


Ciliary receptors and their certain downstream signaling components undergo intraflagellar transport (IFT) as BBSome cargoes to maintain their ciliary dynamics for sensing and transducing extracellular stimuli inside the cell. Cargo-laden BBSomes pass the transition zone (TZ) for ciliary retrieval, but how this passage is controlled remains elusive. Here, we show that phospholipase D (PLD)-laden BBSomes shed from retrograde IFT trains at the proximal ciliary region right above the TZ to act as Arf-like 3 (ARL3) GTPase-specific effectors in Chlamydomonas cilia. Under physiological condition, ARL3GDP binds to the membrane for diffusing into cilia. Following nucleotide exchange, ARL3GTP detaches from the ciliary membrane, binds to retrograde IFT train-shed and PLD-laden BBSomes at the proximal ciliary region right above the TZ, and recruits them to pass the TZ for ciliary retrieval likely via diffusion. ARL3 mediates the ciliary dynamics of certain signaling molecules through facilitating BBSome ciliary retrieval, providing a mechanistic understanding behind why ARL3-related Joubert syndrome shares overlapping phenotypes with Bardet-Biedl syndrome.
Introduction
The cilium refers to the axonemal microtube-based subcellular organelle projecting from the cell surface of most eukaryotic cells. They act as antennas for sensing and transducing the extracellular stimuli into the cell, essential for maintaining many physiological and developmental signaling pathways (Goetz and Anderson, 2010; Nachury and Mick, 2019; Singla and Reiter, 2006). Ciliary malfunction thus causes a group of related genetic disorders collectively named ciliopathies (Hildebrandt et al., 2011). Underlying ciliopathies is the fact that many G protein-coupled receptors (GPCRs), ion channels, and their certain downstream signaling components position to and traffic inside the ciliary membrane by motor protein-driven intraflagellar transport (IFT) trains along the axoneme (Liu et al., 2020; Nachury and Mick, 2019; Schneider et al., 2005; Yeh et al., 2013). During this process, the BBSome composed of multiple BBS proteins links these signaling proteins to IFT trains composed of repeating units of IFT-A (6 subunits) and -B (16 subunits subdivided into IFT-B1 and -B2 entities) complexes by acting as an IFT cargo adaptor (Fan et al., 2010; Jin et al., 2010; Lechtreck et al., 2009; Loktev et al., 2008; Nachury et al., 2007; Nakayama and Katoh, 2020; Taschner and Lorentzen, 2016; Wang et al., 2009). Therefore, defects in assembly, composition, or ciliary turnover of the BBSome all can lead to loss and/or abnormal buildup of these signaling proteins in cilia, eventually causing ciliopathic Bardet-Biedl syndrome (BBS; Chiang et al., 2004; Lechtreck et al., 2009; Loktev et al., 2008; Nachury et al., 2007; Scheidecker et al., 2014; Zhang et al., 2011b). Compared with these signaling molecules, certain lipidated signaling factors (e.g., heterotrimeric G protein transducin [Gα,β,γ], inositol polyphosphate-5-phosphatase [INPP5E], nephronophthisis 3 [NPHP3]) do not count on the IFT/BBS system for shuttling into cilia. They instead use the ADP-ribosylation factor (Arf)-like 3 (ARL3) GTPase to achieve this goal.
As a member of the Arf subfamily of the Ras superfamily of small GTPases, ARL3 is conserved among the ciliated species but absent from the non-ciliated organisms, localizes throughout the cell, and is enriched in cilia (Avidor-Reiss et al., 2004; Efimenko et al., 2005; Pazour et al., 2005). Cross-ciliated species, phosphodiesterase 6 delta subunit (PDE6D), uncoordinated-119A/B (UNC119A/B), and binder of Arl2 (BART)/binder of Arl2-like 1 (BARTL1) have been identified as ARL3-specific effectors in cilia (ElMaghloob et al., 2021; Linari et al., 1999; Lokaj et al., 2015; Wright et al., 2011). Among them, PDE6D and UNC119A/B act as carrier/solubilizing proteins for binding to and shuttling the cytoplasmic lipidated cargoes of different groups into cilia. It was known that PDE6D binds to and transports the C-terminal prenylated (farnesylated or geranylgeranylated) cargoes (e.g., the catalytic α and β subunits of PDE6, INPP5E, transducin γ subunit [Tγ], and rhodopsin kinase [GRK1]) into cilia (Li and Baehr, 1998; Thomas et al., 2014; Zhang et al., 2007; Zhang et al., 2004). UNC119A/B instead binds to and transports the N-terminal myristoylated cargoes (e.g., NPHP3, cystin, and transducin α subunits [GNAT-1 and GNAT-2]) into cilia (Wright et al., 2011; Zhang et al., 2011a). Compared with PDE6D and UNC119A/B, BART/BARTL1 has no cargoes determined thus far, while BART acts as an ARL3-specific co-guanine nucleotide exchange factor (GEF) to contribute to convert GDP-bound ARL3 (ARL3GDP) to GTP-bound ARL3 (ARL3GTP; ElMaghloob et al., 2021). During cargo ciliary targeting, the carrier protein binds to the lipidated cargo in the cytoplasm and the carrier-cargo complex then shuttles toward cilia. Upon arriving at the transition zone (TZ) region, the activated ARL3GTP binds to its carrier protein effector at a site allosterically different from the one for lipidated cargo binding, and this induces conformational changes of the carrier protein, leading to the release of the cargo for binding to the ciliary membrane (Fansa et al., 2016; Ismail et al., 2012; Watzlich et al., 2013). After this, ARL3GTP is bound to and stimulated by retinitis pigmentosa 2 (RP2), the ARL3-specific GTPase-activating protein (GAP), to hydrolyze for releasing the carrier protein from ARL3 (Veltel et al., 2008). ARL3GDP is then reactivated by its ARL13b GEF, and ARL3GTP recycles back to bind to the carrier-cargo complex for cargo releasing in cilia (Gotthardt et al., 2015; Zhang et al., 2016). Thus far, several mutations of ARL3 have been implicated to cause ciliopathic Joubert syndrome (JBTS) likely through disrupting the releasing of cytoplasmic lipidated proteins for binding to the ciliary membrane (Alkanderi et al., 2018; Fu et al., 2021; Ratnapriya et al., 2021; Sheikh et al., 2019).
Besides its role in releasing a variety of lipidated signaling factors for ciliary membrane binding, ARL3 is essential for mouse rhodopsin, human polycystin-1 and -2 (PKD1 and PKD2), and worm PKD1 to target to cilia (Schrick et al., 2006; Su et al., 2014; Zhang et al., 2013). Since they cycle through cilia by IFT through binding to the BBSome, ARL3 could mediate their ciliary turnover through the BBSome (Abd-El-Barr et al., 2007; Liu et al., 2020; Nachury, 2018; Nishimura et al., 2004; Su et al., 2014). If this holds true in humans, it could satisfactorily explain the phenotypical overlap of BBS and ARL3-related JBTS, with the latter being the more severe ciliopathy. In this study, the BBSome was identified to be able to shed from retrograde IFT trains at the proximal ciliary region right above the TZ in an ARL3-independent manner in Chlamydomonas reinhardtii. ARL3 in a GDP-bound state binds to the membrane for diffusing into and residing along the whole length of cilia. Following a nucleotide exchange likely occurring at the proximal ciliary region above the TZ, ARL3GTP binds to the retrograde IFT train-shed and phospholipase D (PLD)-laden BBSome as an ARL3-specific effector and recruits it to move across the TZ and out of cilia. Our finding thus fills a gap in our understanding of how ARL3 mediates BBSome ciliary turnover via promoting BBSome movement across the TZ for ciliary retrieval in C. reinhardtii, shedding lights on the molecular basis of why ARL3 deficiency can cause BBS phenotypes.
Results
ARL3 enters cilia by diffusion and does not affect IFT
Chlamydomonas ARL3 shares significant homology with its orthologues in ciliated species and is more closely related to homologues of worms, Leishmania, and Trypanosoma than mammals and humans phylogenetically (Fig. S1, A and B). ARL3 was shown to be a negative regulator of ciliation in Leishmania and mouse (Cuvillier et al., 2000; Efimenko et al., 2005; Hanke-Gogokhia et al., 2016). In worms, depletion of ARL3 causes IFT-B and KIF17 motor to dissociate through histone deacetylatase 6 (HDAC6)-dependent pathway and then disrupts IFT (Li et al., 2010; Zhang et al., 2013). To clarify whether ARL3 affects IFT and ciliation in C. reinhardtii, we examined the ARL3 CLiP mutant (LMJ.RY0420.182282) that we named arl3-282. The arl3-282 cell contains a 2,217-bp paromomycin gene insertion in the fourth exon of the ARL3 gene (Fig. S2, A–C). With the newly developed ARL3 antibody available, this insertion was verified to prevent ARL3 from being synthesized, as shown by immunoblotting, demonstrating that arl3-282 is an ARL3-null mutant (Fig. S3 A). arl3-282 cells grown cilia of normal length, excluding ARL3 from mediating ciliation (Fig. S3 B). Supportive of this conclusion, arl3-282 cells retained IFT-A subunits IFT43 and IFT139, IFT-B1 subunits IFT22 and IFT70, and IFT-B2 subunits IFT38 and IFT57 at wild-type (WT) levels of CC-125 cells both in whole cell and ciliary samples in the steady state (Fig. 1 A). To examine whether ARL3 affects IFT ciliary dynamics, we generated transgenic strains arl3-282; IFT43:HA:YFP-TG, arl3-282; IFT22:HA:YFP-TG, and arl3-282; IFT38:YFP-TG, which expresses IFT43, IFT22, or IFT38 fused at their C-terminus to hemagglutinin (HA) and/or yellow fluorescent protein (YFP; IFT43-HA-YFP, IFT22-HA-YFP, and IFT38-YFP) in arl3-282 cells. When expressed at the same level as when three HA-YFP/YFP-tagged proteins of different IFT subcomplexes were expressed alone in CC-125 cells (resulting strains ARL3; IFT43:HA:YFP-TG, ARL3; IFT22:HA:YFP-TG, and ARL3; IFT38:YFP-TG; Fig. S3 C), they entered cilia (Fig. 1 B) and underwent typical bidirectional IFT of C. reinhardtii as reflected by total internal reflection fluorescence (TIRF) assays, excluding ARL3 from mediating IFT ciliary dynamics (Fig. 1 C and D; Xue et al., 2020). After knowing this, we asked whether and how Chlamydomonas ARL3 enters cilia. To answer this question, we expressed ARL3-HA-YFP in arl3-282 cells at the endogenous ARL3 level of parental CC-5325 cells (resulting strain arl3-282; ARL3:HA:YFP-TG; Fig. S3 D). The arl3-282; ARL3:HA:YFP-TG cells retained ARL3-HA-YFP in cilia at the endogenous ARL3 level of CC-5325 cells (Fig. 1 E) and ARL3-HA-YFP diffused into cilia to reside along the whole length of cilia as reflected by TIRF assay (Fig. 1 F and Video 1). In summary, ARL3 does not affect IFT in both the steady and dynamic states, and it enters cilia by diffusion.

ARL3 is highly conserved across ciliated species. (A) Sequence alignment of deduced amino acid sequences from 11 invertebrate and vertebrate ARL3 orthologues. Alignments were generated using CLC main workbench (version 6.8); the most conserved residues are shown in black, the least conserved are in red. Dashes indicate gaps introduced to optimize the alignment. ARL3 contains five conserved domains including G1, G2, G3, G4, and G5 as labeled in the boxes. Arrowheads indicate missense mutations created in dominant-negative and constitutive-active ARL3 mutants. (B) The phylogenetic tree of ARL3 proteins from invertebrate and vertebrate species as indicated. The neighbor-joining tree was calculated using the MEGA 7 software. Branch length represents evolutionary relatedness. The ARL3 GenBank accession numbers are as follows: Bostaurus, NP_001033656.1; Ovis aries, NP_001156025.1; Homo sapiens, AAA21654.1; Xenopus laevis, AAH87495.1; Rattus norvegicus, NP_073191.1; Danio rerio, NP_001038373.1; Mus musculus, NP_001342162.1; Caenorhabditis elegans, CAB07583.1; Trypanosoma brucei, AAC32774.1; Leishmania major strain Friedlin, XP_001687211.1; and Chlamydomonas reinhardtii, XP_042925186.1.

