


Exosomes are endosome-derived extracellular vesicles involved in intercellular communication. They are generated as intraluminal vesicles within endosomal compartments that fuse with the plasma membrane (PM). The molecular events that generate secretory endosomes and lead to the release of exosomes are not well understood. We identified a subclass of non-proteolytic endosomes at prelysosomal stage as the compartment of origin of CD63 positive exosomes. These compartments undergo a Rab7a/Arl8b/Rab27a GTPase cascade to fuse with the PM. Dynamic endoplasmic reticulum (ER)-late endosome (LE) membrane contact sites (MCS) through ORP1L have the distinct capacity to modulate this process by affecting LE motility, maturation state, and small GTPase association. Thus, exosome secretion is a multi-step process regulated by GTPase switching and MCS, highlighting the ER as a new player in exosome-mediated intercellular communication.
Introduction
Exosomes are an endosome-derived extracellular vesicle (EV) subclass involved in intercellular communication. Intracellularly, they correspond to the 50–150 nm intraluminal vesicles (ILV) found inside endosomes throughout the cell’s heterogeneous and complex late-endosomal (LE)/lysosomal (L) network (Vacca et al., 2016). Early experiments pointed to multivesicular bodies (MVB) that fuse with the plasma membrane (PM) as the source of exosomes (Raposo et al., 1996). The first step of exosome biogenesis that concerns the inward budding mechanisms to generate ILVs is relatively well understood (van Niel et al., 2018). Likewise, the final steps of MVBs approaching the membrane (Ostrowski et al., 2010; Hsu et al., 2010; Savina et al., 2005; Hyenne et al., 2015) and their SNARE-mediated fusion with the PM (Gross et al., 2012; Verweij et al., 2018; Hyenne et al., 2015; Wei et al., 2017) are gradually becoming more clear. However, despite these advances, it is still largely a mystery when and how MVBs, which are usually fated for lysosomal degradation of their contents, acquire their secretory capacities to release exosomes.
MVB is a generic morphological term that can refer to various ILV-containing compartments that would correspond to subclasses of early and LEs, endolysosomes, and autolysosomes. Indeed, ILV biogenesis is observed as early as in early endosome antigen 1 (EEA1) compartments and further maturation of endosomal carrier vesicles (ECV)/MVB by fusion with endolysosomes provides a multivesicular appearance to endolysosomes albeit with a more pleiomorphic ultrastructure (van Niel et al., 2011; Bissig et al., 2017; van der Beek et al., 2022). The canonical “textbook” MVBs that display a regular ultra-structural morphology correspond to ECVs that are likely acting as intermediate between early-/recycling endosomal and the endolysosomal network (Vacca et al., 2016). Endosomal maturation involves acidification, homo- and heterotypic fusion and fission steps between “younger” and further matured LEs, and eventually a gain in proteolytic activity thanks to an increased interaction with lysosomes or fusion with terminal storage lysosomes (TSL; Gruenberg and Stenmark, 2004). Endosomes undergo an ordered, stepwise membrane identity progression or maturation in which small GTPases are sequentially recruited in a cascade-like manner (Cullen and Carlton, 2012; Jongsma et al., 2020). Precisely which multivesicular endosomal subtype(s) are capable to fuse with the PM to release their ILVs into the extracellular space remains unclear, although many have long suspected PM fusion to be an alternative to fusion with lysosomes.
Apart from homo- and heterotypic interactions within the endosomal system, endosomal maturation is also impacted by its interaction with other organelles, notably the endoplasmic reticulum (ER), one of the major players in inter-organelle membrane contact sites (MCS; Phillips and Voeltz, 2016; van Anken and Sitia, 2016). MCS are defined as close (∼10–30 nm), non-fusogenic appositions between cellular membranes mediated by specific protein tethers (Scorrano et al., 2019) such as protrudin and ORP1L for ER-LE MCS. ER-LE MCS occur throughout the cytoplasm and are known to impact LE motility, lipid and ion exchange, and endosome tubulation/fission (Phillips and Voeltz, 2016; Eden, 2016; Hoyer et al., 2018; Saheki and De Camilli, 2017). Intriguingly, while MCS have a profound impact on LE maturation and fate whether they can modulate the secretory capacity of MVBs is not known.
In this study, we exploit a novel CD63-based quantitative single-cell live-imaging approach (Verweij et al., 2018; Bebelman et al., 2020) to gain insight into the molecular identity of MVBs that fuse with the PM and to investigate the impact of ER-LE MCS on exosome release. We show that (CD63-positive) MVBs competent to fuse with the PM represent a subclass of non-catalytic ECV/MVB distinguished by the presence of (low-level) LAMP1 and association with Rab27 but an absence of Rab5a/7a/11a/35 and Arl8b. Furthermore, we demonstrate that ER-LE MCS have the distinct ability to modulate exosome secretion, both in a stimulatory and inhibitory fashion. Mechanistically, these effects appear to correlate with an impact of these MCS on LE positioning, motility, and maturation, including a final switch to Rab27a to render the MVB PM fusion competent. Altogether our findings provide further insight into the identity of PM-fusing ECV/MVB and highlight a novel role for ER-LE MCS in the regulation of exosome release.
Results
Maturation stage of PM fusing endosomes
EEA1, CD63, and lysosomal associated membrane protein 1 (LAMP1) represent three widely used markers for early and late endosomal compartments with multivesicular appearance. CD63 localization itself spans late-endosomal, lysosomal, and endolysosomal organelles (Kobayashi et al., 2000). To visualize CD63-positive compartments that fuse with the PM, we deployed optical exosome reporters we recently developed, CD63-pHluorin and CD63-pHuji. pHluorin and pHuji are pH-sensitive variants of GFP and mApple, respectively (Miesenbock et al., 1998; Shen et al., 2014), and by inserting them into the first extracellular loop of CD63, the fluorescence of CD63-pHluorin/-pHuji is quenched due to the low pH of late-endo/lysosomal compartments. Once these compartments fuse with the PM, CD63-pHluorin/-pHuji are dequenched by the neutral extracellular pH which results in a burst of fluorescence that can be visualized by live-cell microscopy. Dynamic-correlative light-electron tomography confirmed that these events correspond to multivesicular endosomes fusion with the PM, and the frequency of these events correlated with the release of CD63+ EVs recovered and analyzed by conventional methods (Verweij et al., 2018).
To verify which marker is present in CD63-positive MVBs that fuse with the PM, we visualized MVB/PM fusion events in CD63-pHuji/LAMP1-GFP and CD63-pHuji/EEA1-GFP HeLa cells by live dual-color TIRF microscopy (DC-TIRF-M; Bebelman et al., 2020). This method visualizes MVB/PM fusion event in one channel (red/green), and the protein of interest (POI) tagged with a different color imaged in a second channel. Measuring the signal intensity of the POI at the site of fusion can reveal co-occurrence of the POI at this location (Bebelman et al., 2020). This allows for a detailed characterization of the MVB subpopulation that fuses with the PM, which can subsequently be compared to the overall LE/MVB population present throughout the cell’s cytoplasm. Following this approach, we observed a small increase in LAMP1-GFP signal coinciding with the main CD63-pHuji peak at the sites of fusion (Fig. 1, A and B; and Video 1). By contrast, EEA1, a common marker for early endosomes, was absent (Fig. 1 B and Fig. S1 A).

CD63 compartments fusing with PM represent an early non-degradative stage of late-endosomes. (A) Example of DC-TIRF-M analysis on CD63-(pHuji)fusion event in HeLa cells co-expressing LAMP1-GFP. (B) Fluorescent signal pattern for CD63-pHuji/LAMP1-GFP averaged over >12 events, synchronized using peak-intensity of CD63-pHuji signal at 60 s. Inset is 3.5 µm. (C) Immunofluorescent co-labeling of endogenous CD63 (red) and LAMP1 (green) in HeLa cells. (D) Fluorescent intensity of endogenous CD63 and LAMP1 levels (as in A), plotted for individual endosomes. R-squared shown for linear regression (light blue). (E) Fusion activity of apilimod treated CD63-pHluorin HeLa cells. Analysis >40 cells from n = 3 experiments, using Student’s two-tailed two-sample t test. Red line indicates median. (F) Fluorescent analysis of overlap degradative compartments labeled by MagicRed with late-endo/lysosomal markers LAMP1(-GFP) or CD63(-pHluorin) in HeLa cells untreated or treated with apilimod. (G) Quantification of analysis as in (F). Shown is the ratio of degradative MagicRed compartments that overlap with LAMP1 and CD63, respectively (measured over >12 imaging fields using Student’s two-tailed two-sample t test; red line indicates median). (H) Example of DC-TIRF-M analysis on CD63-(pHluorin)fusion event in HeLa cells incubated with MagicRed. Inset is 3.4 µm. (I) Example of DC-TIRF-M analysis on CD63-(pHuji)fusion event in HeLa cells co-expressing TRPML1-GFP. Inset is 5.6 µm. (J) Example of DC-TIRF-M analysis on CD63-(pHluorin)fusion event in HeLa cells co-expressing LC3B-mRFP. Inset is 3.4 µm. (K) Fluorescent signal patterns for MagicRed, LC3, and TRPML1 averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. Bars: (A, C, F, and H–J) 5 µm. N indicates nucleus.
CD63 compartments fusing with PM represent an early non-degradative stage of late-endosomes. (A) Example of DC-TIRF-M analysis on CD63-(pHuji)fusion event in HeLa cells co-expressing LAMP1-GFP. (B) Fluorescent signal pattern for CD63-pHuji/LAMP1-GFP averaged over >12 events, synchronized using peak-intensity of CD63-pHuji signal at 60 s. Inset is 3.5 µm. (C) Immunofluorescent co-labeling of endogenous CD63 (red) and LAMP1 (green) in HeLa cells. (D) Fluorescent intensity of endogenous CD63 and LAMP1 levels (as in A), plotted for individual endosomes. R-squared shown for linear regression (light blue). (E) Fusion activity of apilimod treated CD63-pHluorin HeLa cells. Analysis >40 cells from n = 3 experiments, using Student’s two-tailed two-sample t test. Red line indicates median. (F) Fluorescent analysis of overlap degradative compartments labeled by MagicRed with late-endo/lysosomal markers LAMP1(-GFP) or CD63(-pHluorin) in HeLa cells untreated or treated with apilimod. (G) Quantification of analysis as in (F). Shown is the ratio of degradative MagicRed compartments that overlap with LAMP1 and CD63, respectively (measured over >12 imaging fields using Student’s two-tailed two-sample t test; red line indicates median). (H) Example of DC-TIRF-M analysis on CD63-(pHluorin)fusion event in HeLa cells incubated with MagicRed. Inset is 3.4 µm. (I) Example of DC-TIRF-M analysis on CD63-(pHuji)fusion event in HeLa cells co-expressing TRPML1-GFP. Inset is 5.6 µm. (J) Example of DC-TIRF-M analysis on CD63-(pHluorin)fusion event in HeLa cells co-expressing LC3B-mRFP. Inset is 3.4 µm. (K) Fluorescent signal patterns for MagicRed, LC3, and TRPML1 averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. Bars: (A, C, F, and H–J) 5 µm. N indicates nucleus.
DC-TIRF-M fusion event example of CD63-pHuji HeLa cell expressing LAMP1-GFP.

