/2/3 KO cells were transfected with TopFlash-luciferase reporter and WT Dvl2-EGFP or its mutants, then treated with or without Wnt3a conditional medium for 12 h and harvested for determination of luciferase activity. The expression level of Dvl2 WT and mutant proteins in the reporter assay was detected with immunoblotting (bottom panel). (F) Working model of Dvl2 LLPS in assembly of the Wnt receptor signalosome and disruption of the β-catenin destruction complex. Statistical analyses were performed with the two-tailed unpaired t test. Data are shown in A, B, C, and E as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars in B, 10 µm; A and C, 2 µm. Source data are available for this figure: SourceData F4.
We then determined whether Dvl2 regulates the recruitment of β-catenin to destruction complex condensates scaffolded by Axin1 LLPS in vivo. β-Catenin-mCherry was coexpressed with untagged Axin1. β-Catenin-mCherry was recruited to the puncta formed by Axin1, but the number of puncta and the intensity of the recruited β-catenin were significantly attenuated by Dvl2 coexpression in Dvl1/2/3 KO HEK293T cells (Fig. 4 B). Moreover, the internal mobility of β-catenin was specifically elevated by WT Dvl2 coexpression, while Dvl2(dRE) had no effect on the intensity and mobility of β-catenin recruited to destruction complex condensates (Fig. S3 J).
We also investigated the effect of Dvl2 on the assembly of the destruction complex in vitro. Each of the five component proteins (Axin1, dAPC, β-catenin, CK1α, and GSK3β) was individually dye-labeled and mixed with the other four unlabeled proteins in the presence or absence of Dvl2 or Dvl2(dRE). Labeled protein was found in the droplets (Fig. 4 C), as shown previously (Nong et al., 2021). WT Dvl2 apparently reduced the fluorescence intensity of labeled protein, whereas Dvl2(dRE) had a weaker effect (Fig. 4 C). Moreover, WT Dvl2 increased the mobility of β-catenin recruited to droplets (Fig. S3 K). These results indicate that Dvl2 disrupts the organization of the destruction complex by reducing Axin1 LLPS and thus attenuating the recruitment of other components into the Axin1 condensates. The inhibitory effect of Dvl2 requires its phase separation property. This conclusion was further supported by the analysis of the in vitro phosphorylation of β-catenin. Purified β-catenin was mixed with Axin1, dAPC2, GSK3β, and CK1α, and phase separation was induced with 2.5% PEG8000 dissolved in phosphorylation assay buffer with or without Dvl2 or Dvl2(dRE) for 30 min. As shown in Fig. 4 D, WT Dvl2 decreased β-catenin phosphorylation at S33, S37, and T41 by GSK3β or at S45 by CK1α, while Dvl2(dRE) had no effect.
The phase separation property of Dvl is essential to mediate Wnt/β-catenin signaling
To determine the function of Dvl2 LLPS in Wnt/β-catenin signaling, we performed the TOPFlash-luciferase reporter assay in Dvl1/2/3 KO HEK293T cells, which do not respond to Wnt3a stimulation. Expression of Dvl2 significantly increased Wnt3a-induced luciferase expression, while Dvl2(D1) and Dvl2(dRE) failed to activate Wnt signaling (Fig. 4 E). In accordance with the above data that the C-terminal domain of TDP43 restored LLPS of Dvl2(D1), D1TDP43 also exhibited partial function in mediating Wnt signaling. The weaker signal activity of D1TDP43 may be due to self-association and protein aggregation with poor dynamics of the CTD domain of TDP43 (Carter et al., 2021). These results indicate that the LLPS property of Dvl2 is critical to mediate Wnt/β-catenin signaling.
Conclusion
Here, we showed that endogenous Dvl2 undergoes phase separation and exhibits an initial and rapid accumulation to the plasma membrane followed by Axin1 recruitment. We also revealed that Dvl2 LLPS is crucial for mediating the assembly of signalosome condensates and disruption of the destruction complex condensates (Fig. 4 F).