ARL3 is highly conserved across ciliated species. (A) Sequence alignment of deduced amino acid sequences from 11 invertebrate and vertebrate ARL3 orthologues. Alignments were generated using CLC main workbench (version 6.8); the most conserved residues are shown in black, the least conserved are in red. Dashes indicate gaps introduced to optimize the alignment. ARL3 contains five conserved domains including G1, G2, G3, G4, and G5 as labeled in the boxes. Arrowheads indicate missense mutations created in dominant-negative and constitutive-active ARL3 mutants. (B) The phylogenetic tree of ARL3 proteins from invertebrate and vertebrate species as indicated. The neighbor-joining tree was calculated using the MEGA 7 software. Branch length represents evolutionary relatedness. The ARL3 GenBank accession numbers are as follows: Bostaurus, NP_001033656.1; Ovis aries, NP_001156025.1; Homo sapiens, AAA21654.1; Xenopus laevis, AAH87495.1; Rattus norvegicus, NP_073191.1; Danio rerio, NP_001038373.1; Mus musculus, NP_001342162.1; Caenorhabditis elegans, CAB07583.1; Trypanosoma brucei, AAC32774.1; Leishmania major strain Friedlin, XP_001687211.1; and Chlamydomonas reinhardtii, XP_042925186.1.
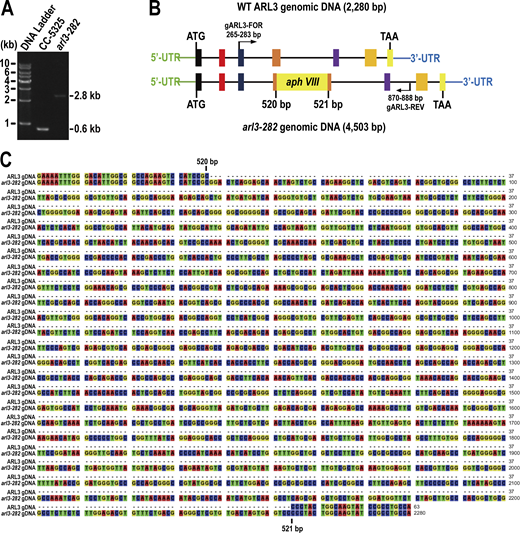
Characterization of the CLiP strain (LMJ.RY0420.182282) named arl3-282. (A) Agarose gel electrophoresis of the PCR products amplified from genomes of CC-5325 and arl3-282 cells. The primer pair gARL3-FOR and gARL3-REV were used to amplify the ARL3 genomic DNA of 624-bp from CC-5325 cells. A single DNA fragment of ∼2.8-kb (2,848-bp) was amplified from the arl3-282 cells by using the same primer pair. (B) Schematic representation showing that a paromomycin-resistant gene (aphVIII) inserted to the fourth exon in ARL3 gene of arl3-282 cells. The boxes with colors and the lines represent the exons and introns of ARL3 gene, respectively. (C) Sequence alignment of the ARL3 genomic DNAs (gDNA) between CC-5325 and arl3-282 cells. The arl3-282 cell contains a 2,284-bp aphVIII insertion between 520- and 521-bp in the ARL3 gDNA. Source data are available for this figure: SourceData FS2.

Characterization of the CLiP strain (LMJ.RY0420.182282) named arl3-282. (A) Agarose gel electrophoresis of the PCR products amplified from genomes of CC-5325 and arl3-282 cells. The primer pair gARL3-FOR and gARL3-REV were used to amplify the ARL3 genomic DNA of 624-bp from CC-5325 cells. A single DNA fragment of ∼2.8-kb (2,848-bp) was amplified from the arl3-282 cells by using the same primer pair. (B) Schematic representation showing that a paromomycin-resistant gene (aphVIII) inserted to the fourth exon in ARL3 gene of arl3-282 cells. The boxes with colors and the lines represent the exons and introns of ARL3 gene, respectively. (C) Sequence alignment of the ARL3 genomic DNAs (gDNA) between CC-5325 and arl3-282 cells. The arl3-282 cell contains a 2,284-bp aphVIII insertion between 520- and 521-bp in the ARL3 gDNA. Source data are available for this figure: SourceData FS2.
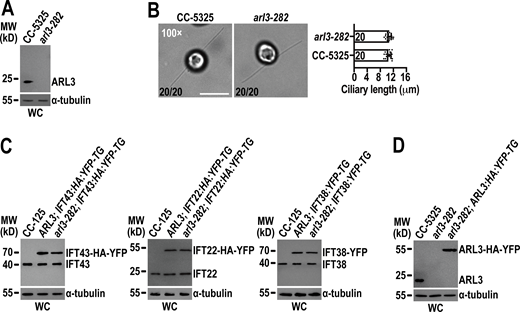
Chlamydomonas ARL3 is dispensable for ciliation and IFT. (A)arl3-282 is an ARL3-null mutant. Immunoblots with the affinity-purified ARL3 antiserum identified ARL3 protein with a size of ∼20 kD in whole cell (WC) samples of CC-5325 but not arl3-282 cells. MW stands for molecular weight. (B)arl3-282 cells assemble cilia with normal length. Representative phase contrast images of CC-5325 and arl3-282 cells were shown (left). The number of cells which have cilia in 20 counted cells was listed for each strain. Inset magnification (100 times) was shown. Scale bar: 10 µm. arl3-282 cells had full-length cilia (10.47 ± 0.82 µm, n = 20) compared to CC-5325 cells (10.57 ± 0.76 µm, n = 20). Mean lengths are listed; error bar indicates SD, and n indicates the number of cilia counted and listed in each bar. ns: non-significance (right). One sample unpaired Student’s t test is indicated. (C) Immunoblots of WC samples of three group cells including CC-125, ARL3; IFT43:HA:YFP-TG, and arl3-282; IFT43:HA:YFP-TG (left); CC-125, ARL3; IFT22:HA:YFP-TG, and arl3-282; IFT22:HA:YFP-TG (middle); and CC-125, ARL3; IFT38:YFP-TG, and arl3-282; IFT38:YFP-TG (right) probed with α-IFT43, α-IFT22, and α-IFT38, respectively. For each group of the cells, the YFP- or HA-YFP-tagged proteins of both CC-125 and arl3-282 background were determined to express at the WT CC-125 protein levels. (D) Immunoblots of WC samples of CC-5325, arl3-282, and arl3-282; ARL3:HA:YFP-TG probed with α-ARL3. For A, C, and D, α-tubulin we used as a loading control. Source data are available for this figure: SourceData FS3.

Chlamydomonas ARL3 is dispensable for ciliation and IFT. (A)arl3-282 is an ARL3-null mutant. Immunoblots with the affinity-purified ARL3 antiserum identified ARL3 protein with a size of ∼20 kD in whole cell (WC) samples of CC-5325 but not arl3-282 cells. MW stands for molecular weight. (B)arl3-282 cells assemble cilia with normal length. Representative phase contrast images of CC-5325 and arl3-282 cells were shown (left). The number of cells which have cilia in 20 counted cells was listed for each strain. Inset magnification (100 times) was shown. Scale bar: 10 µm. arl3-282 cells had full-length cilia (10.47 ± 0.82 µm, n = 20) compared to CC-5325 cells (10.57 ± 0.76 µm, n = 20). Mean lengths are listed; error bar indicates SD, and n indicates the number of cilia counted and listed in each bar. ns: non-significance (right). One sample unpaired Student’s t test is indicated. (C) Immunoblots of WC samples of three group cells including CC-125, ARL3; IFT43:HA:YFP-TG, and arl3-282; IFT43:HA:YFP-TG (left); CC-125, ARL3; IFT22:HA:YFP-TG, and arl3-282; IFT22:HA:YFP-TG (middle); and CC-125, ARL3; IFT38:YFP-TG, and arl3-282; IFT38:YFP-TG (right) probed with α-IFT43, α-IFT22, and α-IFT38, respectively. For each group of the cells, the YFP- or HA-YFP-tagged proteins of both CC-125 and arl3-282 background were determined to express at the WT CC-125 protein levels. (D) Immunoblots of WC samples of CC-5325, arl3-282, and arl3-282; ARL3:HA:YFP-TG probed with α-ARL3. For A, C, and D, α-tubulin we used as a loading control. Source data are available for this figure: SourceData FS3.

ARL3 enters cilia by diffusion and does not affect IFT. (A) Immunoblots of whole cell (WC) samples and cilia of CC-5325 and arl3-282 cells probed for IFT43 and IFT139 (IFT-A), IFT22 and IFT70 (IFT-B1), and IFT38 and IFT57 (IFT-B2). (B) Immunoblots of cilia of three cell groups indicated on the top probed with α-IFT43, α-IFT22, and α-IFT38, respectively. (C) Representative TIRF images and corresponding kymograms of the three cell groups shown in B. Velocities and frequencies of YFP- and HA-YFP–labeled proteins to traffic inside cilia were shown as graphs. Error bar indicates SD n indicates the number of cilia analyzed. For measuring anterograde and retrograde velocities, 50 cilia were analyzed. 20 cilia were measured for determining anterograde and retrograde frequencies. ns indicates non-significance. One sample unpaired Student’s t test is indicated. (D) Velocities and frequencies of YFP- and HA-YFP–tagged proteins to traffic inside cilia in C shown as numbers. (E) Immunoblots of cilia of CC-5325, arl3-282, and arl3-282; ARL3:HA:YFP-TG cells probed with α-ARL3. (F) Representative TIRF image and corresponding kymogram of the arl3-282; ARL3:HA:YFP-TG cell (Video 1, 15 fps). For A, B, and E, α-tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW: molecular weight. For C and D, Ant. and Ret. represent anterograde and retrograde, respectively. For C and F, the time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. Source data are available for this figure: SourceData F1.
ARL3 enters cilia by diffusion and does not affect IFT. (A) Immunoblots of whole cell (WC) samples and cilia of CC-5325 and arl3-282 cells probed for IFT43 and IFT139 (IFT-A), IFT22 and IFT70 (IFT-B1), and IFT38 and IFT57 (IFT-B2). (B) Immunoblots of cilia of three cell groups indicated on the top probed with α-IFT43, α-IFT22, and α-IFT38, respectively. (C) Representative TIRF images and corresponding kymograms of the three cell groups shown in B. Velocities and frequencies of YFP- and HA-YFP–labeled proteins to traffic inside cilia were shown as graphs. Error bar indicates SD n indicates the number of cilia analyzed. For measuring anterograde and retrograde velocities, 50 cilia were analyzed. 20 cilia were measured for determining anterograde and retrograde frequencies. ns indicates non-significance. One sample unpaired Student’s t test is indicated. (D) Velocities and frequencies of YFP- and HA-YFP–tagged proteins to traffic inside cilia in C shown as numbers. (E) Immunoblots of cilia of CC-5325, arl3-282, and arl3-282; ARL3:HA:YFP-TG cells probed with α-ARL3. (F) Representative TIRF image and corresponding kymogram of the arl3-282; ARL3:HA:YFP-TG cell (Video 1, 15 fps). For A, B, and E, α-tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW: molecular weight. For C and D, Ant. and Ret. represent anterograde and retrograde, respectively. For C and F, the time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. Source data are available for this figure: SourceData F1.
TIRF imaging of ARL3-HA-YFP movement in arl3-282; ARL3:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 1 F. Play speed is real-time (15 fps).
TIRF imaging of ARL3-HA-YFP movement in arl3-282; ARL3:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 1 F. Play speed is real-time (15 fps).