Supplemental data related to Fig. 1 . (A) Examples of DC TIRF-M analysis for EEA1-GFP in CD63-pHuji expressing HeLa. (B) Fluorescent analysis of LAMP1(-GFP) and CD63(-mRFP) localization apilimod-treated HeLa cells at different z (indicated in lower panels). (C) CD63-pHluorin fusion-spot size analysis in control (ctrl) or apilimod (apili)-treated condition. N > 100 fusion events from >10 cells in a typical experiment. Bars: 5 µm.

Supplemental data related to Fig. 1 . (A) Examples of DC TIRF-M analysis for EEA1-GFP in CD63-pHuji expressing HeLa. (B) Fluorescent analysis of LAMP1(-GFP) and CD63(-mRFP) localization apilimod-treated HeLa cells at different z (indicated in lower panels). (C) CD63-pHluorin fusion-spot size analysis in control (ctrl) or apilimod (apili)-treated condition. N > 100 fusion events from >10 cells in a typical experiment. Bars: 5 µm.
Although LAMP1 is mainly enriched in TSL and endolysosomes (Bright et al., 2016; Bissig et al., 2017), it can traffic through (early) endosomes to reach lysosomes (Ebrahim and Thilo, 2011; Cook et al., 2004). Conversely, a proportion of endogenous CD63 is also associated with degradative, endolysosomal compartments (Jongsma et al., 2020; Bissig et al., 2017). To get more insight into the global distribution of CD63 and LAMP1 in HeLa cells, we examined their endogenous localization. Consistent with literature, CD63 and LAMP1 showed an extensive overall overlap (Pearson’s r = 0.713), although the relative intensities seemed not to correlate, with perinuclear compartments appearing relatively high in CD63 and low in LAMP1, whereas peripheral compartments appeared high in LAMP1 and low in CD63 (Fig. 1 C). Indeed, plotting LAMP1 vs. CD63 intensity levels per compartment showed a rather low correlation (R2 = 0.29; Fig. 1 D) indicating a discrepancy in intensity between CD63 and LAMP1 that is consistent with their respective enrichment at various endosomal maturation states. To assess whether CD63-pHluorin compartments that fuse with the PM represent endolysosomes or earlier compartments, we incubated cells with PIKfyve inhibitor Apilimod that blocks PtdIns (3,5)P2 synthesis from PI(3)P (Cai et al., 2013). This inhibition blocks lysosomal reformation from endolysosomes, and results in enlarged endolysosomal compartments and a depletion of TSL but no alteration in the number of ECV/MVBs (Bissig et al., 2017). Intracellularly, Apilimod treatment (2 h) resulted in enlarged (low motile) compartments positive for LAMP1 and CD63 (Fig. S1 B and Video 2), whereas CD63 positive endosomes, seemingly unaffected, are observed in the proximity to the PM (Fig. S1 B, red arrowheads; Video 2). Consistent with this, Apilimod treatment for 2 h did not significantly affect CD63-pHluorin fusion activity (Fig. 1 E), nor alter the size of fusion spots (Fig. S1 C).
Short time-lapse of HeLa cell co-expressing LAMP1-GFP and CD63-RFP in untreated condition (left), and upon treatment of apilimod (2 h) in the middle and at the bottom of the cell (middle and right panel, respectively).
Short time-lapse of HeLa cell co-expressing LAMP1-GFP and CD63-RFP in untreated condition (left), and upon treatment of apilimod (2 h) in the middle and at the bottom of the cell (middle and right panel, respectively).
Since endolysosomes are considered the principal site of acid hydrolases activity (Bright et al., 2016), we then assessed the degradative capacity of LAMP1 and CD63 compartments. LAMP1-GFP and CD63-pHluorin expressing cells were probed with MagicRed a (live) fluorescent substrate/reporter for Cathepsin B activity (Bissig et al., 2017). Interestingly, the majority of intracellular MagicRed signal was found within LAMP1-GFP compartments (∼80%), where LAMP1 localized predominantly to the limiting membrane as more clearly shown upon apilimod treatment (Fig. 1 F). By contrast, only a minor fraction of MagicRed signal (∼20%) localized to CD63-pHluorin compartments (Fig. 1, F and G). To directly address whether PM-fusing CD63-pHluorin compartments are catalytically active, we incubated CD63-pHluorin cells with MagicRed for 10 min prior to imaging. DC-TIRF-M analysis of CD63-pHluorin/MagicRed cells revealed that MagicRed was not detectable at the site of fusion (Fig. 1, H and K; and Video 3), although MagicRed positive compartments could clearly be detected in the PM vicinity (Fig. 1 H, red arrowheads). To further exclude the possibility that CD63-pHluorin compartments fusing with the PM represent (endo)lysosomes, we visualized MVB/PM fusion events in CD63-pHuji/Transient Receptor Potential Mucolipin 1 (TRPML1)-GFP expressing cells. TRPML1 is a lysosomal Ca2+ channel important in vesicular trafficking, lysosomal exocytosis, and autophagy (Di Paola et al., 2018). In line with the previous results, we did not detect a TRPML1 signal at the site of MVB/PM fusion (Fig. 1, I and K). Likewise, the key macroautophagy marker LC3B was not detected at fusion sites (Fig. 1, J and K, further excluding auto(phago)lysosomes as a major class of CD63-positive PM-fusing compartments in HeLa cells under these conditions.
DC-TIRF-M fusion event example of CD63-pHluorin HeLa cell incubated with active Cathepsin B dye MagicRed.
DC-TIRF-M fusion event example of CD63-pHluorin HeLa cell incubated with active Cathepsin B dye MagicRed.
Together, these results strongly suggest that (CD63-positive) compartments that fuse with the PM represent multi-vesicular organelles at a pre-endolysosomal maturation stage that are not catalytically active.
Small-GTPase decoration of PM fusing LE
The effect of Apilimod on a significant proportion of the late endosomal/lysosomal population (Fig. 1 F and Fig. S1 B) suggests that the unaffected LE fraction fusing with the PM is a sub-population of ECV/MVBs. ECV act as an intermediate between early-/recycling endosomal and the endolysosomal network (Vacca et al., 2016) and undergo a Rab5/Rab7 conversion mediated by the class C VPS/HOPS complex to access the endolysosomal network (Rink et al., 2005). To characterize CD63-positive MVBs in further detail, we screened a panel of early-, recycling-, and late-endosomal/lysosomal small-GTPases (Rab5a, Rab11a, and Rab35, as well as Rab7a, Arl8b, and Rab27a/b) previously directly or indirectly implicated in the exosomal pathway in order to assess their overlap with CD63. Intracellular co-localization analysis revealed that the majority of CD63 co-localized well with Rab27a and Rab27b and even stronger with Rab7a and Arl8b (Fig. 2, A and B). The levels of colocalization between CD63 and Rab5a, Rab11, and Rab35 on the other hand were weak (PCC < 0.5; Fig. 2, A and B; and Fig. S2 A).

Intracellular overlap CD63 with small-GTPases. (A and B) Fluorescent analysis of Early- (EE), Recycling- (RE), and Late-Endosome (LE)/Lysosome (L)-associated small-GTPases in CD63-pHluorin/pHuji expressing HeLa cells. Representative examples of (A) Rab5a-GFP (EE), Rab11a-mCherry (RE), Rab7a-GFP, Arl8b-mCherry, and Rab27a/b-GFP (LE/L) localization in relation to CD63-pHluorin/pHuji. (B) Quantification of colocalization between CD63 and various small-GTPases as performed in A and B. n ≥ 3 images (3≥ cells per image) analyzed from ≥2 independent experiments. Red line indicates median. (C) Signal pattern for fluorescently tagged RabGTPases averaged over >12 events, synchronized using peak-intensity of the CD63-pHluorin/pHuji signal at 60 s. (D–G) Examples of DC TIRF-M analysis depicted in C for (D) Rab7a-mCherry signal at CD63-pHluorin fusion spot (inset is 8.2 µm), (E) Rab5a-mCherry and Rab11a-mCherry signal at CD63-pHluorin fusion spot (inset sizes are 8.5 and 5.9 µm, respectively), (F) Rab27b-GFP signal at CD63-pHuji fusion spot (inset is 5.1 µm), (G) Arl8b-mCherry and Rab27a-GFP signal at CD63-pHluorin/pHuji fusion spot in HeLa cells (inset sizes are 5.1 µm). Bars: 5 µm.
Intracellular overlap CD63 with small-GTPases. (A and B) Fluorescent analysis of Early- (EE), Recycling- (RE), and Late-Endosome (LE)/Lysosome (L)-associated small-GTPases in CD63-pHluorin/pHuji expressing HeLa cells. Representative examples of (A) Rab5a-GFP (EE), Rab11a-mCherry (RE), Rab7a-GFP, Arl8b-mCherry, and Rab27a/b-GFP (LE/L) localization in relation to CD63-pHluorin/pHuji. (B) Quantification of colocalization between CD63 and various small-GTPases as performed in A and B. n ≥ 3 images (3≥ cells per image) analyzed from ≥2 independent experiments. Red line indicates median. (C) Signal pattern for fluorescently tagged RabGTPases averaged over >12 events, synchronized using peak-intensity of the CD63-pHluorin/pHuji signal at 60 s. (D–G) Examples of DC TIRF-M analysis depicted in C for (D) Rab7a-mCherry signal at CD63-pHluorin fusion spot (inset is 8.2 µm), (E) Rab5a-mCherry and Rab11a-mCherry signal at CD63-pHluorin fusion spot (inset sizes are 8.5 and 5.9 µm, respectively), (F) Rab27b-GFP signal at CD63-pHuji fusion spot (inset is 5.1 µm), (G) Arl8b-mCherry and Rab27a-GFP signal at CD63-pHluorin/pHuji fusion spot in HeLa cells (inset sizes are 5.1 µm). Bars: 5 µm.

Supplemental data related to Fig. 2. (A) Rab35-GFP (recycling endosome) localization in relation to CD63-pHuji. (B–F) Overview panels of the DC TIRF-M analysis for (B) CD63-pHluorin/Rab5a-mCherry, (C) Arl8b-GFP/CD63-pHuji, (D) CD63-pHluorin/Rab11a-mCherry, (E) Rab35-GFP/CD63-pHuji, and (F) Rab27a-GFP/CD63-pHuji, related to Fig. 2.