Three Dvl homologs possess conserved domains that can mediate multiprotein interactions (Shi and Chen, 2021). Both Dvl1 and Dvl3 also include three IDRs. Especially, IDR1 of Dvl1 or Dvl3 contains two arginine-rich domains and one glutamate-rich domain, exhibiting a similar composition to the Dvl2 IDR1 region. Owing to the similarities of IDR region in the three Dvl homologs, Dvl1 and Dvl3 might also undergo LLPS mediated by charged residues. The protein-interacting DIX domain also contributes to Dvl2 LLPS. The DIX domain-mediated polymerization of Dvl2 has been reported to increase the local concentration of membrane Dvl2 to facilitate ligand binding (Schwarz-Romond et al., 2007). However, the endogenous Dvl concentration (0.2 μM) is extremely lower than the one for homomeric DIX–DIX affinity (5–20 μM) as assessed with purified protein (Ma et al., 2020; Schwarz-Romond et al., 2007; Yamanishi et al., 2019). Therefore, the combined structured and weak interactions drive Dvl LLPS, the IDR provides the basis for electrostatic effects and other weak interactions, and modular domains such as DIX mediate direct interactions with high binding avidity (Bienz, 2020).
Several posttranslational modifications (PTMs) including phosphorylation, methylation, acetylation, and ubiquitination have been shown to change the charge, hydrophobicity, structure, and other properties of the proteins and affect the phase separation behavior (Luo et al., 2021). Dvl undergoes multiple PTMs including phosphorylation, ubiquitination, methylation, and acetylation that affect Dvl conformation, subcellular localization, stability, and biological functions (Shi and Chen, 2021), respectively. It is worth noting that some PTMs targeting the charged residues can regulate the polymerization of Dvl homologs. Enhancing K63-linked ubiquitination at its DIX domain can inhibit the polymerization of Dvl, and K68-linked acetylation promotes Dvl2-puncta formation (Shen et al., 2020). Moreover, highly conserved Lys in the DIX and PDZ domains which are subject to posttranslational acetylation is critical for Dvl1 subcellular localization and its protein-binding capacity (Sharma et al., 2021). As we have shown that the electrostatic force created by these charged residues in IDR1 mediates Dvl2 LLPS, whether PTMs affect the phase separation behavior of Dvl2 is an interesting issue for future studies.
Wnt stimulation promotes the disassembly of the destruction complex (Kim et al., 2013; MacDonald and He, 2012). Previously, it was proposed that Dvl blocks the scaffold function of Axin by copolymerizing with Axin to reduce Axin self-association (Fiedler et al., 2011) or by interfering with the recruitment of Axin and APC (Mendoza-Topaz et al., 2011). The cytoplasmic destruction complex is organized by Axin LLPS (Nong et al., 2021). Our data reveal that Dvl2 inhibits Axin1 LLPS and suppresses the recruitment of other components to the destruction complex condensate scaffolded by Axin1. Instead, the signalosome, scaffolded by Dvl, recruits Axin. Compared with the destruction complex, a lower concentration of Axin is required for partitioning into droplets in an in vitro system containing Dvl2 and the intracellular domains of Fzd5 and LRP6. Moreover, Axin in the signalosome is more dynamic than that in the destruction complex condensates in the cytoplasm. It has been reported that Wnt-induced inhibition of GSK3β tips the balance toward Axin dephosphorylation by protein phosphatase 1 (Kim et al., 2013). Dephosphorylated Axin adopts an inactivated (“closed”) state and becomes incompetent for interacting with LRP6, which is thus freed for another round of recruitment of phosphorylated-activated Axin (Kim et al., 2013). Thus, phosphorylated Axin1 may be continuously recruited to the membrane during the signalosome assembly. Consistently, the endogenous assembly of the signalosome visualized by TIRF revealed that Axin1 polymer is unstable and can dissociate from the signalosome. Hence, a high dynamic of Axin1 may contribute to the processes of recruitment to and dissociation from the signalosome. Wnt stimulation may change the scaffolding role of Axin1 in the destruction complex to a flexible client in signalosome assembly along with the modulation of phosphorylation (Alberti et al., 2019). Previous data have shown that Wnt-induced dephosphorylation of Axin by PP2A contributes to the dissociation of Axin from the destruction complex and the release of β-catenin (Kofron et al., 2007; Liu et al., 2005; Mariotti et al., 2017). Phosphorylation may regulate Axin LLPS and result in its relocation from the cytoplasm to the PM. Therefore, the distinct properties of Axin in different condensates provide a mechanism for coordinating the assembly of the Wnt receptor signalosome and the disruption of the β-catenin destruction complex.