ARL3GDP binds the membrane via its N-terminal amphipathic helix for diffusing into cilia
Arf family GTPases associate with the membrane through its N-terminal amphipathic helix (Amor et al., 1994; Liu et al., 2010; Zhang et al., 2011b) and their N-terminal 15 residues are found essential for this association (Jin et al., 2010; Liu et al., 2021; Mourão et al., 2014). In addition, studies have identified the GTP-bound configuration as a prerequisite for certain Arf GTPases, i.e., ARL6/BBS3, to bind the membrane (Liu et al., 2021; Mourão et al., 2014). To dissect whether and how the N-terminal amphipathic helix and the nucleotide state influence the association of ARL3 to the membrane, we expressed ARL3 and its variants harboring the mutation Q70L, T30N, or the N-terminal 15 residue deletion (ΔN15) alone or containing ΔN15 combined with Q70L or T30N in bacteria (Fig. 2 A). Q70L and T30N were introduced in ARL3 or ARL3ΔN15 as they are supposed to represent constitutive-active (Q70L) and dominant-negative (T30N) mutations that retain ARL3 in an active (GTP-bound, ARL3Q70L) and inactive (GDP-bound/nucleotide-free, ARL3T30N) states, respectively (Cuvillier et al., 2000; Li et al., 2010; Veltel et al., 2008). As revealed by liposome flotation assays conducted on bacterially expressed ARL3 preloaded with GTPγS or GDP, which render ARL3 GTP-bound (ARL3GTP) or GDP-bound (ARL3GDP), respectively (Gotthardt et al., 2015; Liew et al., 2014; Tucker et al., 1986), ARL3 efficiently bound to liposomes only when it was preloaded with GDP, supporting a notion that ARL3GDP rather than ARL3GTP binds to liposomes (Fig. 2 A). Reflecting these observations, ARL3Q70L, as expected, was disabled for binding to liposomes (Fig. 2 A). ARL3T30N resembled ARL3GDP to bind to liposomes, identifying ARL3T30N mimics the GDP-bound form of ARL3 in our experimental conditions (Fig. 2 A). Besides, ARL3ΔN15 failed to bind to liposomes and it remained unbound to liposomes even when the T30N mutation was introduced (ARL3ΔNT30N), revealing that the N-terminal amphipathic helix is indispensable for ARL3 to bind to liposomes (Fig. 2 A). We then propose the GDP-bound state and the N-terminal amphipathic helix as two prerequisites for ARL3 to bind to the membrane efficiently.

ARL3GDPbinds the membrane via its N-terminal amphipathic helix for diffusing into cilia. (A) Schematic presentation of bacterially expressed ARL3 and its variants (shown on the left). ΔN15 stands for the N-terminal 15 amino acids of ARL3 deleted. Immunoblots of liposome flotation captured ARL3 preloaded with GTPγS or GDP (shown in the middle) and its variants (shown on the right) with α-ARL3. ARL3 or its variants along was used as an input for evaluating their binding/input ratio shown as percentile. (B) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-ARL3. α-Tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW, molecular weight. (C) Representative TIRF images and corresponding kymograms of cells indicated on the top (Videos 2, 3, and 4, 15 fps). The time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. (D) Immunoblots of ciliary fractions of cells indicated on the bottom probed with α-HA, α-IFT57 (ciliary matrix marker), α-PLD (ciliary membrane marker) and Ac-tubulin (axoneme marker). (E) Schematic presentation of how ARL3GDP binds the membrane for diffusing into cilia prior to converting to ARL3GTP for being enriched at the proximal ciliary region. Source data are available for this figure: SourceData F2.
ARL3GDPbinds the membrane via its N-terminal amphipathic helix for diffusing into cilia. (A) Schematic presentation of bacterially expressed ARL3 and its variants (shown on the left). ΔN15 stands for the N-terminal 15 amino acids of ARL3 deleted. Immunoblots of liposome flotation captured ARL3 preloaded with GTPγS or GDP (shown in the middle) and its variants (shown on the right) with α-ARL3. ARL3 or its variants along was used as an input for evaluating their binding/input ratio shown as percentile. (B) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-ARL3. α-Tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW, molecular weight. (C) Representative TIRF images and corresponding kymograms of cells indicated on the top (Videos 2, 3, and 4, 15 fps). The time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. (D) Immunoblots of ciliary fractions of cells indicated on the bottom probed with α-HA, α-IFT57 (ciliary matrix marker), α-PLD (ciliary membrane marker) and Ac-tubulin (axoneme marker). (E) Schematic presentation of how ARL3GDP binds the membrane for diffusing into cilia prior to converting to ARL3GTP for being enriched at the proximal ciliary region. Source data are available for this figure: SourceData F2.
To discern how membrane binding affects ARL3 to enter cilia, we expressed the above-mentioned ARL3 variants fused at their C-terminus to HA-YFP in arl3-282 cells to generate five strains as shown in Fig. 2 B. When expressed at the endogenous ARL3 level of parental CC-5325 cells, ARL3-HA-YFP resembled ARL3 in entering cilia, while ARL3∆N15-HA-YFP and ARL3∆N15T30N-HA-YFP both failed to do so, revealing that membrane association is a prerequisite for ARL3 to enter cilia under physiological conditions (Fig. 2 B). This notion was verified as ARL3T30N-HA-YFP mimicked ARL3 and ARL3-HA-YFP to enter cilia (Fig. 2 B). Interestingly, ARL3Q70L-HA-YFP and ARL3∆N15Q70L-HA-YFP were also found to enter cilia (Fig. 2 B). Since they are unable to bind to the membrane by containing Q70L alone or both Q70L and ∆N15, the Q70L-induced GTP-bound configuration likely confers ARL3-HA-YFP and even the ∆N15-containing variant to enter cilia through a membrane-independent pathway. Based on these observations, the fact that ARL3∆N15-HA-YFP failed to enter cilia instead reveals that, before entering cilia, ARL3 must exist in a GDP-bound state in cytoplasm under physiological conditions (Fig. 2 B). As reflected by the ARL3T30N-HA-YFP variant, ARL3GDP enters cilia by diffusion, resides in the ciliary membrane fraction, and distributes along the whole length of cilia as shown by TIRF and ciliary fraction assays (Fig. 2, C and D; and Video 2).
TIRF imaging of ARL3T30N-HA-YFP movement in arl3-282; ARL3T30N:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
TIRF imaging of ARL3T30N-HA-YFP movement in arl3-282; ARL3T30N:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
We noticed that ARL3Q70L-HA-YFP and ARL3∆N15Q70L-HA-YFP enter cilia by diffusion and, unlike ARL3T30N-HA-YFP that resides along the whole length of cilia, instead were enriched mostly to the proximal ciliary region (Fig. 2 C and Videos 2, 3, and 4). They resided in the ciliary matrix as they are unable to bind to the ciliary membrane (Fig. 2 D). The mechanism underlying how ARL3, when being in a GTP-bound state, can diffuse into cilia remains unknown, while this endowed us with a tool for carrying out functional rescue with ARL3GTP in cilia (as shown below). It was also noticed that, once inside cilia, ARL3-HA-YFP resembled ARL3T30N-HA-YFP in distributing along the whole length of cilia but, unlike ARL3T30N-HA-YFP that attaches to the ciliary membrane, instead resided in the ciliary matrix (Fig. 1 F; and Fig. 2, C and D; and Videos 1 and 2). This observation suggests that ARL3GDP, upon entering cilia, is likely subject to a rapid nucleotide exchange for converting into ARL3GTP. ARL3GTP then detaches from the ciliary membrane to reside in the ciliary matrix (Fig. 2 E). Eventually, it is ARL3GTP that is somehow enriched to the proximal ciliary region as reflected by the ARL3Q70L-HA-YFP variant (Fig. 2, C and E).
TIRF imaging of ARL3Q70L-HA-YFP movement in arl3-282; ARL3Q70L:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
TIRF imaging of ARL3Q70L-HA-YFP movement in arl3-282; ARL3Q70L:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
TIRF imaging of ARL3ΔN15Q70L-HA-YFP movement in arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
TIRF imaging of ARL3ΔN15Q70L-HA-YFP movement in arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 2 C. Play speed is real-time (15 fps).
ARL3GTP promotes the BBSome to move across the TZ for ciliary retrieval
ARL3 is essential for maintaining ciliary turnover of mouse rhodopsin, human polycystin-1 and -2 (PKD1 and PKD2), and worm PKD1 (Schrick et al., 2006; Su et al., 2014; Zhang et al., 2013). These transmembrane signaling proteins cycle through cilia by IFT through binding to the BBSome directly (Abd-El-Barr et al., 2007; Liu et al., 2020; Nachury, 2018; Nishimura et al., 2004; Su et al., 2014). Given that ARL3 does not affect IFT (Fig. 1, A–D; and Fig. S3 C), we wondered whether ARL3 mediates ciliary turnover of signaling proteins through the BBSome. To answer this question, we examined arl3-282 and the rescuing strains arl3-282;ARL3:HA:YFP-TG, arl3-282;ARL3Q70L:HA:YFP-TG, and arl3-282;ARL3T30N:HA:YFP-TG. As compared to CC-5325 control cells, four strains retained the BBSome (represented by BBS1, BBS4, BBS5, BBS7, and BBS8) at control cell level (Fig. 3 A). Of note, the BBSome was accumulated in arl3-282 cilia to a level approximately four-fold higher than control cell cilia (Fig. 3 A). This buildup was restored back to normal by ARL3-HA-YFP (Fig. 3 A). Together with the observation that ARL3Q70L-HA-YFP but not ARL3T30N-HA-YFP restored the BBSome back to normal, we concluded that ARL3GTP rather than ARL3GDP is required for maintaining BBSome ciliary turnover (Fig. 3 A). This notion was strengthened as ARL3ΔN15Q70L-HA-YFP, which lacks the N-terminal 15 amino acids of ARL3 for membrane association but is supposed to be locked in a GTP-bound state configuration of ARL3, retained the ability to restore BBSome ciliary content (Fig. S4 A). The BBSome undergoes a disassembly/reassembly (remodeling) process at the ciliary tip prior to loading onto retrograde IFT trains for transporting to the ciliary base (Sun et al., 2021). As reflected by sucrose density gradient centrifugation assays, the BBSome remained to be an intact entity in cilia in an ARL3-independent manner, revealing that ARL3 is dispensable for the BBSome to remodel at the ciliary tip (Fig. S4 B). This result also excludes ARL3 from affecting BBSome integrity during bidirectional IFT in cilia (Fig. S4 B). According to immunofluorescence staining assays, the loss of ARL3 or its replacement with ARL3T30N-HA-YFP both caused the BBSome (represented by the endogenous BBS8) to build up at the proximal ciliary region obviously above the BBS8- and IFT81-double labeled basal bodies (Fig. 3 B). In arl3-282; ARL3Q70L:HA:YFP-TG cells, however, a BBSome accumulation at the ciliary base was not observed (Fig. 3 B). To discern if the BBSome ciliary accumulation is caused by its increased ciliary entry, decreased ciliary removal, or both, we generated an ARL3- and BBS8-double null mutant that we named arl3;bbs8 and expressed BBS8-YFP in bbs8 and arl3;bbs8 cells (resulting strains bbs8;BBS8:YFP-TG and arl3;bbs8;BBS8:YFP-TG, respectively) at WT BBS8 level of CC-125 cells (Fig. 3 C; Sun and Pan, 2019). BBS8-YFP resembled BBS8 to enter bbs8; BBS8:YFP-TG cilia and so did for the BBSome subunits BBS1, BBS4, BBS5, and BBS7 (Fig. 3 C). In contrast, ARL3 knockout did not affect cellular level of the BBSome but, as expected, caused it to build up in arl3; bbs8;BBS8:YFP-TG cilia (Fig. 3 C). Since the BBSome (represented by BBS8-YFP) underwent typical bidirectional IFT of the Chlamydomonas BBSome at frequencies and velocities the same in cilia of both cells, ARL3 was excluded from affecting the IFT behavior of the BBSome (Fig. 3, D and E; and Videos 5 and 6; Lechtreck et al., 2009). As compared to bbs8;BBS8:YFP-TG cilia, arl3; bbs8; BBS8:YFP-TG cilia accumulated the BBSome (represented by BBS8-YFP) at the ciliary base defined to the CEP290-labled TZ above the BBS8-YFP labeled basal bodies (Fig. 3 F). We then conclude that the BBSome performs normal IFT and ciliary tip remodeling in an ARL3-independent manner. However, the BBSome fails to pass the TZ for ciliary retrieval in the absence of ARL3 or in the presence of ARL3GDP (Fig. 3, B, F, and G). It is ARL3GTP that promotes the outward movement of the BBSome across the TZ for ciliary retrieval (Fig. 3, B and G).