Supplemental data related to Fig. 2. (A) Rab35-GFP (recycling endosome) localization in relation to CD63-pHuji. (B–F) Overview panels of the DC TIRF-M analysis for (B) CD63-pHluorin/Rab5a-mCherry, (C) Arl8b-GFP/CD63-pHuji, (D) CD63-pHluorin/Rab11a-mCherry, (E) Rab35-GFP/CD63-pHuji, and (F) Rab27a-GFP/CD63-pHuji, related to Fig. 2.
Endosomes are very dynamic organelles and known to undergo identity progression by sequential, step-like recruitment and dissociation of multiple small-GTPases, such as the Rab5/Rab7 conversion (Cullen and Carlton, 2012; Jongsma et al., 2020; Rink et al., 2005). Consequently, while analysis of the overall co-localization between intracellular endosomal markers at “steady state” is highly informative, the subset of endosomes that has acquired the competency to fuse (with the PM) could significantly diverge from our global characterization. To pinpoint the “molecular identity” of the subset of ECV/MVBs fusing with the PM, we performed a manual, targeted DC-TIRF-M screen for the same panel of small-GTPases at the site of fusion. Importantly, we observed that neither Rab5a nor Rab7a were present at fusion sites (Fig. 2, C–E and Fig. S2 B). Rab7a-negative LE moving toward the periphery can also harbor Arl8b (Jongsma et al., 2020; Hofmann and Munro, 2006), which also showed an equally strong overlap with CD63-pHluorin compartments as Rab7a (Fig. 2, A and B). However, DC-TIRF-M revealed that Arl8b was also absent at the site of fusion (Fig. 2, C and G; and Fig. S2 C), strongly suggesting that both GTPases do not mark and/or dissociate from CD63+ compartments that are becoming PM fusion competent. We, therefore, considered a possible involvement of Rab11 or Rab35 that have both been linked to docking to and/or fusion of MVBs with the PM (Savina et al., 2005; Messenger et al., 2018; Hsu et al., 2010). However, DC-TIRF-M revealed that Rab11a and Rab35 were likewise absent from the sites of fusion (Fig. 2, C and E; and Fig. S2, D and E) consistent with the relatively low amount of overlap between Rab11/Rab35 and CD63 compartments (Fig. 2, A and B). Apart from Rab11 and Rab35, Rab27a/b has been implicated in the transport and/or docking of MVBs to the PM (Ostrowski et al., 2010), as well as various other secretory pathways (Fukuda, 2013). Unlike all other small-GTPases tested, Rab27a and Rab27b were readily detected at the site of fusion (Fig. 2, F and G; and Fig. S2 F).
Altogether these data indicate that CD63 compartments are in majority positive for Rab7a and Arl8b, whereas the CD63 subpopulation that fuses with the PM likely represents mature ECV/MVBs at pre-endolysosome stage, negative for Rab7a and Arl8b but positive for Rab27a/b.
ER/LE membrane contact sites impact exosome secretion
Having determined that CD63 compartments that fuse with the PM harbor Rab27a/b GTPases, but lack most other endosomal Rabs, we sought to investigate intracellular mechanisms that could impact such maturation. The striking discrepancy between the complete absence of Rab7a at the site of fusion despite its prior implication in exosome release (Baietti et al., 2012), the overall high amount of CD63/Rab7a overlap in the cell (Fig. 2, A and B), and the clear effect of Rab7a-wt or dominant negative (DN) expression on sEV secretion (Fig. S3 A) would be compatible with a regulatory role upstream of ECV/MVB-PM fusion. Rab7 has several mediators of ER-LE MCS among its effectors and ER-endosome MCSs are known to impact endosome maturation and motility and increase as these endosomes further mature (Friedman et al., 2013; Wu and Voeltz, 2021). Depletion of Rab7a lead to a strong decrease in fusion activity as did RNAi interference with its effectors ORP1L and protrudin previously implicated in ER-LE MCS formation (Fig. 3 A; and Fig. S3, B and C). Protrudin-wt overexpression has been shown to promote fusion of LAMP1 compartments with the PM by facilitating kinesin-1 loading onto the compartment (Raiborg et al., 2015). To broaden the modulatory role of ER-LE MCS in exosome secretion, we expressed protrudin in CD63-pHluorin cells. Indeed, compared to ctrl, overexpression of protrudin-wt increased fusion activity roughly twofold, whereas expression of a point mutant defective in phosphoinositide binding, protrudin-FYVE4A, did not alter fusion activity (Fig. 3, B and C). This increased fusion activity of CD63 compartments under protrudin-wt overexpression could not be attributed to a selective increase in endo-lysosome/PM fusion, as MagicRed signal was still absent at the CD63-pHluorin fusion sites (Fig. S3 C). Thus, protrudin ER-LE MCS known to promote anterograde transport and secretion of LAMP1 compartments also stimulate PM fusion of CD63 compartments in non-neuronal cells.
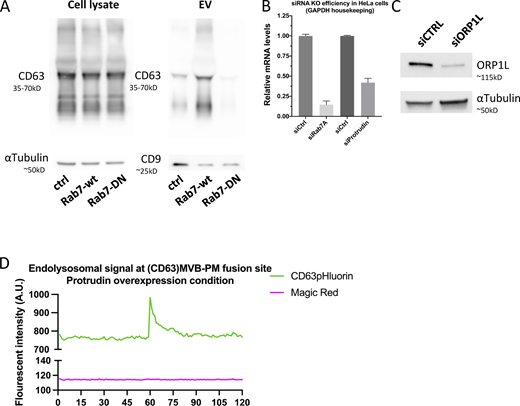
Supplemental data related to Fig. 3. (A) Western blotting analysis of cell lysates and small EVs isolated from CD63-pHluorin HeLa cells transfected with empty vector (control), Rab7a-wt, or Rab7-DN. (B) Analysis of Rab7A and protrudin mRNA levels by quantitative PCR for RNAi-treated HeLa cells. Duplo’s measured from three independent biological replicates, error bar represents standard error of the mean. (C) Western blotting analysis for ORP1L protein levels. (D) Fluorescent signal patterns for CD63-pHluorin/MagicRed in Protrudin-wt overexpressing cells, averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. Source data are available for this figure: SourceData FS3.

Supplemental data related to Fig. 3. (A) Western blotting analysis of cell lysates and small EVs isolated from CD63-pHluorin HeLa cells transfected with empty vector (control), Rab7a-wt, or Rab7-DN. (B) Analysis of Rab7A and protrudin mRNA levels by quantitative PCR for RNAi-treated HeLa cells. Duplo’s measured from three independent biological replicates, error bar represents standard error of the mean. (C) Western blotting analysis for ORP1L protein levels. (D) Fluorescent signal patterns for CD63-pHluorin/MagicRed in Protrudin-wt overexpressing cells, averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. Source data are available for this figure: SourceData FS3.

ER-LE MCSs impact fusion activity—protrudin & U18666A. (A) MVB/PM fusion activity in RNAi-treated CD63-pHluorin HeLa cells depleted for Rab7a, Protrudin or ORP1L. NTC, non-targeting control. Measured over >60 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (B) Fluorescent analysis of protrudin-wt or -FYVE4A (GFP) overexpressing CD63-pHuji HeLa cells after fixation. (C) MVB/PM fusion activity in protrudin-wt or -FYVE4A overexpressing CD63-pHuji HeLa cells compared to control. Measured over >30 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (E) Confocal analysis of CD63, NPY- and VAMP2-pHluorin HeLa cells treated with 2 μg/ml U18666A (inset sizes are 6.1, 6.7 and 3.8 µm for control, and 3.7, 4.7 and 3.6 µm for U18666A condition, respectively). (F) Effect of U18666A treatment on PM fusion activity of VAMP2/NPY transport vesicles (left) and MVBs (right). Measured over >40 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (G) Graph showing the number of acidic vesicles close to the PM in control and U18666A-treated cells. Measured over >4 cells in n = 2 experiments using Student’s two-tailed two-sample t test. Red line indicates median. Bars: 5 µm.
ER-LE MCSs impact fusion activity—protrudin & U18666A. (A) MVB/PM fusion activity in RNAi-treated CD63-pHluorin HeLa cells depleted for Rab7a, Protrudin or ORP1L. NTC, non-targeting control. Measured over >60 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (B) Fluorescent analysis of protrudin-wt or -FYVE4A (GFP) overexpressing CD63-pHuji HeLa cells after fixation. (C) MVB/PM fusion activity in protrudin-wt or -FYVE4A overexpressing CD63-pHuji HeLa cells compared to control. Measured over >30 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (E) Confocal analysis of CD63, NPY- and VAMP2-pHluorin HeLa cells treated with 2 μg/ml U18666A (inset sizes are 6.1, 6.7 and 3.8 µm for control, and 3.7, 4.7 and 3.6 µm for U18666A condition, respectively). (F) Effect of U18666A treatment on PM fusion activity of VAMP2/NPY transport vesicles (left) and MVBs (right). Measured over >40 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (G) Graph showing the number of acidic vesicles close to the PM in control and U18666A-treated cells. Measured over >4 cells in n = 2 experiments using Student’s two-tailed two-sample t test. Red line indicates median. Bars: 5 µm.
To further implicate ER-LE MCS in ECV/MVB-PM fusion, we opted to modulate cholesterol levels. Many of the ER-LE MCS described in literature, including ORP1L, ORP5, ORP6, and STARD3, are implicated in cholesterol shuttling between the ER and the LE (Ridgway and Zhao, 2018), and for one in particular, ORP1L, cholesterol levels are mechanistically coupled to CD63/LAMP1 endosomal positioning (Rocha et al., 2009). LE cholesterol levels can be increased by blocking of cholesterol exporter Niemann-Pick 1 using U18666A treatment, inducing ORP1L-mediated LE transport away from the PM (Wijdeven et al., 2016). Indeed, treatment of CD63-pHluorin expressing cells with U18666A resulted in strong perinuclear clustering of CD63 compartments, whereas NPY-pHluorin and VAMP2-pHluorin (soluble- and transmembrane protein cargo secretory vesicles, respectively [Verweij et al., 2018]) compartments were much less affected (Fig. 3 E). In accordance with this, U18666A treatment did not affect fusion activity of VAMP2- or NPY-compartments, whereas CD63(-pHluorin) fusion activity was decreased by ∼3.7-fold (Fig. 3 F); this was accompanied by a ∼fourfold reduction in CD63(-pHluorin) vesicles close to the (basolateral) PM measured by TIRFM after NH4+ treatment that neutralizes endosomal pH (Fig. 3 G), further indicating that indeed the ORP1L machinery has an impact on ECV/MVB positioning as opposed to bona fide secretory transport vesicles marked by VAMP2 and NPY. U18666A treatment thus leads to a drastic retreat of CD63 vesicles from the peripherally located PM (Fig. 3, E and G), and as a consequence strongly impacts their secretory capacity.
ORP1L ER-LE MCS and exosome secretion
Thanks to its oxysterol-binding protein (OSBP)-related ligand-binding domain (ORD), ORP1L senses (membrane) cholesterol. The presence of cholesterol induces a conformational change in ORP1L that shields an FFAT motif, preventing MCS formation, and simultaneously allows the recruitment of dynein to the ORP1L/Rab7-RILP complex, inducing LE distribution toward the nucleus (Rocha et al., 2009). The absence of cholesterol on the other hand results in an “open” ORP1L conformation that exposes its FFAT motif domain, allowing interaction with VAP-A, an ER resident protein. This latter conformation thereby induces the formation of MCS while releasing the dynein motor complex. These two conformational states are mimicked by ORP1L mutants -∆ORDPHDPHD and -∆ORD resulting in a perinuclear or scattered LE localization, respectively (Rocha et al., 2009). To further confirm that this mechanism is likewise active on CD63-pHluorin compartments, ORP1L-wt and mutants were expressed in CD63-pHluorin cells. Indeed, expression of ORP1L-∆ORDPHDPHD resulted in perinuclear clustering, whereas expression of ORP1L-∆ORD resulted in a scattered distribution of CD63-pHluorin, consistent with literature (Fig. 4 A; and Fig. S4, A and B). To validate that these altered distributions correlated with an absence or increase of MCS of CD63-pHluorin compartments, we performed colocalization and correlative light-electron microscopy (CLEM) on ORP1L-wt/mutants expressing CD63-pHluorin HeLa cells. Strikingly, while the CD63-pHluorin compartments in ORP1L-wt condition did not show any increased association with ER, CD63-pHluorin compartments in ORP1L-∆ORD were tightly enwrapped in ER (Fig. 4 B; and Fig. S4, B and C). The “closed” conformation of ORP1L also leads to the recruitment of HOPs complex and fusion of Rab7 compartment with lysosomes (Wijdeven et al., 2016). We performed colocalization analysis between CD63 and LAMP1 in ORP1L-wt, -∆ORDPHDPHD, and -∆ORD expressing cells. Indeed, the expression of ORP1L-∆ORDPHDPHD increased overlap between LAMP1 and CD63, which could be suggestive of further maturation into endolysosomes. The expression of ORP1L-∆ORD was associated with a decrease in overlap between LAMP1 and CD63 (Fig. 4, C and D) suggesting that we prevented the maturation of ECV/MVB into endolysosomes. Strikingly, live TIRF-M revealed that this phenotype correlated with a stark (∼3.4×) decrease in fusion activity for ORP1L-∆ORD expressing cells while ORP1L-∆ORDPHDPHD expressing cells showed an (∼1.7×) increase in CD63-pHluorin fusion activity with the PM (Fig. 4 E). This suggested that forced (-∆ORD) ER-LE MCS do not only prevent fusion with (endo)lysosomes but also with the PM, analogous to forced (-∆ORDPHDPHD) perinuclear clustering promoting CD63 compartment fusion with (endo)lysosomes but also with the PM (Fig. 4, C and D). The same trend was observed by Western blotting analysis on the 100,000 ×g EV pellets of ORP1L wt, -∆ORD, and -∆ORDPHDPHD expressing cells, whereas the CD63 levels in cell lysates remained unaltered (Fig. 4, F and G). CD9 levels, on the other hand, remained largely unaltered, consistent with its relative enrichment on the PM and microvesicles (Mathieu et al., 2021), that are presumably not directly affected by ORP1L localization and function.