In this study, the in vitro LLPS assays did not include all known components of the signalosome and the destruction complex. We also realized that it is hard to clarify the driving force of the droplets induced in the mixture containing both Axin1 and Dvl2, and detect whether the formation of the droplets represents the disassembling destruction complex or the newly assembled signalosome. Thus, the in vitro simple reconstitution by phase separation system should be used in conjunction with cell-based assays to determine the process of destruction complex disassembly and signalosome assembly at the endogenous level.
Materials and methods
Antibodies
The antibodies used in this study were purchased from the indicated suppliers: rabbit monoclonal anti-Dvl2 (3216; Cell Signaling Technology), mouse monoclonal anti-Dvl1 (sc-8025; Santa Cruz Technology), mouse monoclonal anti-Dvl3 (sc-8027; Santa Cruz Technology), mouse monoclonal anti-β-catenin (SC7963; Santa Cruz Technology), rabbit monoclonal anti-CK1α (ab108296; Abcam), rabbit monoclonal anti-GSK3β (12456; Cell Signaling Technology), rabbit monoclonal anti-β-catenin (S45; 9564; Cell Signaling Technology), rabbit monoclonal anti-β-catenin (S33,37,T41; 2009; Cell Signaling Technology), rabbit monoclonal anti-Axin1 (2074; Cell Signaling Technology), mouse monoclonal anti-flag (bsm-33346M; Bioss Antibodies), and mouse monoclonal anti-GFP (scc8334; Santa Cruz Technology).
Cell culture, plasmids, and transfection
Human embryonic kidney HEK293T cells were obtained from ATCC (CRL-3216TM). HEK293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS; Gibco) at 37°C in a humidified 5% CO2 incubator. Cell transfection was conducted with VigoFect (Vigorous Biotechnology) or Lipofectamine 2000 (Invitrogen). All plasmids used in this study were generated by subcloning corresponding cDNAs into HA-pcDNA3.1 (Invitrogen) or pETMBP.3C vectors (a gift from Dr. Hong Zhang, Institute of Biophysics, Chinese Academy of Sciences).
Gene editing with CRISPR/Cas9
To create Dvl1/2/3 KO cell lines, HEK293T cells were transfected with PX459 (#48139; Addgene)-based plasmid encoding Cas9 and sgRNAs 5′-CTACATTGGCTCCATCATGA-3′ targeting Dvl1, 5′-TCGGCGATTTCAAGAGCGTC-3′ targeting Dvl2, and 5′-GTCACCTTGGCGGACTTTAA-3′ targeting Dvl3. Dvl1/2/3 KO clones were screened by immunoblotting and confirmed by genomic DNA sequencing. Two clones (KO1 and KO2) were obtained and used in this study.
To create Dvl2-mEGFP knock-in cells lines, HEK293T cells were transfected with PX459-based plasmids encoding Cas9 and sgRNAs 5′-GAGCAAGCACATGACGGCC-3′ and 5′-GTGCTTGCTCTTACAGTGCC-3′ targeting the 3′ UTR region of Dvl2. Dvl2-mEGFP KI clones were screened by immunoblotting and confirmed by genomic DNA sequencing. Two clones (KI1 and KI2) were obtained and used in this study.
To create the Axin1-Flag-tdtomato knock-in cell line, HEK293T cells were transfected with PX459-based plasmids encoding Cas9 and the sgRNA 5′-CATCGGCAAAGTGGAGAAGG-3′ targeting the stop codon region of Axin1. Axin1-tdtomato KI clones were screened by immunoblotting and confirmed by genomic DNA sequencing. One clone was obtained and used in this study.
Immunoblotting, coimmunoprecipitation, immunofluorescence, pulldown, and reporter assays
These experiments were performed as previously reported (Gao et al., 2010). All genes were PCR-amplified and cloned into the HA-pcDNA3.1 vector (for mammalian cell expression) to produce HA-tagged recombinant proteins. To express EGFP- or mCherry-fused proteins, the EGFP or mCherry cDNA was fused to the 3′ end of target cDNAs, and therefore, the expressed proteins were tagged with HA at the N-terminus and EGFP or mCherry at the C-terminus. TDP43 CTD domain (267–414aa) were PCR-amplified and cloned into HA-D1-GFP-pcDNA3.1 vector to produce TDP43 CTD domain-fused D1 proteins (D1TDP43). Dvl1/2/3 deletions (D1 and dRE) were introduced (D1: del 141–259; dRE: del 160–167, 210–215, and 235–240 within 97–267aa from IDR1).