ARL3GTPpromotes the BBSome to move across the TZ for ciliary retrieval. (A) Immunoblots of WC samples and cilia of cells indicated on the top probed for the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8. The numbers below the blots indicate band intensities (CC-5325 as 1). The values were normalized based on the α-tubulin (WC) and Ac-tubulin (cilia) signaling. (B) Cells indicated on the left stained with α-BBS8 (red) and α-IFT81 (green). BBS8 and IFT81 both are basal body markers. (C) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-BBS8, α-ARL3, α-BBS1, α-BBS4, α-BBS5, and α-BBS7. MW, molecular weight. (D) Representative TIRF images and corresponding kymograms of bbs8; BBS8:YFP-TG and arl3;bbs8; BBS8:YFP-TG cells (Videos 5 and 6, 15 fps). The time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. Velocities and frequencies of BBS8-YFP to traffic inside cilia were shown as graphs. Error bar indicates SD; n indicates the number of cilia analyzed. For measuring anterograde and retrograde velocities, 50 cilia were analyzed. 20 cilia were measured for determining anterograde and retrograde frequencies. ns indicates non-significance. One sample unpaired Student’s t test is indicated. (E) Velocities and frequencies of BBS8-YFP to traffic inside cilia of bbs8;BBS8:YFP-TG and arl3;bbs8;BBS8:YFP-TG cells shown as numbers. (F) CC-125, bbs8;BBS8:YFP-TG, and arl3;bbs8;BBS8:YFP-TG cells stained with α-CEP290 (red) and α-YFP (green). CEP290 and BBS8-YFP serve as a TZ and basal body marker, respectively. (G) Schematic representation of how the loss of ARL3 and the presence of ARL3GDP but not ARL3GTP blocks outward movement of the BBSome across the TZ for ciliary retrieval. For A and C, α-tubulin and Ac-tubulin were used to adjust the loading of WC samples and cilia, respectively. For D and E, Ant. and Ret. represent anterograde and retrograde, respectively. For B and F, phase contrast (PC) images of cells were also shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnifications (100 times) were shown. Scale bars, 10 µm. Source data are available for this figure: SourceData F3.
ARL3GTPpromotes the BBSome to move across the TZ for ciliary retrieval. (A) Immunoblots of WC samples and cilia of cells indicated on the top probed for the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8. The numbers below the blots indicate band intensities (CC-5325 as 1). The values were normalized based on the α-tubulin (WC) and Ac-tubulin (cilia) signaling. (B) Cells indicated on the left stained with α-BBS8 (red) and α-IFT81 (green). BBS8 and IFT81 both are basal body markers. (C) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-BBS8, α-ARL3, α-BBS1, α-BBS4, α-BBS5, and α-BBS7. MW, molecular weight. (D) Representative TIRF images and corresponding kymograms of bbs8; BBS8:YFP-TG and arl3;bbs8; BBS8:YFP-TG cells (Videos 5 and 6, 15 fps). The time and transport lengths are indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. Velocities and frequencies of BBS8-YFP to traffic inside cilia were shown as graphs. Error bar indicates SD; n indicates the number of cilia analyzed. For measuring anterograde and retrograde velocities, 50 cilia were analyzed. 20 cilia were measured for determining anterograde and retrograde frequencies. ns indicates non-significance. One sample unpaired Student’s t test is indicated. (E) Velocities and frequencies of BBS8-YFP to traffic inside cilia of bbs8;BBS8:YFP-TG and arl3;bbs8;BBS8:YFP-TG cells shown as numbers. (F) CC-125, bbs8;BBS8:YFP-TG, and arl3;bbs8;BBS8:YFP-TG cells stained with α-CEP290 (red) and α-YFP (green). CEP290 and BBS8-YFP serve as a TZ and basal body marker, respectively. (G) Schematic representation of how the loss of ARL3 and the presence of ARL3GDP but not ARL3GTP blocks outward movement of the BBSome across the TZ for ciliary retrieval. For A and C, α-tubulin and Ac-tubulin were used to adjust the loading of WC samples and cilia, respectively. For D and E, Ant. and Ret. represent anterograde and retrograde, respectively. For B and F, phase contrast (PC) images of cells were also shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnifications (100 times) were shown. Scale bars, 10 µm. Source data are available for this figure: SourceData F3.
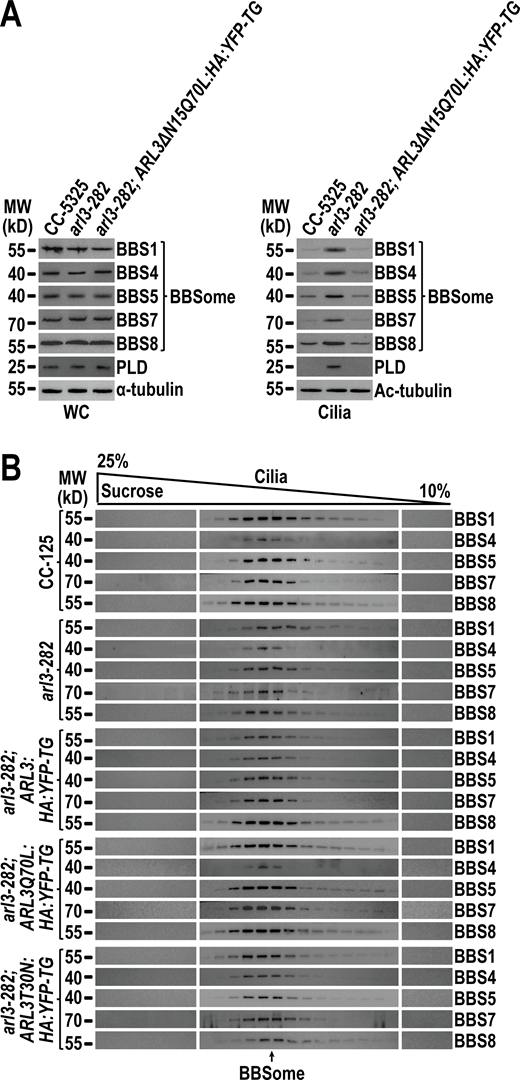
N-terminal 15 amino acids are dispensable for ARL3Q70Lto maintain the ciliary content of the BBSome and PLD and ARL3 does not affect BBSome integrity in cilia. (A) Immunoblots of WC samples and cilia of CC-5325, arl3-282, arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cells probed for the BBSome subunits BBS1, BBS4, BBS5, BBS7, BBS8, and PLD. α-tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. (B) Immunoblots of sucrose density gradients of CC-5325, arl3-282, arl3-282;ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282;ARL3T30N:HA:YFP-TG cilia probed for BBS1, BBS4, BBS5, BBS7, and BBS8. These BBSome proteins co-sedimented completely. Source data are available for this figure: SourceData FS4.

N-terminal 15 amino acids are dispensable for ARL3Q70Lto maintain the ciliary content of the BBSome and PLD and ARL3 does not affect BBSome integrity in cilia. (A) Immunoblots of WC samples and cilia of CC-5325, arl3-282, arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cells probed for the BBSome subunits BBS1, BBS4, BBS5, BBS7, BBS8, and PLD. α-tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. (B) Immunoblots of sucrose density gradients of CC-5325, arl3-282, arl3-282;ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282;ARL3T30N:HA:YFP-TG cilia probed for BBS1, BBS4, BBS5, BBS7, and BBS8. These BBSome proteins co-sedimented completely. Source data are available for this figure: SourceData FS4.
TIRF imaging of BBS8-YFP movement in bbs8; BBS8:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 3 D. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement in bbs8; BBS8:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 3 D. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement in arl3;bbs8; BBS8:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 3 D. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement in arl3;bbs8; BBS8:YFP-TG cilia. A frame from this video and kymograph are shown in Fig. 3 D. Play speed is real-time (15 fps).
The BBSome can shed from retrograde IFT trains at the TZ region to act as an ARL3 effector
Different from ARL3GDP that binds the ciliary membrane, ARL3GTP resembles the BBSome to reside in the ciliary matrix (Fig. 2 D). ARL3GDP resides along the whole length of cilia, while GTP loading restricts ARL3 to the proximal ciliary region (Fig. 2 C). The BBSome accumulates at the TZ region in the absence of ARL3 or in the presence of ARL3GDP (Fig. 3, B and F), proposing that ARL3GTP and the BBSome may distribute to the same TZ region. Since ARL3GTP promotes outward movement of the BBSome across the TZ (Fig. 3), it may interact with the BBSome at the TZ region for achieving this goal. We failed to visualize ARL3 co-localization with the BBSome at the TZ region by immunostaining as ARL3-HA-YFP and its variants cannot be detected even when the commercial α-YFP and α-HA were used (Fig. S5, A and B). However, this notion was strengthened because ARL3-HA-YFP and ARL3Q70L-HA-YFP, although only partial of them, co-sedimented with the BBSome in sucrose density gradients of arl3-282; ARL3:HA:YFP-TG and arl3-282; ARL3Q70L:HA:YFP-TG cilia (Fig. 4 A). In contrast, ARL3T30N-HA-YFP remained to be separated from the BBSome in arl3-282; ARL3T30N:HA:YFP-TG cilia (Fig. 4 A). Our previous study has shown that HMEKN buffer confers the IFT-A, IFT-B1, and IFT-B2 subcomplex components of IFT trains to separate from one another, while the BBSome remains to associate with IFT-B1 (Sun et al., 2021). Of note, the addition of excessive GTPγS conferred ARL3-HA-YFP to immunoprecipitate the BBSome but not IFT-B1 (represented by IFT22 and IFT70) in arl3-282; ARL3:HA:YFP-TG cilia (Fig. 4 B). In contrast, neither the BBSome nor IFT-B1 were recovered in the presence of excessive GDP, revealing that a reservoir of the BBSome, autonomous of IFT-B1 association, exists in cilia for ARL3GTP to interact with (Fig. 4 B). Supportive of this notion, ARL3Q70L-HA-YFP but not ARL3T30N-HA-YFP recovered the BBSome and none of them can immunoprecipitate IFT-B1 (Fig. 4 B). Furthermore, ARL3Q70L-HA-YFP failed to immunoprecipitate the BBSome and IFT-B1 in cell body samples even in the presence of dithiothreitol (DTT) that confers the BBSome to separate from IFT-B1 (Sun et al., 2021), revealing that ARL3GTP interacts with the BBSome independent of IFT-B1 association in cilia but not in cytoplasm (Fig. S5 C). As identified by in vitro protein interaction assays, BBS1 and BBS5 were the BBSome subunits efficiently captured by ARL3Q70L, revealing that ARL3GTP binds the BBSome directly (Fig. 4 C). As reflected by the facts that ARL3 does not affect the IFT behavior of the BBSome (Fig. 3 C–E; and Videos 5 and 6); the BBSome accumulates above the TZ region in the absence of ARL3 (Fig. 3, B and F) or in the presence of ARL3GDP (Fig. 3 B); and a reservoir of BBSomes, autonomous of IFT-B1 association, exists in cilia in the presence of ARL3GTP (Fig. 4 B), the BBSome must be able to shed from retrograde IFT trains at the TZ region in an ARL3-indepdendent manner. It is the retrograde IFT train-shed BBSome that can bind to ARL3 as an ARL3 effector at the TZ region.