ER-LE MCSs impact fusion activity—ORP1L. (A) Confocal analysis of ORP1L -wt, -∆ORDPHDPHD or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells. (B) Correlative Light-Electron Microscopy (CLEM) on section on CD63-pHluorin cells transfected with ORP1L -wt or -∆ORD and post-stained with DAPI. Top panel: large overview, with squares indicating zoom areas depicted right below. Subsequent panels below: zoom of boxes indicated in top panel and Fig. S1, B and C, respectively, with below same pictures including transparent orange and blue masks indicate ER and MVB, respectively. (C) Immunofluorescent labeling for LAMP1 (blue) in CD63-pHluorin (green) HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. (D) Quantification of CD63 overlaps with LAMP1 in cells as in C. Measured over >4 cells in n = 3 experiments using Mander’s Overlap Coefficient. (E) Fusion activity of CD63-pHluorin HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Measured over >60 cells from n > 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (Fand G) Western blotting analysis of cell lysates and small EVs isolated from CD63-pHluorin HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Multiple comparisons using ordinary one-way ANOVA. (H) Still of Video 4 of CD63-eGFP HeLa cells transfected with ORP1L-∆ORD-RFP. (I) Motility of CD63 compartments in CD63-eGFP HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Student’s two-tailed two-sample t test, red line indicates median. n = 40–70 organelles. (J) Fusion activity of CD63-pHluorin HeLa cells transfected with HA-ORP1L-wt, -FFAT or control (ctrl). Measured over >100 cells from n > 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (K and L) Fusion activity of CD63-pHluorin HeLa cells overexpressing RILP (K) or SKIP (L). Measured over >37 cells from n > 2 independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. Bars: 5 µm, unless stated otherwise. Source data are available for this figure: SourceData F4.
ER-LE MCSs impact fusion activity—ORP1L. (A) Confocal analysis of ORP1L -wt, -∆ORDPHDPHD or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells. (B) Correlative Light-Electron Microscopy (CLEM) on section on CD63-pHluorin cells transfected with ORP1L -wt or -∆ORD and post-stained with DAPI. Top panel: large overview, with squares indicating zoom areas depicted right below. Subsequent panels below: zoom of boxes indicated in top panel and Fig. S1, B and C, respectively, with below same pictures including transparent orange and blue masks indicate ER and MVB, respectively. (C) Immunofluorescent labeling for LAMP1 (blue) in CD63-pHluorin (green) HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. (D) Quantification of CD63 overlaps with LAMP1 in cells as in C. Measured over >4 cells in n = 3 experiments using Mander’s Overlap Coefficient. (E) Fusion activity of CD63-pHluorin HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Measured over >60 cells from n > 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (Fand G) Western blotting analysis of cell lysates and small EVs isolated from CD63-pHluorin HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Multiple comparisons using ordinary one-way ANOVA. (H) Still of Video 4 of CD63-eGFP HeLa cells transfected with ORP1L-∆ORD-RFP. (I) Motility of CD63 compartments in CD63-eGFP HeLa cells transfected with ORP1L -wt, -∆ORDPHDPHD or -∆ORD. Student’s two-tailed two-sample t test, red line indicates median. n = 40–70 organelles. (J) Fusion activity of CD63-pHluorin HeLa cells transfected with HA-ORP1L-wt, -FFAT or control (ctrl). Measured over >100 cells from n > 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (K and L) Fusion activity of CD63-pHluorin HeLa cells overexpressing RILP (K) or SKIP (L). Measured over >37 cells from n > 2 independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. Bars: 5 µm, unless stated otherwise. Source data are available for this figure: SourceData F4.

Supplemental data related to Fig. 4. (A) Confocal analysis of ORP1L -wt, -∆ORDPHDPHD or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells, as in Fig. 4 A with single channel images in grey with inverted LUT. (B) Fluorescent analysis of VAPA-GFP (green) HeLa cells co-expressing mCherry-ORP1L-wt, -∆ORD, or ∆ORDPHDPHD (red), and labeled for endogenous CD63 (blue; inset sizes are 3.1, 3.4 and 3.4 µm, respectively). (C) Large overview of Correlative Light-Electron Microscopy (CLEM) on section on CD63-pHluorin cells transfected with ORP1L-∆ORD and post-stained with DAPI. (D) Fluorescent analysis of CD63 localization in CD63-pHluorin HeLa co-expressing HA-SKIP or HA-RILP. N, nucleus. (E) Fluorescent analysis of CD63 localization in CD63-pHluorin HeLa co-expressing GFP-SKIP, GFP-RILP, GFP-RILP-∆N, or GFP-p50 with CD63-pHuji. (F) Fusion activity of CD63-pHuji HeLa cells overexpressing GFP-RILP-∆N or GFP-p50. Control (ctrl) has filler plasmid. N, nucleus.

Supplemental data related to Fig. 4. (A) Confocal analysis of ORP1L -wt, -∆ORDPHDPHD or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells, as in Fig. 4 A with single channel images in grey with inverted LUT. (B) Fluorescent analysis of VAPA-GFP (green) HeLa cells co-expressing mCherry-ORP1L-wt, -∆ORD, or ∆ORDPHDPHD (red), and labeled for endogenous CD63 (blue; inset sizes are 3.1, 3.4 and 3.4 µm, respectively). (C) Large overview of Correlative Light-Electron Microscopy (CLEM) on section on CD63-pHluorin cells transfected with ORP1L-∆ORD and post-stained with DAPI. (D) Fluorescent analysis of CD63 localization in CD63-pHluorin HeLa co-expressing HA-SKIP or HA-RILP. N, nucleus. (E) Fluorescent analysis of CD63 localization in CD63-pHluorin HeLa co-expressing GFP-SKIP, GFP-RILP, GFP-RILP-∆N, or GFP-p50 with CD63-pHuji. (F) Fusion activity of CD63-pHuji HeLa cells overexpressing GFP-RILP-∆N or GFP-p50. Control (ctrl) has filler plasmid. N, nucleus.
To further decipher the impact of ORP1L ER-LE MCS on fusion activity, we first investigated their impact on the motility of CD63 compartments. To visualize the movements of CD63 compartments in living cells, we swapped the pH-sensitive pHluorin moiety in the first extracellular loop (Verweij et al., 2018) for eGFP. Consistent with previous reports on the effect of ORP1L on LE transport and motility (Rocha et al., 2009; Vihervaara et al., 2011), our analysis by LSC-microscopy revealed that ORP1L-∆ORD/CD63-eGFP endosomes had a markedly decreased motility compared to those in ORP1L-wt or -∆ORDPHDPHD expressing cells (Fig. 4, H and I), although CD63-eGFP compartments in ORP1L-∆ORD expressing cells that were not associated with ORP1L-∆ORD still had a high motility (Fig. 4 H and Video 4). This suggested that forced MCS inhibit MVB-PM fusion by acting primarily on their capacity to move. LE motility in ORP1L-∆ORDPHDPHD expressing cells was predominantly observed in vesicles trafficking toward and from the peri-nuclear endosomal cluster (Video 5), linking both anterograde and retrograde transports to increased fusion activity in this condition. To further explore the counterintuitive observation that both centripetal and centrifugal movements stimulate MVB-PM fusion, we first stimulated centripetal LE movement toward the MTOC by RILP overexpression (Jongsma et al., 2020) and observed an increase in ECV/MVB-PM fusion activity (∼1.6×; Fig. 4 K), comparable to ORP1L-∆ORDPHDPHD overexpression (Fig. 4 E). We confirmed the role of RILP and associated retrograde transport (Fig. S4 D) by overexpressing RILP-ΔN and p50/dynamityn. Overexpression of the p50 fragment acts as a DN to p150Glued (Jacquot et al., 2010) and RILP-ΔN can still associate with endosomes, but cannot recruit any of its effectors, thereby functioning as a DN. RILP-ΔN and p50 fragment overexpression both reduced centripetal movement of CD63 compartments compared to RILP-wt overexpression and reduced fusion activity (Fig. S4, D, E, and F). Stimulation of peripheral LE movement by SKIP overexpression on the other hand (Fig. S4 D) did result in an ∼80% increase of fusion activity (Fig. 4 L).
Short time-lapse of CD63-eGFP HeLa cells transfected with ORP1L-∆ORD-RFP (Fig. 4 H ), showing from left to right the overlay, the single/predominantly ORP1L vesicles (red), the ORP1L/CD63 vesicles (yellow) and the single/predominantly CD63 vesicles (green).
Short time-lapse of CD63-eGFP HeLa cells transfected with ORP1L-∆ORD-RFP (Fig. 4 H ), showing from left to right the overlay, the single/predominantly ORP1L vesicles (red), the ORP1L/CD63 vesicles (yellow) and the single/predominantly CD63 vesicles (green).
Short time-lapse of CD63-eGFP endosomes in HeLa cells co-transfected with ORP1L-∆ORDPHDPHD.
Short time-lapse of CD63-eGFP endosomes in HeLa cells co-transfected with ORP1L-∆ORDPHDPHD.
To mechanistically uncouple the forced dynein recruitment from the block in ER-LE MCS formation by ORP1L-∆ORDPHDPHD, we expressed an ORP1L-FFAT mutant (where Y477 and D478 in the FFAT motif are mutated to alanines) that inhibits MCS formation but can still induce dynein recruitment (Wijdeven et al., 2016). ORP1L-FFATydaa overexpression showed moderate clustering of MVB (Fig. S5 A) but reduced fusion activity by almost twofold compared to ORP1L-wt (Fig. 4 J). This suggests that both dynein recruitment and ORP1L-mediated ER-LE MCS are promoting MVB-PM fusion.