For immunoblotting, the cells were lysed on ice with lysis solution (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40, 2 mM EDTA, 1 mM NaVO3, 10 mM NaF and protease inhibitors) and rotated for 30 min at 4°C. The lysates were subjected to SDS-PAGE, and immunoblotting was performed with primary antibodies as indicated, and secondary anti-rabbit (31460; Thermo Fisher Scientific) or anti-mouse (31430; Thermo Fisher Scientific) antibodies conjugated to horseradish peroxidase, followed by detection with enhanced chemiluminescent substrate (Pierce) according to the manufacturer’s instructions, making use of the BioTrace NT Nitrocellulose Transfer Membrane (66485; Pall Life Sciences). For immunofluorescence, cells transfected with the indicated plasmids were washed two times with PBS, fixed with 4% formaldehyde solution for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked in 5% BSA in PBS for 30 min at room temperature. Then 4′, 6-diamidino-2-phenylindole (DAPI) was added for 1 h, and the images were obtained using the NIKON A1 HD25 microscope equipped with a 100X/1.45 NA oil objective at 1,024 × 1,024 pixels. Images were analyzed with NIS-Elements AR Analysis. For reporter assays, cells were transfected with TopFlash-luciferase reporter (D. Xi He) and Renilla (Promega) as the internal control and treated with or without Wnt3a conditional medium for 24 h. Luciferase activity was measured after 36 h with the dual reporter assay system (Promega) following the manufacturer’s protocol. The experiments were repeated in triplicate, and the data are presented as the mean ± SD after normalization to Renilla luciferase activity.
Fluorescence microscopy of living cells
Live-imaging fluorescence microscopy was performed using the NIKON A1 HD25 microscope equipped with a 100X/1.45 NA oil objective at 1,024 × 1,024 pixels. Cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco) at 37°C in a humidified 5% CO2 incubator. The FWHM (full width at half maximum) of the puncta was measured to define the thresholding values in algorithms for subsequent number, size, and intensity measurements by NIS-Elements AR Analysis.
Protein expression and purification
All genes were PCR-amplified and cloned into the pETMBP.3C vector to produce MBP-tag-fused recombinant proteins. Deletions were introduced by the site-directed mutagenesis approach. All recombinant proteins used in this study were expressed in E. coli BL21-CodonPlus (DE3) with induction by 1 M IPTG for 16 h at 18°C. Following induction, E. coli cells were resuspended in binding buffer (50 mM Tris-Cl, pH 7.9, 1 M NaCl, and 10 mM imidazole), lysed with a high-pressure homogenizer, and sedimented at 18,000 rpm for 30 min. The supernatant lysates were purified on Amylose Resin (NEB). After extensive washing with binding buffer, the proteins were eluted with MBP elution buffer (50 mM Tris-Cl, pH 7.9, 1 M NaCl, and 10 mM imidazole for MBP-tagged proteins), then purified with a HiPrep 26/60 Sephacryl S-200 HR column (17-1195-01; GE Healthcare) on an AKTA purifier (GE Healthcare), and eluted with a buffer containing 20 mM HEPES, pH 7.4, 500 mM NaCl.
In vitro phase separation assay
Proteins dissolved in a buffer containing 20 mM HEPES, pH 7.4, 500 mM NaCl were mixed, and the concentration of NaCl was adjusted to 150 mM with a buffer containing 20 mM HEPES, pH 7.4. The mixture was treated immediately with PEG8000 or PreScission protease (a gift from Dr. Hong Zhang, Institute of Biophysics, Chinese Academy of Sciences), and the concentration of NaCl was further adjusted to 150 mM NaCl and then droplet formation was examined. For imaging, droplets were observed either on a glass slide or in a glass-bottom cell culture dish for differential interference contrast (DIC) or fluorescence imaging. Imaging was with a NIKON A1 HD25 microscope equipped with a 100X/1.45 NA oil objective (1,024 × 1,024 pixels) at room temperature. NIS-Elements AR analysis was used to analyze these images. The FWHM (Full width at half maximum) of the droplets was measured to define the thresholding values in algorithms for subsequent size measurement by NIS-Elements AR Analysis.