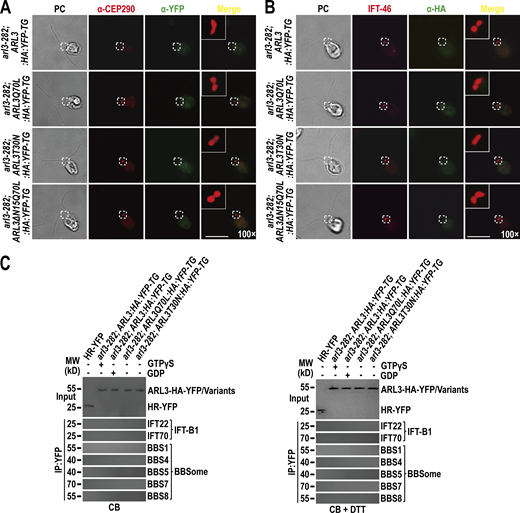
ARL3 is undetectable at the TZ and basal bodies by immunostaining nor binds the IFT-B1-shed BBSome in cytoplasm. (A)arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, arl3-282; ARL3T30N:HA:YFP-TG, and arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cells stained with α-CEP290 (red) and α-YFP (green). The CEP290-labeled TZ was shown. α-YFP fails to detect ARL3-HA-YFP and its variants. (B) Same cells as shown in A stained with α-IFT46 (red) and α-HA (green). IFT46-labeled basal bodies were shown. α-HA fails to detect ARL3-HA-YFP and its variants. (C) Immunoblots of α-YFP-captured proteins from cell body (CB) samples of HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells probed for the IFT-B1 subunits IFT22 and IFT70 and the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8 in the absence of DTT (left) and in the presence of DTT (right). Input was quantified with α-YFP by immunoblotting. MW stands for molecular weight. ARL3-HA-YFP fails to recover the IFT-B1-shed BBSome in the cell body. For A and B, phase contrast (PC) images of cells were shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnifications (100 times) were shown. Scale bars, 10 µm. Source data are available for this figure: SourceData FS5.

ARL3 is undetectable at the TZ and basal bodies by immunostaining nor binds the IFT-B1-shed BBSome in cytoplasm. (A)arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, arl3-282; ARL3T30N:HA:YFP-TG, and arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cells stained with α-CEP290 (red) and α-YFP (green). The CEP290-labeled TZ was shown. α-YFP fails to detect ARL3-HA-YFP and its variants. (B) Same cells as shown in A stained with α-IFT46 (red) and α-HA (green). IFT46-labeled basal bodies were shown. α-HA fails to detect ARL3-HA-YFP and its variants. (C) Immunoblots of α-YFP-captured proteins from cell body (CB) samples of HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells probed for the IFT-B1 subunits IFT22 and IFT70 and the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8 in the absence of DTT (left) and in the presence of DTT (right). Input was quantified with α-YFP by immunoblotting. MW stands for molecular weight. ARL3-HA-YFP fails to recover the IFT-B1-shed BBSome in the cell body. For A and B, phase contrast (PC) images of cells were shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnifications (100 times) were shown. Scale bars, 10 µm. Source data are available for this figure: SourceData FS5.

The BBSome can shed from retrograde IFT trains at the TZ region to act as an ARL3 effector. (A) Immunoblots of sucrose density gradients of arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cilia probed with α-BBS1, α-BBS4, α-BBS5, α-BBS7, α-BBS8, and α-ARL3. (B) Immunoblots of α-YFP-captured proteins from HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of excessive GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cilia probed for the IFT-B1 subunits IFT22 and IFT70 and the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8. HMEKN stands for the buffer used for solving cilia. Input was quantified with α-YFP by immunoblotting. A schematic representation of how a reservoir of the BBSome independent of IFT-B1 association exists in HMEKN buffer was shown on the right. (C) Bacterially expressed MBP, MBP-BBS1, MBP-BBS2, MBP-BBS4, MBP-BBS5, MBP-BBS7, MBP-BBS8, and MBP-BBS9 (left) were mixed with ARL3Q70L or ARL3T30N (second to the left) and complexes recovered on amylose beads were resolved by SDS-PAGE followed by Coomassie staining and immunoblotting with α-ARL3 (second to the right and right, respectively). A schematic representation of direct interactions of ARL3Q70L with BBS1 and BBS5 of the BBSome was shown (lower left). MW, molecular weight. Source data are available for this figure: SourceData F4.
The BBSome can shed from retrograde IFT trains at the TZ region to act as an ARL3 effector. (A) Immunoblots of sucrose density gradients of arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cilia probed with α-BBS1, α-BBS4, α-BBS5, α-BBS7, α-BBS8, and α-ARL3. (B) Immunoblots of α-YFP-captured proteins from HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of excessive GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cilia probed for the IFT-B1 subunits IFT22 and IFT70 and the BBSome subunits BBS1, BBS4, BBS5, BBS7, and BBS8. HMEKN stands for the buffer used for solving cilia. Input was quantified with α-YFP by immunoblotting. A schematic representation of how a reservoir of the BBSome independent of IFT-B1 association exists in HMEKN buffer was shown on the right. (C) Bacterially expressed MBP, MBP-BBS1, MBP-BBS2, MBP-BBS4, MBP-BBS5, MBP-BBS7, MBP-BBS8, and MBP-BBS9 (left) were mixed with ARL3Q70L or ARL3T30N (second to the left) and complexes recovered on amylose beads were resolved by SDS-PAGE followed by Coomassie staining and immunoblotting with α-ARL3 (second to the right and right, respectively). A schematic representation of direct interactions of ARL3Q70L with BBS1 and BBS5 of the BBSome was shown (lower left). MW, molecular weight. Source data are available for this figure: SourceData F4.
Retrograde IFT train-shed BBSomes pass the TZ for ciliary retrieval likely via diffusion
As shown above, ARL3GTP promotes outward movement of retrograde IFT train-shed BBSomes across the TZ (Fig. 3). To have a full review on how ARL3 and the BBSome interplay, we ought to dissect if ARL3 ciliary turnover is mediated by the BBSome. To answer this question, we expressed ARL3-HA-YFP, ARL3Q70L-HA-YFP, and ARL3T30N-HA-YFP in arl3; bbs8 cells to generate strains arl3; bbs8; ARL3:HA:YFP-TG, arl3; bbs8; ARL3Q70L:HA:YFP-TG, and arl3; bbs8; ARL3T30N:HA:YFP-TG. By doing so, the BBSome is assured to be absent from cilia of these cells as BBS8 knockout disrupts BBSome assembly in cytoplasm, making the BBSome unavailable for entering cilia (Fig. 5 A). When expressed at the WT ARL3 level of CC-125 cells, three ARL3 recombinant proteins all entered cilia by diffusion (Fig. 5 B) and retained at the WT ARL3 level in cilia (Fig. 5 A). They showed ciliary distribution and fraction patterns the same as in arl3-282 cilia (Fig. 5, B and C; also see Fig. 1 F; and Fig. 2, C and D), excluding the BBSome from mediating ARL3 ciliary turnover.

Retrograde IFT train-shed BBSomes pass the TZ for ciliary retrieval likely via diffusion. (A) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-ARL3, α-BBS8, α-BBS1, α-BBS4, α-BBS5, and α-BBS7. α-Tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW, molecular weight. (B) Representative TIRF images and corresponding kymograms of arl3; bbs8; ARL3:HA:YFP-TG, arl3; bbs8; ARL3Q70L:HA:YFP-TG, and arl3; bbs8; ARL3T30N:HA:YFP-TG cells. The time and transport lengths were indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. (C) Immunoblots of ciliary fractions of arl3; bbs8; ARL3:HA:YFP-TG, arl3; bbs8; ARL3Q70L:HA:YFP-TG, and arl3; bbs8; ARL3T30N:HA:YFP-TG cells probed with α-HA, α-IFT57 (ciliary matrix marker), α-PLD (ciliary membrane marker) and Ac-tubulin (axoneme marker). (D) Schematic representation of how IFT trains and the BBSome cycle between basal body and cilia. The diffusion of retrograde IFT train-shed BBSomes through the TZ for ciliary retrieval was shown. (E) Representative TIRF images and corresponding kymograms of ift46-1; IFT46:YFP-TG, bbs8; BBS8:YFP-TG, and arl3; bbs8; BBS8:YFP-TG cells (Videos 7, 8, and 9, 15 fps). Blue braces indicate the diffusion pattern of certain BBS8-YFP inside the TZ region of bbs8; BBS8:YFP-TG cells (the middle panel) and BBS8-YFP accumulation right above the TZ of arl3; bbs8; BBS8:YFP-TG cells (the right panel). The time was indicated on the bottom. The ciliary base (base) and tip (tip), the transition zone (TZ) and the basal body (BB) were shown. The corresponding schematic representation of how IFT46-YFP and BBS8-YFP cycle between the basal body and cilia was also shown. Source data are available for this figure: SourceData F5.
Retrograde IFT train-shed BBSomes pass the TZ for ciliary retrieval likely via diffusion. (A) Immunoblots of WC samples and cilia of cells indicated on the top probed with α-ARL3, α-BBS8, α-BBS1, α-BBS4, α-BBS5, and α-BBS7. α-Tubulin and acetylated (Ac)-tubulin were used to adjust the loading of WC samples and cilia, respectively. MW, molecular weight. (B) Representative TIRF images and corresponding kymograms of arl3; bbs8; ARL3:HA:YFP-TG, arl3; bbs8; ARL3Q70L:HA:YFP-TG, and arl3; bbs8; ARL3T30N:HA:YFP-TG cells. The time and transport lengths were indicated on the right and on the bottom, respectively. The ciliary base (base) and tip (tip) were shown. (C) Immunoblots of ciliary fractions of arl3; bbs8; ARL3:HA:YFP-TG, arl3; bbs8; ARL3Q70L:HA:YFP-TG, and arl3; bbs8; ARL3T30N:HA:YFP-TG cells probed with α-HA, α-IFT57 (ciliary matrix marker), α-PLD (ciliary membrane marker) and Ac-tubulin (axoneme marker). (D) Schematic representation of how IFT trains and the BBSome cycle between basal body and cilia. The diffusion of retrograde IFT train-shed BBSomes through the TZ for ciliary retrieval was shown. (E) Representative TIRF images and corresponding kymograms of ift46-1; IFT46:YFP-TG, bbs8; BBS8:YFP-TG, and arl3; bbs8; BBS8:YFP-TG cells (Videos 7, 8, and 9, 15 fps). Blue braces indicate the diffusion pattern of certain BBS8-YFP inside the TZ region of bbs8; BBS8:YFP-TG cells (the middle panel) and BBS8-YFP accumulation right above the TZ of arl3; bbs8; BBS8:YFP-TG cells (the right panel). The time was indicated on the bottom. The ciliary base (base) and tip (tip), the transition zone (TZ) and the basal body (BB) were shown. The corresponding schematic representation of how IFT46-YFP and BBS8-YFP cycle between the basal body and cilia was also shown. Source data are available for this figure: SourceData F5.