Supplemental data related to Fig. 4. (A) Confocal analysis of ORP1L-∆ORDPHDPHD, -FFATydaa, or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells with single channel images in gray with inverted LUT. (B and C) Fluorescent signal patterns for CD63-pHluorin/mCherry-ORP1L-wt and -∆ORDPHDPHD averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. DC, Dual Color.

Supplemental data related to Fig. 4. (A) Confocal analysis of ORP1L-∆ORDPHDPHD, -FFATydaa, or -∆ORD (red) expression in CD63-pHluorin (green) HeLa cells with single channel images in gray with inverted LUT. (B and C) Fluorescent signal patterns for CD63-pHluorin/mCherry-ORP1L-wt and -∆ORDPHDPHD averaged over >12 events, synchronized using peak-intensity of CD63 signal at 60 s. DC, Dual Color.
Finally, to assess if ORP1L (as cholesterol-dependent molecular switch between dynein transport and ER-LE MCS formation) is still associated with MVBs that fuse with the PM, we performed DC TIRF-M on ORP1L expressing CD63-pHluorin HeLa cells. In agreement with the absence of Rab7 at the site of fusion (Fig. 2, C and D), neither ORP1L-wt nor ORP1L-∆ORDPHDPHD could be detected at the site of fusion (Fig. S5, B and C), suggesting the ORP1L regulation is an active upstream of fusion. Altogether this demonstrates that dynamic ORP1L-mediated ER-LE MCS have a stimulatory effect on MVB/PM fusion activity by affecting the motility and maturation of LE.
ORP1L ER-LE MCS and GTPase switching
Our results indicated that Rab7 is implicated in exosome secretion upstream of MVB-PM fusion by recruitment of ER-LE MCS mediator ORP1L while MVBs that fuse with the PM are negative for Rab7 and instead are only positive for Rab27 (Fig. 2 C). To strengthen the involvement of Rab27a in MVB-PM fusion, we first overexpressed WT and DN forms of Rab27a with CD63-pHluorin, resulting in a 2.8-fold increase and a 2.3-fold decrease in fusion activity, respectively (Fig. 5 A). This suggests that MVBs primed for fusion with the PM have exchanged Rab7a for Rab27a, either directly or through an intermediate step. To test this hypothesis, we co-expressed Rab27a with Rab7a and Arl8b, the two small-GTPases that showed the strongest overlap with CD63 compartments in the cell (Fig. 2, A and B). Close observation of subcellular compartments revealed that Rab27a endosomes did not show substantial overlap with Rab7a, whereas Rab27a decorated numerous Arl8b compartments (Fig. 5, B and D), consistent with a switch from Rab7a to Arl8b (Jongsma et al., 2020) prior to Rab27a conversion. As Arl8b has been previously involved in lysosome and lysosome-related organelle (LRO) transport toward and fusion with the PM (Tuli et al., 2013; Michelet et al., 2018), we tested if Arl8b could likewise be involved in ECV/MVB-PM fusion activity. Strikingly, knockdown of Arl8b resulted in an almost fourfold decrease in fusion activity (Fig. 5 E and Fig. S6 A), whereas overexpression of Arl8b-wt roughly doubled MVB-PM fusion activity, and overexpression of Arl8b-DN reduced fusion activity by 2.5-fold compared to WT (Fig. 5 F), further supporting a role for Arl8b in exosome secretion. To further solidify these results, we focused on endosomal Rab27a recruitment in cells overexpressing Arl8b, Rab5a, or Rab7a. Strikingly, Arl8b but not Rab5a or Rab7a overexpressing cells showed a pronounced recruitment of Rab27a to intracellular compartments (Fig. 5 G; and Fig. S6, B and D).

ORP1L ER-LE MCSs impact small GTPase switching. (A) Fusion activity of CD63-pHluorin HeLa cells overexpressing Rab27a-wt or Rab27a-DN. Measured over >70 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (B and C) Fluorescent analysis of HeLa cells co-expressing Rab27a-GFP and Rab7a-RFP (C) or co-expressing Rab27a-GFP and Arl8b-RFP. Green and red arrowheads indicate (some of the) non co-localizing endosomes, white arrowheads indicate co-recruitment to endosomes. (D) Overlap coefficient of Rab7a-RFP and Arl8b-RFP with Rab27a-GFP expressing HeLa cells. n ≥ 3 images (2≥ cells per image) analyzed from ≥2 independent experiments. Red line indicates median. (E) Fusion activity of CD63-pHluorin HeLa cells treated with siArl8b. siNTC, non-targeting control. Measured over >60 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (F) Fusion activity of CD63-pHluorin HeLa cells overexpressing Arl8b-wt or DN. Measured over >40 cells from n = 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (G) Fluorescent analysis of Rab27a-GFP localization in HeLa overexpressing Rab5a, Rab7a, or Arl8b. (H) Fluorescent analysis of Rab5a-, Rab7a-, or Arl8b-GFP HeLa cells co-expressing mCherry-ORP1L-wt, -∆ORD, or ∆ORDPHDPHD (red). (I) Co-localization analysis of H. PCC, Pearson’s Correlation Coefficient. Measured over n ≥ 4 images (2≥ cells per image) analyzed from three independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. (J) Fusion activity of CD63-pHluorin HeLa cells treated overexpressing ORP1L-∆ORDPHDPHD without or with Arl8b-DN co-expression. Measured over >70 cells from n = 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (K) Co-localization analysis of Rab27a-GFP/CD63-pHuji co-expressing HeLa, treated with siORP1L. To the right: quantification of Rab27a-GFP/CD63-pHuji overlap in ctrl (siNTC) or ORP1L (siORP1L) depleted condition. Measured over n ≥ 5 images (2≥ cells per image) analyzed from three independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. DN, dominant negative. Bars: 5 µm.
ORP1L ER-LE MCSs impact small GTPase switching. (A) Fusion activity of CD63-pHluorin HeLa cells overexpressing Rab27a-wt or Rab27a-DN. Measured over >70 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (B and C) Fluorescent analysis of HeLa cells co-expressing Rab27a-GFP and Rab7a-RFP (C) or co-expressing Rab27a-GFP and Arl8b-RFP. Green and red arrowheads indicate (some of the) non co-localizing endosomes, white arrowheads indicate co-recruitment to endosomes. (D) Overlap coefficient of Rab7a-RFP and Arl8b-RFP with Rab27a-GFP expressing HeLa cells. n ≥ 3 images (2≥ cells per image) analyzed from ≥2 independent experiments. Red line indicates median. (E) Fusion activity of CD63-pHluorin HeLa cells treated with siArl8b. siNTC, non-targeting control. Measured over >60 cells from n = 3 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (F) Fusion activity of CD63-pHluorin HeLa cells overexpressing Arl8b-wt or DN. Measured over >40 cells from n = 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (G) Fluorescent analysis of Rab27a-GFP localization in HeLa overexpressing Rab5a, Rab7a, or Arl8b. (H) Fluorescent analysis of Rab5a-, Rab7a-, or Arl8b-GFP HeLa cells co-expressing mCherry-ORP1L-wt, -∆ORD, or ∆ORDPHDPHD (red). (I) Co-localization analysis of H. PCC, Pearson’s Correlation Coefficient. Measured over n ≥ 4 images (2≥ cells per image) analyzed from three independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. (J) Fusion activity of CD63-pHluorin HeLa cells treated overexpressing ORP1L-∆ORDPHDPHD without or with Arl8b-DN co-expression. Measured over >70 cells from n = 4 experiments using Student’s two-tailed two-sample t test. Red line indicates median. (K) Co-localization analysis of Rab27a-GFP/CD63-pHuji co-expressing HeLa, treated with siORP1L. To the right: quantification of Rab27a-GFP/CD63-pHuji overlap in ctrl (siNTC) or ORP1L (siORP1L) depleted condition. Measured over n ≥ 5 images (2≥ cells per image) analyzed from three independent experiments using Student’s two-tailed two-sample t test. Red line indicates median. DN, dominant negative. Bars: 5 µm.

Supplemental data related to Fig. 5. (A) Analysis of Arl8b mRNA levels by quantitative PCR for RNAi-treated HeLa cells. Duplo’s measured from three independent experiments; error bar represents SEM. (B) Fluorescent analysis of Rab27a-GFP localization in HeLa overexpressing Rab5a, Rab7a, or Arl8b, as in Fig. 5 G. (C) Graphs showing quantification of number of Rab27a-GFP endosomes (left) and intensity of Rab27a-GFP endosomes (right) of small-GTPase expressing HeLa cells. n ≥ 3 images (2≥ cells per image) analyzed from ≥2 independent experiments. (D) Overlay images of Fig. 5 G, including the GTPase-RFP channels.

Supplemental data related to Fig. 5. (A) Analysis of Arl8b mRNA levels by quantitative PCR for RNAi-treated HeLa cells. Duplo’s measured from three independent experiments; error bar represents SEM. (B) Fluorescent analysis of Rab27a-GFP localization in HeLa overexpressing Rab5a, Rab7a, or Arl8b, as in Fig. 5 G. (C) Graphs showing quantification of number of Rab27a-GFP endosomes (left) and intensity of Rab27a-GFP endosomes (right) of small-GTPase expressing HeLa cells. n ≥ 3 images (2≥ cells per image) analyzed from ≥2 independent experiments. (D) Overlay images of Fig. 5 G, including the GTPase-RFP channels.
This could indicate that in order to recruit Rab27a to endosomes, a Rab7a-to-Arl8b identity switch (Jongsma et al., 2020) needs to occur and would then simultaneously promote dissociation of (Rab7a effector) ORP1L. To study if ORP1L ER-LE MCS impacts small-GTPase switching, we co-expressed ORP1L-wt and mutants with Rab5a, Rab7a, and Arl8b (Fig. 5 H). Overlap between ORP1L and Rab5a was always low, whereas ORP1L overlap with Rab7a was consistently high (Fig. 5, H and I). This can be likely explained by the fact that ORP1L is a direct effector of and binds to Rab7a through its ankyrin domain (Johansson et al., 2005), whereas Rab5a acts upstream of ORP1L. Interestingly, ORP1L overlap with Arl8b varied depending on the conformational mutant of ORP1L. Whereas the ∆ORD mutant showed a low level of overlap with Arl8b, overlap with ORP1L-∆ORDPHDPHD was high (Fig. 5, H and I), correlating with the effects of the ORP1L mutants on ECV/MVB-PM fusion activity (Fig. 4 E). This suggests that ORP1L-∆ORDPHDPHD could stimulate fusion activity in an Arl8b-dependent manner, by promoting the Rab7a-to-Arl8b handover. Indeed, the increase in fusion activity observed for (Fig. 4 E) was decreased to normal levels in combination with Arl8b-DN overexpression (Fig. 5 J). Remarkably, overexpression of ORP1L mutants did not affect Rab27a localization to the same extent as it did for Rab7a and Arl8b even though for a number of cells expressing the ORP1L-∆ORDPHDPHD mutant, we observed an increased Rab27a recruitment toward endosomes (Fig. S7, A–C). These endosomes were, however, in majority not positive for ORP1L-∆ORDPHDPHD, suggesting that the Rab7a-to-Arl8b identity switch it promotes (Jongsma et al., 2020) also results in a net loss of ORP1L-∆ORDPHDPHD association with Arl8b compartments. Finally, to validate if interference with ORP1L and Arl8b impaired Rab27a recruitment to CD63 compartments, we depleted both targets using RNAi (Fig. S3 B and Fig. S6 A), and indeed observed a significant decrease in overlap between Rab27a and CD63 upon ORP1L (Fig. 5 K) or Arl8b (Fig. S7 D) depletion. Together, these results indicate that to render the fusion competent, ECV/MVBs undergo a small-GTPase cascade, switching from Rab7a to Arl8b to Rab27a. The first conversion from Rab7a to Arl8b is promoted by ORP1L, facilitating the dynein-mediated ECV/MVBs centripetal movement. After the Rab7a-to-Arl8b switch is completed (Jongsma et al., 2020), Arl8b (kinesin)-mediated ECV/MVBs centrifugal movement allows for a switch to Rab27a, losing Arl8b and rendering the ECV/MVBs PM fusion competent.