Turbidity measurement
Turbidity was quantified by the absorbance of light at a wavelength of 600 nm for proteins diluted with 20 mM HEPES, pH 7.4, and different concentrations of NaCl. Phase separation was initiated by the addition of PEG8000. Turbidity was monitored by VARIOSKAN FLASH (Thermo Fisher Scientific). All measurements were performed in triplicate with three repeats of each group, and similar results were obtained.
Fluorophore labeling of proteins
Alexa Fluor 488 NHS ester (Thermo Fisher Scientific), Sulfo-Cyanine3 maleimide (Lumiprobe), and Alexa Fluor 647 NHS ester (Thermo Fisher Scientific) were dissolved in DMSO and incubated with the corresponding protein at room temperature for 1 h (fluorophore to protein molar ratio was 1:1). The reaction was quenched by 20 mM HEPES, pH 7.4, and 500 mM NaCl. The fluorophores and other small molecules were removed from the proteins by passing the reaction mixture through a desalting column (89882; Thermo Fisher Scientific) with buffer containing 20 mM HEPES, pH 7.4, 500 mM NaCl. In imaging assays, fluorophore-labeled proteins were further diluted with the corresponding unlabeled proteins in the same buffer.
Fluorescence recovery after photobleaching (FRAP) analysis
FRAP was performed with confocal microscopy (NIKON A1 HD25) equipped with a 100X/1.45 NA oil objective (1,024 × 1,024 pixels) at room temperature. The concentration of the fluorescently labeled protein was adjusted to 1% by diluting the labeled protein into the unlabeled one. Defined regions were photobleached at a specific wavelength and the fluorescence intensities in these regions were collected every 3 s (for in vitro droplets) and normalized to the initial intensity before bleaching. Image intensity was measured by mean ROI and further analyzed by Prism (GraphPad).
Quantification of protein concentration in the condensed phase droplets
Only one component of the condensate was labeled with Alexa FluorTM 488 NHS ester (Thermo Fisher Scientific). To generate a standard calibration curve, the fluorescence intensity of the protein at different concentrations was measured by confocal microscopy (NIKON A1 HD25) equipped with a 100X/1.45 NA oil objective (1,024 × 1,024 pixels) at room temperature and analyzed by ImageJ software. The fluorescence intensity of the protein within droplets was measured and the concentration was obtained from the standard calibration curve.
In vitro phosphorylation assay
The purified proteins were mixed in 40 μl of reaction buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 5 mM ATP for 30 min at room temperature. The reaction was stopped by adding the SDS loading buffer, and proteins were analyzed by immunoblotting.
Quantification of endogenous protein concentrations by LC-MS/MS
For the quantitative analysis of human Dvl2 protein, cells are lysed by lysis buffer containing 8 M urea and protease inhibitor (Roche) in PBS. 100 μg protein was reduced, alkylated, digested by trypsin, and desalted. The tryptic peptides and standard synthetic peptides (Dvl2: EEISDDNAR, TSGIGDSR and VIYHLDEEETPYLVK) were labeled by TMT reagent and mixed together. The mixture was desalted using C18 stage-tips.
For LC-MS/MS analysis, the peptides were separated by a C18 column (75 μm inner-diameter, 150 mm length, 5 μm, 300 Å) with a Thermo-Dionex Ultimate 3000 HPLC system, which was directly connected with a Thermo Fisher Scientific Q-Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer. A series of adjusted linear gradients according to the hydrophobicity of fractions with a flow rate of 300 nl/min was applied. The mass spectrometer was programmed to acquire in the data-dependent acquisition mode. The survey scan was from m/z 300 to 1,800 with a resolution of 60,000 at m/z 400. After one microscan, the top 40 most intense peaks with charge state 2 and above were dissociated by normalized collision energy of 32%. The isolation window was set at 0.7 Da width and the dynamic exclusion time was 15 s. The MS2 spectra were acquired with a resolution of 15,000, AGC target of 1e5, and maximum injection time (IT) of 50 ms.