ARL3 does not affect IFT and ARL3GTP promotes the outward movement of retrograde IFT train-shed BBSomes across the TZ for ciliary retrieval, raising a question of how the BBSome autonomous of retrograde IFT train association passes the TZ and out of cilia (Fig. 1, A–D; Fig. 3; and Fig. S3 C). Theoretically, this could be achieved for them to reload onto retrograde IFT trains, to diffuse without relying on IFT, or both. Our biochemical data do not support the former as ARL3Q70L-HA-YFP fails to immunoprecipitate the BBSome and IFT-B1 simultaneously in cilia, thus excluding ARL3GTP/BBSome from being able to reload onto retrograde IFT trains for ciliary retrieval through IFT (Fig. 4 B). This result cannot rule out that ARL3GTP could convert back to ARL3GDP at the ciliary base, releasing the BBSome for reloading onto retrograde IFT trains for ciliary retrieval through IFT. However, the facts that ARL3GDP, upon entering cilia, convert to ARL3GTP for residing in ciliary matrix mostly at the ciliary base and the presence of the ARL3T30N-HA-YFP variant cannot lower the BBSome content at the ciliary base back to normal, largely disapproving this possibility (Fig. 2 D; and Fig. 3, A and B). As a result, the retrograde IFT train-shed BBSome is proposed to pass the TZ and out of cilia through diffusion (Fig. 5 D). To test this hypothesis, we examined ift46-1; IFT46:YFP-TG, bbs8; BBS8:YFP-TG, and arl3; bbs8; BBS8:YFP-TG cells (Lv et al., 2017). TIRF assays identified retrograde IFT trains (represented by IFT46-YFP) transported from the ciliary tip all the way to the basal bodies, revealing that they move across the TZ for ciliary retrieval through IFT (Fig. 5 E and Video 7). The BBSome (represented by BBS8-YFP) performed normal retrograde IFT for trafficking from the ciliary tip to base in bbs8; BBS8:YFP-TG cilia (Fig. 5 E and Video 8). Upon reaching the ciliary base, BBS8-YFP continued to move across the TZ for ciliary retrieval through IFT (Fig. 5 E and Video 8). Certain BBS8-YFP was indeed observed to stop IFT prior to shifting to a diffusion pattern inside the TZ region (Fig. 5 E and Video 8). In the absence of ARL3, BBS8-YFP remained to undergo normal retrograde IFT, while some were suspended for passing the TZ for ciliary retrieval but accumulated right above the TZ (Fig. 5 E and Video 9). Consistent with the biochemical data shown above (Fig. 4), our in vivo imaging data thus endorsed a notion that only a partial but not all BBSomes can shed from retrograde IFT trains at the ciliary base right above the TZ in an ARL3-independent manner. After their shedding, they likely diffuse through the TZ for ciliary retrieval. BBSomes unshed from retrograde IFT trains keep passing the TZ for ciliary retrieval through IFT.
TIRF imaging of IFT46-YFP movement across ift46-1; IFT46:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
TIRF imaging of IFT46-YFP movement across ift46-1; IFT46:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement across bbs8; BBS8:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement across bbs8; BBS8:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement across arl3; bbs8; BBS8:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
TIRF imaging of BBS8-YFP movement across arl3; bbs8; BBS8:YFP-TG transition zone. A frame from this video and kymograph are shown in Fig. 5 E. Play speed is real-time (15 fps).
ARL3GTP recruits the retrograde IFT train-shed and PLD-laden BBSome to move across the TZ for ciliary retrieval. Our previous study and others have shown that the ciliary membrane anchored PLD can load onto the BBSome all along the cilium but likely with a peak at the proximal ciliary region (Liu and Lechtreck, 2018; Liu et al., 2021). As compared to parental CC-5325 cells, arl3-282 cells contained PLD at the endogenous protein level of CC-5325 cells but had it accumulated in cilia (Fig. 6 A). Like the BBSome, PLD can be restored back to normal in arl3-282 cilia by ARL3Q70L-HA-YFP but not ARL3T30N-HA-YFP (Fig. 6 A). Supportive of this observation, ARL3ΔN15Q70L-HA-YFP also, as expected, rescued PLD back to normal in arl3-282 cilia, revealing that ARL3GTP rather than ARL3GDP is required for maintaining PLD ciliary turnover (Fig. S4 A). Notably, PLD ciliary distribution is hardly visualized in CC-5325, arl3-282; ARL3:HA:YFP-TG, and arl3-282; ARL3Q70L:HA:YFP-TG cells by immunostaining due to its low ciliary abundance (Liu and Lechtreck, 2018), while it was easily visualized to build up at the proximal ciliary region above the IFT81-labeled basal bodies in the absence of ARL3 or in the presence of ARL3T30N-HA-YFP (Fig. 6 B). Given that PLD behaves the same as the BBSome in cilia in response to ARL3 (Fig. 3, A, B, and F), ARL3GTP may promote PLD-laden BBSomes to move across the TZ for ciliary retrieval. Supportive of this notion, ARL3-HA-YFP immunoprecipitated both the BBSome and PLD but not IFT-A, IFT-B1, and IFT-B2 in the presence of excessive GTPγS (Fig. 6 C). In the presence of excessive GDP, ARL3-HA-YFP recovered none of these proteins (Fig. 6 C). Consistent with this, ARL3Q70L-HA-YFP but not ARL3T30N-HA-YFP immunoprecipitated both the retrograde IFT train-shed BBSome and PLD (Fig. 6 C). In summary, PLD remains to be a cargo of the retrograde IFT train-shed BBSome. ARL3GTP binds to and recruits the retrograde IFT train-shed and PLD-laden BBSome to move across the TZ for ciliary retrieval.

ARL3GTPrecruits the retrograde IFT train-shed and PLD-laden BBSome to move across the TZ for ciliary retrieval. (A) Immunoblots of WC samples and cilia of CC-5325, arl3-282, arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells probed for PLD. α-Tubulin and acetylated-α-tubulin (Ac-tubulin) were used as a loading control for WC samples and cilia, respectively. (B) CC-5325, arl3-282, arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells stained with α-PLD (red) and α-IFT81 (green). PC images of cells were shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnification (100 times) was shown. Scale bar, 10 µm. (C) Immunoblots of α-YFP-captured proteins from HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3:HA:YFP-TG cilia probed for the IFT-B1 subunits IFT22 and IFT70, the IFT-B2 subunits IFT38 and IFT57, the IFT-A subunits IFT43 and IFT139, the BBSome subunits BBS1 and BBS4, and PLD. HMEKN stands for the buffer used for solving cilia. Input was quantified with α-YFP by immunoblotting. MW, molecular weight. Source data are available for this figure: SourceData F6.
ARL3GTPrecruits the retrograde IFT train-shed and PLD-laden BBSome to move across the TZ for ciliary retrieval. (A) Immunoblots of WC samples and cilia of CC-5325, arl3-282, arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells probed for PLD. α-Tubulin and acetylated-α-tubulin (Ac-tubulin) were used as a loading control for WC samples and cilia, respectively. (B) CC-5325, arl3-282, arl3-282; ARL3:HA:YFP-TG, arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3T30N:HA:YFP-TG cells stained with α-PLD (red) and α-IFT81 (green). PC images of cells were shown. Inset shows the proximal ciliary region and the basal bodies. Inset magnification (100 times) was shown. Scale bar, 10 µm. (C) Immunoblots of α-YFP-captured proteins from HR-YFP (HA-YFP-expressing CC-125 cells), arl3-282; ARL3:HA:YFP-TG (in the presence of GTPγS or GDP), arl3-282; ARL3Q70L:HA:YFP-TG, and arl3-282; ARL3:HA:YFP-TG cilia probed for the IFT-B1 subunits IFT22 and IFT70, the IFT-B2 subunits IFT38 and IFT57, the IFT-A subunits IFT43 and IFT139, the BBSome subunits BBS1 and BBS4, and PLD. HMEKN stands for the buffer used for solving cilia. Input was quantified with α-YFP by immunoblotting. MW, molecular weight. Source data are available for this figure: SourceData F6.
Discussion
As an Arf-like small GTPase, Chlamydomonas ARL3 relies on GDP and its N-terminal amphipathic helix for membrane association, which is a prerequisite for ARL3 to diffuse into cilia, and to reside along the whole length of cilia by anchoring to the ciliary membrane. After a rapid activation process, ARL3GDP is converted to become ARL3GTP for releasing from the ciliary membrane. Following ciliary cycling, the BBSome can shed from retrograde IFT trains at the proximal ciliary region right above the TZ. By acting as a major ARL3 effector, retrograde IFT train-shed and PLD-laden BBSomes are bound to and recruited by ARL3GTP to move across the TZ, likely through diffusion, for ciliary retrieval (Fig. 7). Our data show that ARL3 maintains BBSome ciliary turnover by mediating its movement across the diffusion barrier at the TZ for ciliary retrieval, closing a gap in our understanding of how ARL3 participates in exporting BBSome cargoes out of cilia in C. reinhardtii.

Hypothetical model for how Chlamydomonas ARL3 promotes diffusion of the retrograde IFT train-shed and PLD-laden BBSome through the TZ for ciliary retrieval. IFT22/BBS3, when both in a GTP-bound state (IFT22GTP/BBS3GTP), recruits the BBSome from cytoplasm to the basal body for integrating into anterograde IFT trains (Xue et al., 2020). Upon reaching the ciliary tip via anterograde IFT, the BBSome remodels with the aid of the IFT-B1-shed IFT25/27 for promoting its reassembly (Sun et al., 2021). The reassembled BBSome interacts with its cargo PLD in a BBS3-dependent manner (Liu et al., 2021). This enables the PLD-laden BBSome to U turn in cilia for transporting to the ciliary base via retrograde IFT. ARL3GDP binds to the ciliary membrane for diffusing into cilia and is activated to become ARL3GTP by an unknown mechanism (?). ARL3GTP detaches from the ciliary membrane and resides in the ciliary matrix by concentrating at the proximal ciliary region right above the TZ. Following the transportation from the ciliary tip to base, the PLD-laden BBSome separates from retrograde IFT trains at the proximal ciliary region right above the TZ and is bound to ARL3GTP as an ARL3 effector. ARL3GTP then facilitates the retrograde IFT train-shed and PLD-laden BBSome to diffuse through the TZ for ciliary retrieval.
Hypothetical model for how Chlamydomonas ARL3 promotes diffusion of the retrograde IFT train-shed and PLD-laden BBSome through the TZ for ciliary retrieval. IFT22/BBS3, when both in a GTP-bound state (IFT22GTP/BBS3GTP), recruits the BBSome from cytoplasm to the basal body for integrating into anterograde IFT trains (Xue et al., 2020). Upon reaching the ciliary tip via anterograde IFT, the BBSome remodels with the aid of the IFT-B1-shed IFT25/27 for promoting its reassembly (Sun et al., 2021). The reassembled BBSome interacts with its cargo PLD in a BBS3-dependent manner (Liu et al., 2021). This enables the PLD-laden BBSome to U turn in cilia for transporting to the ciliary base via retrograde IFT. ARL3GDP binds to the ciliary membrane for diffusing into cilia and is activated to become ARL3GTP by an unknown mechanism (?). ARL3GTP detaches from the ciliary membrane and resides in the ciliary matrix by concentrating at the proximal ciliary region right above the TZ. Following the transportation from the ciliary tip to base, the PLD-laden BBSome separates from retrograde IFT trains at the proximal ciliary region right above the TZ and is bound to ARL3GTP as an ARL3 effector. ARL3GTP then facilitates the retrograde IFT train-shed and PLD-laden BBSome to diffuse through the TZ for ciliary retrieval.
How does ARL3 mediate BBSome turnover in cilia?