Supplemental data related to Fig. 5. (A–C) Fluorescent analysis for Rab27a-GFP (green) HeLa co-expressing (A) mCherry-ORP1L-wt, (B) -∆ORD, or (C) -∆ORDPHDPHD (red). (D) Co-localization analysis of Rab27a-GFP/CD63-pHuji co-expressing HeLa, treated with siArl8b. To the right: quantification of Rab27a-GFP/CD63-pHuji overlap in ctrl (siNTC) or Arl8b (siArl8b) depleted condition. Bars: 5 µm.

Supplemental data related to Fig. 5. (A–C) Fluorescent analysis for Rab27a-GFP (green) HeLa co-expressing (A) mCherry-ORP1L-wt, (B) -∆ORD, or (C) -∆ORDPHDPHD (red). (D) Co-localization analysis of Rab27a-GFP/CD63-pHuji co-expressing HeLa, treated with siArl8b. To the right: quantification of Rab27a-GFP/CD63-pHuji overlap in ctrl (siNTC) or Arl8b (siArl8b) depleted condition. Bars: 5 µm.
Discussion
Exosome secretion is a highly dynamic process, but how intracellular mechanisms control their release is not well understood. Using a recently developed TIRF-based dual-color live-imaging approach (Bebelman et al., 2020), we interrogated the molecular identity of PM-fusing CD63-positive LE/MVBs, and investigated how this process is impacted by key mediators of MCS between these LE compartments and the ER. Our data show that PM-fusing LEs represent an acidic, (low-level) LAMP1-positive but non-proteolytic sub-class of ECV/MVBs. These compartments lack typical early/recycling endosome markers such as Rab5a/11/35 and EEA1, but instead undergo a Rab7-Arl8b/Rab27a cascade to fuse with the PM. LE VAP-A/B tethering partners protrudin and ORP1L showed a distinct capacity to affect ECV/MVB-PM fusion activity. Intracellularly, this correlated with an altered LE motility and small-GTPase maturation state. Altogether our results implicate that ER-LE MCS as novel players in the regulation of exosome release where they control a sequential retrograde-anterograde transport maneuver coordinated with small-GTPase switching to promote MVB secretion.
Our research mainly focused on exosome secretion under normal “steady-state” cell-culture conditions from CD63 positive ECV/MVBs that contained low levels of LAMP1 but were not proteolytically active. Although CD63 is considered as a broad marker for MVBs, we therefore cannot exclude that the ECV/MVB PM-fusing population we studied represents a subpopulation of MVBs different from those low- or negative in CD63 (Austin et al., 2004; Fan et al., 2020,Preprint) or those formed upon stimulation (Eden et al., 2016; Fan et al., 2020,Preprint) or later endosomal structures that are secreted in a protrudin-dependent manner (Raiborg et al., 2015).
LAMP1-positivity has long been used as “classical” marker for lysosomes although its targeting to these compartments requires its trafficking through early and LEs to reach lysosomes (Cook et al., 2004; Ebrahim and Thilo, 2011), indicating LAMP1 transits through ECV/MVB (Cheng et al., 2018). Another study indicated that endolysosomes are the main site of hydrolase activity, whereas terminal storage lysosomes are cathepsin inactive and have a non-acidic pH (Bright et al., 2016). In our study, we could not detect proteolytic activity in PM-fusing MVBs (Fig. 1, H and K), neither did impairment of endolysosomal membrane trafficking by Apilimod affect MVB/PM fusion activity (Fig. 1 E), consistent with the absence of TRPML1 (Fig. 1, I and K). This strongly suggests CD63 positive PM fusing compartments represent ECV/MVBs at pre-endolysosomal stage. This is in line with some viral proteins that effectively escape lysosomal degradation by associating with CD63 to ensure exosomal targeting, while similarly for other proteins association with CD63 prevents them from ESCRT-dependent sorting for degradation (Verweij et al., 2011; van Niel et al., 2011).
To better characterize the PM-fusing CD63+ ECV/MVB compartments, we performed a small-scale DC-TIRF-M screen for the presence of small GTPases at the site of fusion. While the unique properties of the evanescent excitation field of TIRF-M have been exploited before to study docking of LE compartments to the PM (Ostrowski et al., 2010; Hsu et al., 2010), the combination with a pH-sensitive MVB marker such as CD63-pHluorin adds the distinct capacity to study precisely the MVBs that fuse with the PM (Verweij et al., 2018). The various small GTPases we tested were selected based on literature (Blanc and Vidal, 2018). The absence of Rab11 and Rab35 (Fig. 2 C) indicates that in HeLa under steady-state conditions, CD63 ECV/MVB fusion is distinct from lysosomal secretion and exosome secretion in oligodendrocytes (Escrevente et al., 2021; Hsu et al., 2010), even though we cannot formally rule out an indirect role (van der Sluijs et al., 2013). While the absence of Rab5a at the site of fusion is not surprising, the absence of Rab7a is (Fig. 2, C and D). Indeed, siRNA interference with Rab7 decreased CD63, syntenin, and syndecan in small EVs (Baietti et al., 2012), and in the present study significantly impaired CD63-pHluorin fusion activity (Fig. 3 A). This suggests that despite its absence at the site of fusion, Rab7a plays an important role in exosome biogenesis. ORP1L and protrudin, which exert their effect on LE transport in a Rab7-dependent manner (Wijdeven et al., 2015), were likewise capable of modulating exosome secretion (Fig. 3, A and C; and Fig. 4), further consolidating this notion. Similar to Rab7a, Arl8b had a significant overlap with CD63 inside the cell (Fig. 2, A and B) but was absent at the site of fusion (Fig. 2, C and G), whereas both knockdown and overexpression of Arl8b had a substantial impact on CD63-pHluorin fusion activity (Fig. 5, E and F). Yet, we were only able to detect Rab27a and Rab27b at the site of PM fusion. Interestingly, Rab27 is implicated in (docking and) the secretion of MVBs as well as various other secretory pathways (Fukuda, 2013; Ostrowski et al., 2010). This suggests that, akin to various LRO, secretory MVBs undergo a handover from Rab7a through Arl8b (Jongsma et al., 2020) to Rab27a when switching from the microtubule to the cortical actin network (Pu et al., 2016; Ostrowski et al., 2010). In this scenario, Rab7a will already have dissociated from LE, consistent with a SKIP/HOPS-mediated inactivation of Rab7 from LE membranes during the Rab7-to-Arl8b handover (Jongsma et al., 2020) and the absence of ORP1L at the site of fusion (Fig. S4, H and I). While ORP1L (mutant) overexpression efficiently redistributed Rab7a and Arl8b endosomes (Fig. 5 H), it had a much less pronounced effect on Rab27a (Fig. S7, A–C). Concomitantly, Rab27a showed more interaction with Arl8b than Rab7a (Fig. 5, B–D and G; and Fig. S6, B–D). Likewise, overexpression of Arl8b but not Rab7a resulted in a more pronounced recruitment of Rab27a to endosomal compartments, reminiscent of the effects on Rab27a recruitment observed for Von Willebrand factor overexpression (Hannah et al., 2003). Whether all MVBs that fuse with the PM initially were positive for Rab7a cannot be stated with absolute certainty, but multiple lines of evidence point in this direction. Firstly, RNA interference with Rab7a as well as Rab7a-wt and -DN overexpression directly impacted MVB/PM fusion, which was also confirmed by Western blot (Fig. S3 A). Secondly, ORP1L is a direct effector of Rab7a, and both proteins show a high level of overlap (PCC ∼0.8, Fig. 5, H and I). While interference with Rab7a could potentially be explained by indirect/secondary effects, ORP1L-∆ORD OE directly immobilizes CD63/Rab7a(+/+) endosomes (Fig. 4 I) and simultaneously strongly inhibit MVB/PM fusion and EV release (Fig. 4, E and F). Altogether our data support a small-GTPase cascade model where (CD63) ECV/MVBs subsequently undergo a Rab7a to Arl8b to Rab27a conversion to acquire secretory capacity and could be considered as LRO.
Our data highlight how the ER impacts exosome secretion through the formation of MCS between the ER and LE/MVBs that result in the gain of secretory capacities for MVBs. We studied the impact of two ER-LE tethers previously described in literature, protrudin and ORP1L that, respectively, promote LE translocation toward the periphery by kinesin loading (Raiborg et al., 2015) or cluster LE by recruiting dynein in its closed conformation (Rocha et al., 2009). To our surprise, both machineries but also overexpression of RILP, promoting dynein recruitment and perinuclear translocation of LE (Jongsma et al., 2020), as well as stimulation of kinesin-mediated movement of LE toward the periphery by overexpression of Arl8b or SKIP, all had a stimulatory effect on fusion activity (Fig. 3 C; and Fig. 4, E, K, and L; and Fig. 5 F). Whether these mechanisms act simultaneously, separately, or sequentially on the same endosome remains to be determined, but our results support a sequential model, where Rab7a/ORP1L/dynein acts upstream of Arl8b/kinesin and Rab27a. This combines a movement toward the MTOC (dynein) impacted by ORP1L ER-LE MCS, followed by a movement away from the MTOC (kinesin) toward the PM through Arl8b (Fig. 6). Forced dynein recruitment by ORP1L-∆ORDPHDPHD caused a remarkable peri-nuclear accumulation of LE that nevertheless stimulated fusion activity. Altogether this suggests that dynein-mediated accumulation of MVBs in the perinuclear region expedites their acquisition of PM-fusion competency, likely by facilitating the recruitment of Arl8b, in turn promoting recruitment of Rab27a (Fig. 5, B–D; and Fig. S6, B and C). A recent insightful study on the interplay between Rab7a and Arl8b (Jongsma et al., 2020) observed an effect of endosomal positioning both by SKIP and RILP overexpression on endosomal maturation at ultrastructural level. An earlier study showed that peripherally localizing endosomes are less acidic and have less degradative properties (Johnson et al., 2016). As these SKIP and RILP machineries also act on the lysosome positioning and activity, it is tempting to speculate that the sequential and coordinated process of retrograde/anterograde transport coupled to Rab-GTPase switching will control not only exosome secretion but also lysosomal targeting of MVBs. Precisely how perinuclear localization facilitates this recruitment warrants further exploration, as it would add important new knowledge to our mechanistic understanding of the switch from Rab7a to Arl8b.