The generated MS/MS spectra were searched against the database containing sequences of target proteins by using the SEQUEST search engine in Proteome Discoverer 2.2 software. The search criteria were as follows: full tryptic specificity was required, one missed cleavage was allowed, carbamidomethylation on cysteine and TMT-6plex on lysine/peptide N-terminal were set as the fixed modifications, oxidation on methionine was set as the variable modification, precursor ion mass tolerances were set at 20 ppm for all MS acquired in an Orbitrap mass analyzer, and the fragment ion mass tolerance was set to 20 mmu for all MS2 spectra acquired. Peptide spectral matches (PSM) were validated using Fixed Value PSM Validator provided by Proteome Discoverer software based on q-values at a 1% false discovery rate (FDR).
GI-SIM/TIRF-SIM imaging
Conventional images of Dvl2-mEGFP KI cells and Axin1-tdTomato KI cells were acquired on a multi-SIM imaging system (NanoInsights) with a 100X/1.49 NA oil objective (Nikon CFI SR HP Apo), solid-state single mode lasers (488, 561, 640 nm), and a COMS (Complementary Metal-Oxide-Semiconductor) camera (ORCA-Fusion C14440-20UP, HAMAMATSU). To obtain optimal images, immersion oils with refractive indices of 1.518 were used for Dvl2-mEGFP KI cells and Axin1-tdTomato KI cells on glass coverslips. Dvl2-mEGFP KI cells were imaged once every 30 s for 40 min, with an exposure time of 25 ms, and Axin1-tdTomato KI cells were imaged once every 2 for 40 min, with an exposure time of 50 ms. The microscope is routinely calibrated with 100 nm fluorescent spheres to calculate both the lateral and axial limits of image resolution. Conventional image stacks were processed by deconvolution methods using SI-Recon 2.11.19 (NanoInsights) with the following method: five times Richardson–Lucy (RL) deconvolution. Pixel registration was corrected to be <1 pixel for all channels using 100 nm fluorescent beads (Guo et al., 2018).
Quantitative analysis of Dvl and Axin by TIRF
Quantification of Dvl and Axin was performed as described (Kan et al., 2020; Ma et al., 2020). Briefly, the knock-in cell lines were imaged with an NA value of 1.49 and a pixel resolution of 30.6 nm. Protein tracking analysis utilized a custom MATLAB algorithm u-Track to extract individual puncta (Jaqaman et al., 2008). Gaussian standard deviation was set as 1.4, 0.05 for alpha, and 1 for fit window size. The output from the u-track was analyzed with homemade MATLAB codes. The amplitude of all the traces was fitted with the Gaussian mixture model and the Bayesian information criterion (BIC) was applied for model selection. The amplitude level of the first selected Gaussian model was considered as the single-molecule intensity.
Mathematical modeling
Related physical interactions are listed in Fig. S2 D for the formation of signalosome. ODE equations were solved with 100,000 groups of randomly varied parameters over the physiologically expected range in vivo. The total level of Fizzled, Dvl, and Axin for modeling input was quantified based on the mass spectrometry analysis. To compare with the TIRF experimental data, the mathematical model was solved every 2 min until 22 min.
Quantification and statistical analysis
Statistical analyses were performed with the two-tailed unpaired t test. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered statistically significant. For the bar charts, data were plotted as mean ± SD from at least three independent experiments. For immunoblot gel quantification, the gels were scanned and the band intensities were quantified with ImageJ. The band intensity of total proteins was normalized to the loading control (GAPDH), and the band intensity of phosphorylated proteins was normalized to the total proteins. Graphs were generated in GraphPad Prism 8.
Online supplemental material
Fig. S1 shows that Dvl2 undergoes phase separation and the IDR1 (141–259aa), especially the charged residues in this region, is critical for Dvl2 LLPS. Fig. S2 shows the endogenous signalosome assembly process at the single-molecule level by TIRF. Fig. S3 shows that Dvl2 LLPS regulates the mobility of Axin1 and β-catenin both in vivo and in vitro.
Data availability
MATLAB code for our implementation of the computational model will be available upon request.
Acknowledgments
We would like to thank staff in the Center for Biological Imaging (CBI), Institute of Biophysics, Chinese Academy of Sciences, for help with multi-SIM analysis.
This work was supported by grants from the National Natural Science Foundation of China (31988101 and 31730056 to Y.-G. Chen).
The authors declare no competing financial interests.
Author contributions: K. Kang, Q. Shi, and Y.-G. Chen designed the study and analyzed the data; K. Kang and Q. Shi performed the experiments; X. Wang performed modeling; K. Kang, Q. Shi, X. Wang, and Y.-G. Chen wrote the manuscript.