Small GTPases function as molecular switches for controlling BBSome ciliary turnover in various BBSome ciliary cycling steps. In Chlamydomonas, IFT22/BBS3, the heterodimer composed of Rab-like 5 (RABL5) GTPase IFT22 and Arf-like 6 (ARL6) GTPase BBS3, binds the BBSome through a direct interaction between BBS3 and the BBSome (Xue et al., 2020). IFT22/BBS3 recruits the BBSome to the basal bodies when they both are GTP-bound, thus controlling BBSome ciliary turnover by deciding its basal body amount available for entering cilia (Xue et al., 2020; Fig. 7). Chlamydomonas BBS3 enters cilia in a GTP-bound form (Liu et al., 2021). Inside cilia, BBS3 promotes PLD association with the BBSome but does not affect BBSome ciliary turnover (Liu et al., 2021; Fig. 7). This may not be the case in mammals. Mammalian Bbs3, like its Chlamydomonas counterpart, enters cilia in a GTP-bound form but followed by GTPase cycling with the aid of RABL4 GTPase Ift27 as a Bbs3-specific GEF (Liew et al., 2014). At the ciliary tip, Bbs3 reloaded with GTP in turn binds to and loads GPCR-laden BBSomes, as its effectors, onto retrograde IFT trains for trafficking to the ciliary base (Liew et al., 2014). It has been known that Ift27, by binding to its stabilizing partner Ift25 to form an Ift25/27 heterodimer, cycles off IFT-B1 at the ciliary tip cross-species (Liew et al., 2014; Sun et al., 2021; Wang et al., 2009). Unlike mammalian Ift27, Chlamydomonas IFT27 does not act as a BBS3 GEF in cilia but promotes BBSome reassembly during its remodeling process at the ciliary tip, critical for making the intact BBSome ready for loading onto the retrograde IFT trains (Sun et al., 2021; Fig. 7). Rather than these, leucine zipper transcription factor-like 1 acts as an upstream player in cytoplasm to mediate BBSome recruitment to the basal body through BBS3 and its ciliary tip reassembly through IFT27 simultaneously, controlling BBSome ciliary turnover in C. reinhardtii (Sun et al., 2021). As for Chlamydomonas ARL3, it mimics BBS3 to bind the membrane for diffusing into cilia and resides along the whole length of cilia by attaching to the ciliary membrane (Fig. 2; Jin et al., 2010; Liu et al., 2021). Different from BBS3 that in a GTP-bound state binds the membrane, Chlamydomonas ARL3 relies on GDP for membrane association (Fig. 2; Jin et al., 2010; Liu et al., 2021). This is easy to understand as the green algae cell may have developed an elaborated system to restrict ARL3 to bind to its BBSome effector only at the TZ region where ARL3GTP concentrates (Figs. 2 and 5). Upon reaching the TZ region through the retrograde IFT, the BBSome can drop-off retrograde IFT trains through a mechanism that remains unknown yet (Fig. 3). GTP loading then enables ARL3 to bind to and recruit the retrograde IFT train-shed and PLD-laden BBSome, as an ARL3-specific effector, to move across the diffusion barrier at the TZ for ciliary retrieval (Fig. 7). We currently do not know if this holds true for mammalian and human Arl3, while Chlamydomonas ARL3 participates in mediating BBSome ciliary turnover through promoting its movement across the TZ for ciliary removal.
The BBSome acts as an ARL3 effector only when they both position to the TZ region
Cross-ciliated species, PDE6D, UNC119A/B, and BART/BARTL1 have been identified as ARL3 effectors with PDE6D and UNC119A/B known as carrier/solubilizing proteins for binding and shuttling cytoplasmic-lipidated signaling protein cargoes into cilia (Linari et al., 1999; Lokaj et al., 2015; Wright et al., 2011). Once at the proximal ciliary region, Arl3 uses RP2 and Arl13b as its GAP and GEF, respectively, and is catalyzed to convert between being GTP- and GDP-bound (Gotthardt et al., 2015; Veltel et al., 2008; Zhang et al., 2016). This Arl3-Arl13b-RP2 cascade works efficiently for releasing the cytoplasmic lipidated cargoes to bind to the ciliary membrane through their lipidated moieties, revealing that ARL3 is critical for importing certain ciliary proteins into cilia. In this study, the BBSome was identified to be a major ARL3 effector in C. reinhardtii, uncovering ARL3’s role, for the first time, in exporting certain ciliary proteins (i.e., PLD), as BBSome cargoes, out of cilia through promoting BBSome movement across the TZ for ciliary retrieval. Chlamydomonas ARL3 uses the BBSome as its effector only when they both position to the TZ region but not in cytoplasm (Figs. 3 and 4; and Fig. S5 C). We now know that ARL3 likely retains in a GDP-bound state in cytoplasm, disabling it to bind to the BBSome (Fig. 2). It was already known that IFT22/BBS3 binds the BBSome in the cell body and recruits the BBSome to the basal bodies only when they both are in a GTP-bound state (Xue et al., 2020). This explains well why ARL3, even when being in a GTP-bound state, fails to bind to the BBSome in cytoplasm as the cell may direct BBSome trafficking in different cellular compartments through applying distinct GTPase pathways (Fig. 4 and Fig. S5 C).
How is ARL3 activated to promote outward BBSome movement across the TZ?
Chlamydomonas ARL3 in a GDP-bound state enters cilia but relies on GTP for binding to and recruiting the BBSome to move across the TZ for ciliary retrieval, suggesting that ARL3 must have to convert from being GDP-bound to being GTP-bound in cilia. Cross-ciliated species, Arl13b, acts as an Arl3 GEF to catalyze the conversion of Arl3GDP to Arl3GTP in cilia and the Arl3-Arl13b cascade works efficiently for releasing the cytoplasmic lipidated signaling proteins to bind to the ciliary membrane (Gotthardt et al., 2015; Linari et al., 1999; Lokaj et al., 2015; Wright et al., 2011; Zhang et al., 2016). Interestingly, knockout of human ARL13b alters BBSome ciliary turnover but by reducing its ciliary content, a result excluding ARL13b from promoting outward BBSome movement across the TZ for ciliary retrieval through ARL3 (Fujisawa et al., 2021). In addition, although they both are required for targeting INPP5E to the ciliary membrane in human cells, ARL3 and ARL13b instead participate in distinct steps of this event, further revealing their functional discrepancy in vivo (Fujisawa et al., 2021). This raised an interesting question, namely, which factor other than ARL13b, if desirable, contributes to activate ARL3 specifically for promoting outward BBSome passage through the TZ. We currently had no answer for this question, while murine Rabl2 only in a GTP-bound state causes the BBSome to cease for moving out of cilia but to accumulate at the proximal region right above the TZ, an intraciliary trafficking defect pattern the same as shown by ARL3 knock-out in C. reinhardtii (Duan et al., 2021). This observation provides a clue, from the functional review, that RABL2 could be an ARL3 GEF candidate in C. reinhardtii cilia. It could be the RABL2-ARL3 cascade that mediates outward BBSome movement across the TZ for ciliary retrieval in C. reinhardtii, though his hypothesis remains to be confirmed.
Implications for the molecular basis of the BBS phenotypes caused by ARL3 mutations
According to our biochemical and living cell imaging data, only partial BBSomes can jump off retrograde IFT trains at the ciliary base right above the TZ (Fig. 4 B and Fig. 5 E). For these unshed from retrograde IFT trains, they seem to move across the TZ for ciliary retrieval through IFT, consistent with the model proposed for murine cells (Fig. 5 E; Duan et al., 2021; Ye et al., 2018). Instead, the reservoir of retrograde IFT train-shed BBSomes pass the TZ for ciliary retrieval likely through diffusion in C. reinhardtii (Fig. 5 E). In mammals and humans, GPCRs are supposed to retain in the membrane following their ciliary retrieval. Uncoupling of the GPCR-laden BBSome with the IFT machinery could occur before they pass the diffusion barrier at the TZ for ciliary retrieval. This hypothesis can well explain why PLD loads onto the BBSome during its ciliary retrieval as PLD, unlike the IFT machinery, anchors to the membrane system during its ciliary export through the ARL3GTP/BBSome pathway (Liu and Lechtreck, 2018; Liu et al., 2021). If this holds true for human ARL3, ARL3-dependent BBSome ciliary retrieval also can satisfactorily explain the features of ARL3 deficiency in ciliary signaling, i.e., hedgehog singling, which is causative of BBS disorders (Duan et al., 2021; Eguether et al., 2014; Liew et al., 2014; Wiens et al., 2010; Ye et al., 2018). This could provide the molecular basis for why ARL3-related JBTS shares overlapping phenotypes with BBS disorders in humans.
Materials and methods
Antibodies, Chlamydomonas strains, and culture conditions
Antibodies used in this study are listed in Table 1. Rabbit-originated antibodies against ARL3 and CEP290 were produced by Beijing Protein Innovation, LLC (Beijing). The Chlamydomonas strains used in this study are listed in Table 2. CC-5325 and the arl3-282 (LMJ.RY0420.182282) mutants were available from the Chlamydomonas Library Project (CLiP, https://www.chlamylibrary.org/allMutants; Li et al., 2019). bbs8 has been reported previously (Sun and Pan, 2019). If not otherwise specialized, strains were grown in Tris acetic acid phosphate (TAP) or minimal 1 (M1) medium in a continuous light with constant aeration at room temperature. Depending on a specific strain, cells were cultured with or without the addition of 20 µg/ml paromomycin (Sigma-Aldrich), 15 µg/ml bleomycin (Invitrogen), or both antibiotics with 10 µg/ml paromomycin and 5 µg/ml bleomycin.
List of primary antibodies used in this study
| Name | IB | IS | Host | Reference or source |
|---|---|---|---|---|
| Anti-ARL3 | 1:250 | N/A | Rb | This study |
| Anti-CEP290 | 1:250 | 1:50 | Rb | This study |
| Anti-IFT22 | 1:1,000 | N/A | Rb | (Xue et al., 2020) |
| Anti-IFT38 | 1:1,000 | N/A | Rb | (Sun et al., 2021) |
| Anti-IFT46 | N/A | 1:100 | Rb | (Dong et al., 2017a) |
| Anti-IFT57 | 1:500 | N/A | Rb | (Dong et al., 2017a) |
| Anti-IFT43 | 1:500 | N/A | Rb | (Zhu et al., 2017) |
| Anti-IFT70 | 1:1,000 | N/A | Rb | (Dong et al., 2017a) |
| Anti-IFT81 | N/A | 1:200 | Mo | (Fan et al., 2010) |
| Anti-IFT139 | 1:1,000 | N/A | Rb | (Dong et al., 2017a) |
| Anti-BBS1 | 1:1,000 | N/A | Rb | (Xue et al., 2020) |
| Anti-BBS4 | 1:500 | N/A | Rb | (Liu et al., 2021) |
| Anti-BBS5 | 1:1,000 | N/A | Rb | (Xue et al., 2020) |
| Anti-BBS7 | 1:500 | N/A | Rb | (Liu et al., 2021) |
| Anti-BBS8 | 1:500 | 1:50 | Rb | (Sun et al., 2021) |
| Anti-PLD | 1:1,000 | N/A | Rb | (Liu et al., 2021) |
| Anti-α-tubulin (B512) | 1:10,000 | N/A | Mo | Sigma-Aldrich (T5168) |
| Anti-acetylated-tubulin (6-11B-1) | 1:10,000 | N/A | Mo | Sigma-Aldrich (T7451) |
| Anti-YFP (7.1 and 13.1) | 1:1,000 | 1:50 | Mo | Roche (11814460001) |
| Anti-HA (3F10) | 1:1,000 | 1:50 | Rt | Roche (11867423001) |
| HRP-conjugated goat anti-rabbit IgG | 1:10,000 | N/A | Gt | The Jackson Lab. (111035003) |
| HRP-conjugated goat anti-mouse IgG | 1:10,000 | N/A | Gt | The Jackson Lab. (115035003) |
| HRP-conjugated goat anti-rat IgG | 1:10,000 | N/A | Gt | The Jackson Lab. (112035003) |
| Alexa-Fluor 594-conjugated goat anti-rabbit IgG | N/A | 1:400 | Gt | Molecular probes (A11012) |
| Alexa-Flour 488-conjugated goat anti-mouse IgG | N/A | 1:400 | Gt | Molecular probes (A10680) |
| Alexa-Flour 488-conjugated goat anti-rat IgG | N/A | 1:400 | Gt | Molecular probes (A11006) |
Rb, rabbit; Mo, mouse; Rt, rat; Gt, goat; IB, immunoblotting; IS, immunostaining.