Proposed model for LE/MVB trafficking prior to exosome release under steady state conditions. Dynamic ORP1L-mediated ER-LE/MVB membrane contact sites ensure maturation and RILP/dynein-mediated transport of non-catalytic CD63/LAMP1/Rab7+ ECV/MVB toward the MTOC. This centripetal movement subsequently facilitates the switch from Rab7 to Arl8b, which in turn allows kinesin-mediated transport toward the periphery and a switch from Arl8b to Rab27a, bypassing lysosomal compartments, and rendering CD63/LAMP1+ LE competent to fuse with the plasma membrane (PM). EE, early endosome; ECV, Endocytic Carrier Vesicles; MVB, multivesicular bodies; LE, late endosome; L, lysosome; E/L, endolysosome; MTOC, microtubule-organizing center. Microtubules are depicted in green.
Proposed model for LE/MVB trafficking prior to exosome release under steady state conditions. Dynamic ORP1L-mediated ER-LE/MVB membrane contact sites ensure maturation and RILP/dynein-mediated transport of non-catalytic CD63/LAMP1/Rab7+ ECV/MVB toward the MTOC. This centripetal movement subsequently facilitates the switch from Rab7 to Arl8b, which in turn allows kinesin-mediated transport toward the periphery and a switch from Arl8b to Rab27a, bypassing lysosomal compartments, and rendering CD63/LAMP1+ LE competent to fuse with the plasma membrane (PM). EE, early endosome; ECV, Endocytic Carrier Vesicles; MVB, multivesicular bodies; LE, late endosome; L, lysosome; E/L, endolysosome; MTOC, microtubule-organizing center. Microtubules are depicted in green.
Protrudin-mediated ER-LE MCS are dynamic in nature (Raiborg et al., 2015). Our data on ORP1L(-∆ORD) show a tight ER enwrapment of the LE, with significant effects on their motility and maturation. While solely based on these data, it would be tempting to speculate that ORP1L ER-LE MCS have an inhibitory effect on exosome biogenesis, our data as a whole argue for a more dynamic, positive regulatory mechanism. First, ORP1L by nature is a dynamic regulatory protein (based on its conformational state). Second, the depletion of ORP1L using RNAi impedes MVB-PM fusion activity (Fig. 3 A). Third, ORP1L-wt overexpression stimulates it (Fig. 4, E–G). Fourth, and by contrast, overexpression of ORP1L-FFATydaa, still allowing RILP-mediated dynein recruitment (Wijdeven et al., 2016) but blocked in its capacity to establish ER-LE MCS, inhibited MVB-PM fusion activity (Fig. 4 J). Altogether, this strongly suggests that dynamic ER-LE MCS have a positive impact on MVB-PM fusion activity. This notion is further supported by recent studies indicating that ER-LE interactions can be both highly dynamic and even reciprocal as LE/Lys now emerge as shaping ER structure and distribution (Guo et al., 2018; Lu et al., 2020 Preprint).
Thus, by combining literature with our results there emerges a picture of an ER-LE interplay that, based on distinct combinations of molecular tethers, dynamically and diversely regulates ECV/MVB transport and maturation including GTPase switching (Fig. 6). These processes combined have the capacity to tune exosome secretion. Intriguing questions that arise from these data are what consequences ER stress (e.g., in cancer) have on exosome secretion, and how this might impact their maturation and content. Indeed, Unfolded Protein Response- and cancer-related ER stress have been reported to augment MVB formation, exosome release, as well as modify exosomal protein, miRNA, and lipid composition (Kanemoto et al., 2016; Liu et al., 2019; Kakazu et al., 2016; Collett et al., 2018; Barman et al., 2022). Besides cancer, MCS dysfunction is thought to play important roles in a number of diseases, including diabetes, neurodegenerative disorders such as Alzheimer’s Disease, and viral and bacterial infections (Prinz et al., 2020; van der Schaar et al., 2016). Interestingly, MCS formation seems amenable to drug-targeting, as for example our data indicate that (drug targeting) of LE cholesterol status affects exosome secretion from cells. The tight link between ER-MCS and LE maturation indicates that we might also affect exosomes qualitatively. Targeting ER-LE MCS could therefore contribute to the development of novel approaches to target and manipulate exosomal communication in disease. We expect that future research will further uncover the full extent into which ER-LE MCS impact all these aspects that are central to the physiology and pathology of exosomes.
Materials and methods
Cell lines
HeLa cells were cultured in DMEM supplemented with 10% FBS (Perbio Sciences; HyClone), 100 U/ml penicillin G, 100 mg/ml streptomycin sulfate, and 2 mM glutamine, split 1/10 at sub-confluency levels (85–90%) and maintained at 37°C and 5% CO2.
Plasmids
CD63-pHluorin (ID: 130901; Addgene) and CD63-pHuji (ID: 130902; Addgene) plasmids were described before (Verweij et al., 2018; Bebelman et al., 2020). LAMP1-GFP was a kind gift from Esteban Dell’Angelica (University of California, Los Angeles, Los Angeles, CA; ID 34831; Addgene). The CD63-mRFP plasmid was a kind gift of Cynthia A. Leifer (Cornell University, Ithaca, NY). The Rab5a-mCherry constructs were a kind gift from Bruno Goud (Institut Curie, Paris, France). Rab7a-mCherry constructs were obtained from Sabrina Simoes (Columbia University, New York, NY). Rab11a-mCherry was obtained from Jean Salamero (Institut Curie, Paris, France). Rab27a-GFP, Rab27b-GFP, and mutant constructs were a kind gift from Robert Ballotti (C3M—Inserm U1065, Centre Méditerranéen de Médecine Moléculaire, Nice, France). Arl8b-GFP and -mCherry constructs were a gift from Volker Haucke (FMP Berlin, Berlin, Germany). Protrudin-wt (myc/GFP), -FYVE4A (myc/GFP), and -∆ FYVE (myc/GFP) were a kind gift from Harald Stenmark and Camilla Raiborg (Oslo University Hospital, Oslo, Norway). All ORP1L plasmids (mCherry-ORP1L-wt, mCherry-ORP1L-∆ORD, mCherry-ORP1L-∆ORDPHDPHD, HA-ORP1L-wt, HA-ORP1L-∆ORD, HA-ORP1L-∆ORDPHDPHD, and HA-ORP1L-FFAT/-∆ORDydaa) were kind gifts from Jacques Neefjes (Leiden University Medical Center, Leiden, The Netherlands). VAMP2-pHluorin and NPY-pHluorin were gifts from J. Rothman (Yale University, New Haven, CT) and R. Holz (University of Michigan, Ann Arbor, MI). EEA1-GFP was a kind gift from Subbarao Gangisetty (Indian Institute of Science, Bengaluru, India). TRPML1-GFP was a kind gift from Sandip Patel (University College London, London, UK). LC3B-RFP was a kind gift from Sébastien Nola (Institute for Psychiatry and Neurosciences of Paris, Paris, France). VAPA-GFP was a kind gift from Francesca Giordano (Institut de Biologie Integrative de la Cellule, Gif-sur-Yvette, France). All plasmids were transfected using jetPRIME (PolyPlus) transfection reagent using the manufacturer’s recommendations at ∼ 0.5–0.75× the suggested amounts of DNA.
siRNA sequences and protocol
ON-TARGETplus siRNA pools were obtained from Horizon Discovery LTD, targeting Human OSBPL1A (Cat ID 114876, L-008350-00-0005), Rab7a (Cat ID 7879, L-010388-00-0005), ZFYVE27 (Cat ID 118813, L-016349-01-0005), and Arl8b (Cat ID 55207, L-020294-01-0005), and were resuspended and transfected at 30 nM according to the manufacturer’s instruction. For live-cell quantification of MVB-PM fusion events, cells were seeded on coverslips after 24 h of siRNA transfection, transfected after 2 d with CD63-pHluorin, and imaged by TIRF microscopy 16–24 h later.
RT-PCR
HeLa cells transfected once with the indicated that siRNAs were collected after 3 d. The RNA was extracted using the NucleoSpin RNA, Mini kit (Macherey-Nagel), and the cDNA was generated from 0.3 µg of RNA using the SuperScript First-Strand Synthesis System (Invitrogen) following manufacturer’s protocols. RT-PCR was performed using the LightCycler 480 SYBR Green I Master (Roche) on plate-based RT-PCR amplification and detection instrument LightCycler 480 (Roche). GAPDH was used as an endogenous normalizer.
The primer sequences (95% of efficiency) are: 5′-ATGCCTTTGTGTTCCTTGCTG-3′ and 5′-GAGTACCATGCACCCTCATTC-3′ for Protrudin; 5′-AAGCATGTGGGAGCGGTAT-3′ and 5′-CGATCTGCAGCATCTATCATGT-3′ for Arl8b and 5′-CATCCTGGGAGATTCTGGAGTC-3′ and 5′-TGTGTCCCATATCTGCATTGTG-3′ for Rab7a. Experiments were performed with at least three biological replicates. The method ΔΔCT was used to obtain the relative expression levels and the ratio between the control and gene of interest was calculated with the formula 2−ΔΔCT.
Drugs
U18666A (Merck) was used at 2 μg/ml overnight (16 h). Apilimod (STA 5326; Axon MedChem) was used at 0.2 μM for 2 h.
Antibodies/reagents
Mouse monoclonal antibodies against CD63 (H5C6; BD; clone TS63b, gift of Eric Rubinstein, Centre d'Immunologie et des Maladies Infectieuses, Paris, France) were used at 1:200 and 1:2,000, respectively, and mouse monoclonal against CD9 (clone TS9 36; gift of Eric Rubinstein) was used 1:2,000. Polyclonal antibody against LAMP1 (PA1-654A; Thermo Fisher Scientific) was used 1:100. Rabbit monoclonal antibody against ORP1L (ab132265; Abcam) was used 1:1,000. Rabbit polyclonal antibody against alpha-Tubulin (ab4074; Abcam) was used 1:4,000. MagicRed (ICT 938; Bio-Rad) was reconstituted according to the manufacturer’s recommendation and added to the cells at a final dilution of 1:2,600 5 min before live-cell imaging. For immune-fluorescence, cells were seeded on coverslips, fixed in 2% PFA (in PBS) for 20 min at RT, incubated with blocking/washing buffer (1X PBS/1% BSA/0.1% Saponin) for 1 h at RT. The slides were incubated with primary antibody for 45 min, secondary antibody (AlexaFluor conjugates, Thermo Fisher Scientific) at 1:2,500 for 45 min at RT, mounted with the Vectashield reagent (Vector Laboratories), and sealed with nail polish.
Light microscopy
Cells were grown in 35-mm imaging chambers (FluoroDish WPI; IBIDI). Dual-color TIRF imaging was performed as previously described (Bebelman et al., 2020). In short, a Ti-E-inverted microscope setup was used with a laser bench (Roper Technologies) coupled into a 100 × CFI Apochromat TIRF, oil, 1.49/0.12-mm objective (Nikon). Images were acquired at 2 Hz unless noted otherwise and were acquired with MetaMorph software (Universal Imaging; Molecular Devices). A barrel pipette (ALpA-VM4) was used to apply NH4+ solution to the cells to deacidify intracellular pH and visualize (CD63-pHluorin/-pHuji) endosomes near the coverslip using standard Tyrode’s solution formulation where 50 mM NH4Cl was exchanged for NaCl. Live-imaging experiments were performed in culture medium at 37°C and 5% CO2, unless indicated otherwise. For live-acquisition of endosomal motility and appearance, a spinning-disk (Fig. S1 B) and a Leica SP5 Confocal (Fig. 4 H) setup were used and equipped with a 60× CFI Plan Apo VC NA 1.4/0.13 mm (Nikon) and a 63× NA 1.4/0.1 mm (Leica) objective, respectively, acquired using Leica Application Suite software. The spinning-disk and TIRF setups were both equipped with an Evolve (Photometrics) 512 × 512 EMCCD camera.
For fixed imaging (all localization studies, except TIRF experiments and the experiments shown in Fig. 4, C and H; and Fig. S4, B, D, and F), cells were analyzed on a confocal (Leica SP5) as mentioned above. For Fig. 4 C and Fig. S4 B, a Nikon Ni-U wide-field setup (100× Plan Apo VC 1.4/0.13 mm) equipped with a photometric CCD Camera CoolSNAP HQ2 was used, acquired with MetaMorph software. The images in Fig. S4, D, and F were acquired with a Zeiss LSM 700 (AxioObserver Z1) equipped with a 63 × 1.4/0.19 mm oil objective using Zen 2,011 software (Zeiss). Fixed samples were imaged at room temperature.
Image analysis
To visualize fusion events of MVBs with the PM, we expressed the live-cell optical exosome reporters consisting of the EV-enriched tetraspanin CD63 fused to (super ecliptic) pHluorin- or pHuji pH-sensitive fluorescent proteins in its first extracellular loop (EC1; Bebelman et al., 2020; Verweij et al., 2018; Shen et al., 2014; Miesenbock et al., 1998). The acidic late-endosomal pH underlies the reason that PM fusion results in a burst of fluorescence from CD63-pHluorin, which can be observed by live microscopy approaches, including spinning-disk microscopy and TIRF as used in this study.
Here, fusion events are defined as sudden increases in fluorescent intensity and were analyzed as before (Verweij et al., 2018; Bebelman et al., 2020), i.e., manually and using the AMvBE macro for quality control (Bebelman et al., 2020). Fusion activity was defined as the number of events over the course of a time-lapse experiment, which was kept consistent within experiments and replicates, but varied between experiments, but was typically 3 min. On average, ≥15 cells were imaged per condition in ≥5 different imaging windows in ≥3 independent replicates.
To determine the apparent size (radius) of fusion events, we first determined the lateral position of the event using Fiji (ImageJ) software. In short, we performed a wavelet transform to remove noise. Subsequently, we identified fusion events that we corrected in time and space by determining max intensity pixel value to reposition the XYT position to of the vesicle seen as a spot. When the XYT position is identified, a line passing through the position XY was drawn and a 1D Gaussian was fitted to extract the radius. We repeated the fitting procedure for each 30° angle rotation of the line and kept only the best goodness-of-fit (R2) as a reference for the radius. Subsequently, we proceeded with a 2D Gaussian fit to extract the radius.
To assess the overlap ratio between MagicRed and CD63/LAMP, MagicRed compartments were randomly selected from single-channel images and saved as ROIs. Subsequently, the CD63/LAMP1 channels were loaded and ROIs were scored for the presence (1) or absence (0) of CD63/LAMP1 endosomes overlapping with or encircling the ROI. This approach was chosen as LAMP1 compartments appear as circles (“donut shapes”) that do not physically overlap with the MagicRed signal, complicating conventional co-localization analysis.
Acidic vesicles are manually counted upon superfusion of the cells with a NH4+ solution using a barrel pipette as described above. Subtraction of pre- and post-superfusion images reveals the number of acidic vesicles near the PM, using the “subtract” operation in the Image calculator tool of Fiji (ImageJ) software, as previously performed (Verweij et al., 2018).
To analyze vesicle motility (Fig. 4 I), we used the TrackMate plugin of Fiji with identical settings for all conditions, according to the author’s instructions (Ershov et al., 2021 Preprint). Colocalization analysis was performed by determining the PCC or Mander’s Coefficient (as specified in the figures and/or figure legends) using Fiji plugins on ≥9 individual cells in ≥3 independent experiments with identical parameters (Costes’ automated thresholding) for each experimental set of conditions that was compared.
Electron microscopy/CLEM on section
CLEM was performed largely as described before (Verweij et al., 2018). In short, HeLa cells were seeded on sapphire discs coated with 20-nm carbon and finder grids, and transfected with CD63-pHluorin- and ORP1L-encoding plasmids 24 h later. The following day, sapphire discs were transferred to 3-mm carrier discs and high-pressure frozen using (HPM Live μ, CryoCapCell). After cryo-immobilization, the specimens were freeze substituted with Lowicryl (HM20) in an AFS-2 as previously described (Heiligenstein et al., 2014). The blocks were trimmed and sectioned in 250- and 90-nm sections, after which the sections were recovered on grids, labeled with Hoechst, and imaged on a wide-field microscope at different magnifications to facilitate correlation with EM data. Grids were contrasted and examined with a Tecnai Spirit electron microscope (FEI Company), and digital acquisitions were made with a numeric camera (Quemesa; Soft Imaging System). Correlations between light- and electron microscopy data were performed using eC-CLEM (Paul-Gilloteaux et al., 2017).
Small EV isolation
Exosomes and other small extracellular vesicles (sEVs) were prepared from the supernatant of 24-h-cultured HeLa cells as previously described (Verweij et al., 2018). In brief, sEVs were purified from the cultured media with sEV-free serum. Differential centrifugations at 500 g (2 × 10 min), 2,000 g (2 × 15 min), and 10,000 g (2 × 30 min) eliminated cellular debris, and centrifugation at 100,000 g (60 min) pelleted sEVs. The sEV pellet was washed once in a large volume of PBS, centrifuged at 100,000 g for 1 h, and resuspended in 100 μl PBS or directly in lysis buffer.
Western blotting
For Western blotting, cells or sEVs were lysed in a 1% SDS buffer, and equal amounts of protein were loaded onto an SDS-PAGE gel. Only gels for CD63 and CD9 detection were run under non-reducing conditions. Western blots were imaged by ChemiDoc (Biorad Laboratories) or the Amersham ImageQuant 800 (GE Healthcare), and quantified using Image Lab v 6.0.1 software.
Image analysis
Pearson’s correlation coefficient between two channels was quantified using JACoP plugin of ImageJ Fiji software.
Statistical analysis
Statistical data are presented as mean ± SEM. Statistics were calculated using unpaired Student’s t test, or ordinary one-way ANOVA (Fig. 4, F, G, I, and J) using GraphPad Prism 9. For all parametric tests, data distribution was assumed to be normal but this was not formally tested. For ANOVA multiple comparisons, Tukey post hoc tests were used. Significant differences between control and treated samples are indicated (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05). Only P < 0.05 was considered as statistically significant.
Online supplemental materials
Fig. S1 shows supplemental data related to Fig. 1. Fig. S2 shows supplemental data related to Fig. 2. Fig. S3 shows supplemental data related to Fig. 3. Fig. S4 shows supplemental data related to Fig. 4. Fig. S5 shows supplemental data related to Fig. 4. Fig. S6 shows supplemental data related to Fig. 5. Fig. S7 shows supplemental data related to Fig. 5. Video 1 shows DC-TIRF-M fusion event example of CD63-pHuji HeLa cell expressing LAMP1-GFP. Video 2 shows short time-lapse of HeLa cell co-expressing LAMP1-GFP and CD63-RFP in untreated condition (left), and upon treatment of apilimod (2 h) in the middle and at the bottom of the cell (middle and right panel, respectively). Video 3 shows DC-TIRF-M fusion event example of CD63-pHluorin HeLa cell incubated with active Cathepsin B dye MagicRed. Video 4 shows short time-lapse of CD63-eGFP HeLa cells transfected with ORP1L-∆ORD-RFP (Fig. 4 H), showing from left to right the overlay, the single/predominantly ORP1L vesicles (red), the ORP1L/CD63 vesicles (yellow), and the single/predominantly CD63 vesicles (green). Video 5 shows short time-lapse of CD63-eGFP endosomes in HeLa cells co-transfected with ORP1L-∆ORDPHDPH.
Acknowledgments
We greatly acknowledge the Nikon Imaging Centre at Institut Curie, the Cell and Tissue Imaging Centre (PICT-IBiSA), the Center for Neurogenetics and Cognitive Research imaging platform of the Neuroscience Campus Amsterdam, and the NeurImag facility of the Institute of Psychiatry and Neuroscience of Paris. We thank Philippe Bun (NeurImag) for assistance with image analysis. We thank Camilla Raiborg and Jacques Neefjes for constructs and advice, and Stéphanie Miserey-Lenkei for insightful discussions.
This article was funded by a European Molecular Biology Organization grant (EMBO ALTF 1383-2014), a Fondation ARC pour la Recherche sur le Cancer fellowship (PJA 20161204808), and a Dutch Cancer Fund (KWF-YIG 12849) to F.J. Verweij, a Dutch Organizations for Scientific Research–Amsterdam Institute for Molecules, Medicines, and Systems STAR Graduate Program grant (022.005.031) and a Cancer Center Amsterdam travel grant to M.P. Bebelman, a Dutch Cancer Fund (KWF-5510) and a Cancer Center Amsterdam–VU University Medical Center grant to D.M. Pegtel, and a Fondation pour la Recherche Médicale grant (AJE20160635884) to G. van Niel and an Institut National Du Cancer grant (INCA N°2019-125 PLBIO19-059) to F.J. Verweij and G. van Niel. The Nikon Imaging Centre at Institut Curie is member of the French National Research Infrastructure France-BioImaging (ANR10-INBS-04).
The authors declare no competing financial interests.
Author contributions: F.J. Verweij, M.P. Bebelman, A.E. George, D.M. Pegtel, and G. van Niel designed, performed, and analyzed experiments. F.J. Verweij and G. van Niel wrote the paper. D.M. Pegtel, M.P. Bebelman, and G. Raposo edited the manuscript. M.P. Bebelman, X. Heiligenstein, and G. van Niel performed the CLEM experiments. Julia Sires-Campos performed qPCR experiments. M. Couty, A. E. George, A. Bécot, and R. Palmulli performed Western blotting experiments. F.J. Verweij and G. van Niel supervised the study.