List of Chlamydomonas stains used in this study
| Name | Genotype | Reference or source |
|---|---|---|
| CC-125 (WT) | nit1; nit2; mt+ | CRC |
| CC-5325 | cw15; mt− | CLiP (Li et al., 2019) |
| arl3-282 | cw15; arl3::aphVIII; mt− | CLiP (LMJ.RY0420.182282) (Li et al., 2019) |
| ARL3; IFT43:HA:YFP-TG | cw15; mt−; IFT43:HA:YFP-TG | This study |
| ARL3; IFT22:HA:YFP-TG | cw15; mt−; IFT22:HA:YFP-TG | This study |
| ARL3; IFT38:YFP-TG | cw15; mt−; IFT38:YFP-TG | This study |
| arl3-282; IFT43:HA:YFP-TG | cw15; arl3::aphVIII; mt−; IFT43:HA:YFP-TG | This study |
| arl3-282; IFT22:HA:YFP-TG | cw15; arl3::aphVIII; mt−; IFT22:HA:YFP-TG | This study |
| arl3-282; IFT38:YFP-TG | cw15;arl3::aphVIII; mt−; IFT38:YFP-TG | This study |
| arl3-282; ARL3:HA:YFP-TG | cw15; arl3::aphVIII; mt−;ARL3:HA:YFP-TG | This study |
| arl3-282; ARL3Q70L:HA:YFP-TG | cw15; arl3::aphVIII; mt−;ARL3Q70L:HA:YFP-TG | This study |
| arl3-282; ARL3T30N:HA:YFP-TG | cw15; arl3::aphVIII; mt−; ARL3T30N:HA:YFP-TG | This study |
| arl3-282; ARL3ΔN15:HA:YFP-TG | cw15;arl3::aphVIII;mt−; ARL3ΔN15:HA:YFP-TG | This study |
| arl3-282; ARL3ΔN15Q70L:HA:YFP-TG | cw15; arl3::aphVIII; mt−; ARL3ΔN15Q70L:HA:YFP-TG | This study |
| arl3-282; ARL3ΔN15T30N:HA:YFP-TG | cw15; arl3::aphVIII;mt−; ARL3ΔN15T30N:HA:YFP-TG | This study |
| bbs8 | bbs8; mt+ | (Sun and Pan, 2019) |
| bbs8; BBS8:YFP-TG | bbs8; mt+; BBS8:YFP-TG | This study |
| arl3; bbs8 | cw15;arl3::aphVIII;bbs8 | This study |
| arl3; bbs8; ARL3:HA:YFP-TG | cw15;arl3::aphVIII; bbs8; ARL3:HA:YFP-TG | This study |
| arl3; bbs8; ARL3Q70L:HA:YFP-TG | cw15; arl3::aphVIII;bbs8;ARL3Q70L:HA:YFP-TG | This study |
| arl3; bbs8; ARL3T30N:HA:YFP-TG | cw15; arl3::aphVIII;bbs8; ARL3T30N:HA:YFP-TG | This study |
| arl3; bbs8; BBS8:YFP-TG | cw15; arl3::aphVIII; bbs8; BBS8:YFP-TG | This study |
| ift46-1; IFT46:YFP-TG | ift46; mt+; IFT46:YFP-TG | (Lv et al., 2017) |
| HR:YFP-TG | nit1;nit2;mt+; HA:YFP-TG | (Dong et al., 2017b) |
CC numbers refer to the Chlamydomonas stock collection number; TG, transgenes; CRC, Chlamydomonas Resource Center.
Vectors and transgenic strain generation
Expression vectors were constructed on pBKS-gBBS3-HA-YFP-Ble that contained HA-YFP coding sequences followed immediately downstream by a sequence encoding the Rubisco 3′-UTR and the bleomycin cassette (Ble, zeocine resistant gene; (Dong et al., 2017a). To generate ARL3-HA-YFP-expressing vector, a 3.4-kb ARL3 fragment consisting of the 1.0-kb promoter sequence and its coding region was amplified from genomic DNA using the primer pair gARL3-FOR1 and gARL3-REV1 as listed in Table 3 and inserted into the XbaI and EcoRI sites of pBKS-gBBS3-HA-YFP-Ble, resulting in pBKS-gARL3-HA-YFP-Ble. To generate ARL3ΔN15-HA-YFP-expressing vector, two fragments were amplified using primer pairs gARL3-FOR1 and gARL3ΔN15-REV and gARL3ΔN15-FOR and gARL3-REV1 as listed in Table 3 and inserted into the XbaI and EcoRI sites of pBKS-gBBS3-HA-YFP-Ble by three-way ligation, resulting in pBKS-gARL3ΔN15-HA-YFP-Ble. The desiring mutations T30N and Q70L were introduced into pBKS-gARL3-HA-YFP-Ble and pBKS-gARL3ΔN15-HA-YFP-Ble by site-directed mutagenesis using the primer pairs ARL3T30N-FOR and ARL3T30N-REV; ARL3Q70L-FOR and ARL3Q70L-REV, respectively, as listed in Table 3. Afterward, the mutated DNAs were inserted into the XbaI and EcoRI sites of pBKS-gARL3-HA-YFP-Ble, resulting in pBKS-gARL3T30N-HA-YFP-Ble, pBKS-gARL3Q70L-HA-YFP-Ble, pBKS-gARL3ΔN15T30N-HA-YFP-Ble, and pBKS-gARL3ΔN15Q70L-HA-YFP-Ble. To express IFT43-HA-YFP, a 2,800-bp DNA fragment composing of a 1,000-bp promoter and IFT43 coding sequence was amplified from genomic DNA using the primer pair gIFT43-FOR and gIFT43-REV as listed in Table 3, and inserted into NotI and EcoRI sites of pBluescript II KS(+) vector, resulting in pBKS-gIFT43. Afterwards, gIFT43 sequence was cut from pBKS-gIFT43 by NotI and EcoRI sites, HA-YFP-Ble sequence was cut from pBKS-gARL3-HA-YFP-Ble by EcoRI and KpnI, and inserted those two fragments into the NotI and KpnI sites of pBluescript II KS(+) vector by three-way ligation, resulting in pBKS-gIFT43-HA-YFP-Ble. To express IFT38-YFP, a 3,875-bp DNA fragment composing of a 1,000-bp promoter sequence and the IFT38 coding sequence was amplified from genomic DNA using the primer pair gIFT38-FOR and gIFT38-REV as listed in Table 3 and inserted into the BamHI and EcoRI sites of pBluescript II KS(+) vector, resulting in pBKS-gIFT38. Next, IFT38 sequence was cut from pBKS-gIFT38, and inserted into the BamHI and EcoRI sites of pBKS-gBBS5-YFP-Paro and pBKS-gBBS5-YFP-Ble, resulting in pBKS-gIFT38-YFP-Paro and pBKS-gIFT38-YFP-Ble, respectively. pBKS-gIFT22-HA-GFP-Paro and pBKS-gIFT22-HA-GFP-Ble have been described previously (Xue et al., 2020). To express IFT22-HA-YFP, the HA-GFP fragment was replaced with the HA-YFP fragment obtained from pBKS-gARL3-HA-YFP-Ble by EcoRI and XhoI digestion, resulting in pBKS-gIFT22-HA-YFP-Paro and pBKS-gIFT22-HA-YFP-Ble, respectively. To express BBS8-YFP, a 4,804-bp DNA fragment composing of 1,000-bp promoter and 3,804-bp BBS8 coding sequence was amplified from genomic DNA using the primer pair gBBS8-FOR and gBBS8-REV as listed in Table 3, and inserted into XbaI and EcoRI sites of pBluescript II KS(+) vector, resulting in pBKS-gBBS8. Then, BBS8 sequence cut from pBKS-gBBS8 by XbaI and EcoRI sites and YFP-Ble sequence cut from pBKS-gBBS5-YFP-Ble by EcoRI and KpnI sites were inserted into the XbaI and KpnI sites of pBluescript II KS(+) vector by three-way ligation, resulting in pBKS-gBBS8-YFP-Ble. After the verification by direct nucleotide sequencing, the new constructs were transformed into Chlamydomonas strain by electroporation and screening of the positive transformants was done on TAP plates containing 20 µg/ml paromomycin, 15 µg/ml bleomycin (Invitrogen), or both antibiotics with 10 µg/ml paromomycin and 5 µg/ml bleomycin.
List of primers used in this study
| Name | Nucleotide sequence |
|---|---|
| Primers used to amplify the aphVIII gene insertion in ARL3 genomic DNA | |
| gARL3-FOR | 5′-CCTCTAGAACATCACCACTATCACGC-3′ |
| gARL3-REV | 5′-CCGAATTCCCAAAGCTCCGAAGCCAC-3′ |
| Primers used to clone target genes | |
| gARL3-FOR1 | 5′-CCTCTAGAAGCTCCGACATGAGCGCC-3′ |
| gARL3-REV1 | 5′-CCGAATTCCTTGACCTGCTTCATCATC-3′ |
| gARL3ΔN15-FOR | 5′-CCGGATCCGTGACATGTGCTCGCAGC-3′ |
| gARL3ΔN15-REV | 5′-CCGGATCCCTGTGGCGAGCACACCAG-3′ |
| gIFT38-FOR | 5′-CCGGATCCATTTAGCCCCAAATTATG-3′ |
| gIFT38-REV | 5′-CCGAATTCGAAGTCATTGTCATCCTC-3′ |
| gIFT43-FOR | 5′-ACGCGGCCGCATGGTGTCCTACCTGGGC-3′ |
| gIFT43-REV | 5′-GTGAATTCCAGCTTGATGGGCATGG-3′ |
| gBBS8-FOR | 5′-GGACTAGTTGCAGCAGCAAGTAATGCAAC-3′ |
| gBBS8-REV | 5′-GGAATTCCAGCATAGTGAAATGGGCC-3′ |
| cARL3-FOR1 | 5′-GGCCATGGATGGGCCTCTTGTCGCTG-3′ |
| cARL3-REV1 | 5′-CGGCGGCCGCCTTGACCTGCTTCATC-3′ |
| cARL3ΔN15-FOR | 5′-CCCCATGGATGGCCCGCATCCTGGTC-3′ |
| cBBS1-FOR | 5′-CCGGATCCATGCTGCCATCAGTCAAG-3′ |
| cBBS1-REV | 5′-CCAAGCTTTTACTCCACCTCCTCCGG-3′ |
| cBBS2-FOR | 5′-CCGGATCCATGCTCGTGCCGGCCTTC-3′ |
| cBBS2-REV | 5′-CCAAGCTTTCACACCGGCCCGTCGCC-3′ |
| cBBS4-FOR | 5′-CAGCAAATGGGTCGGGATCCATGTCGTCATTAGCGCAG-3′ |
| cBBS4-REV | 5′-TCGACGGAGCTCGAATTCTTATCACATGCCCAGCAGCT-3′ |
| cBBS5-FOR | 5′-GACAGCAAATGGGTCGGGATCCATGGCTGACCTCTTTG-3′ |
| cBBS5-REV | 5′-CGACGGAGCTCGAATTCTTATCACAGCACGCTCCACAG-3′ |
| cBBS7-FOR | 5′-GGACAGCAAATGGGTCGGGATCCATGGAGCTGGAACTATTTC-3′ |
| cBBS7-REV | 5′-GTCGACGGAGCTCGAATTCTTACTACCGCTGCCCCAGCAG-3′ |
| cBBS8-FOR | 5′-GGACAGCAAATGGGTCGGGATCCATGCAGCAGCCACAGCAAG-3′ |
| cBBS8-REV | 5′-GTCGACGGAGCTCGAATTCTTATCACAGCATAGTGAAATG-3′ |
| Primers used to do site-directed mutagenesis | |
| ARL3Q70L-FOR | 5′-GGACATTGGCGGCCTGAAGTCCAT-3′ |
| ARL3Q70L-REV | 5′-AGGCCGCCAATGTCCCAAATTTTC-3′ |
| ARL3T30N-FOR | 5′-ATAACGCTGGTAAAAACACCATCCTG-3′ |
| ARL3T30N-REV | 5′-TTTTTACCAGCGTTATCCAGTCCCA-3′ |
Mating experiments
To generate arl3; bbs8 double mutant, 20 ml of each of arl3-282 and bbs8 cells were grown to a final concentration of 2 × 106 cells/ml in M1 medium under continuous light at room temperature. The cells were collected by centrifugation at 1,000 × g for 15 min and washed with M1-N (without nitrogen) medium. Afterwards, the cells were transferred to 20 ml of M1-N medium and aerated for 12 h under continuous light. After that, gametes of two opposite mating types were mixed in flask and incubated under light for 2 h. The cells sitting on the flask bottom were transferred to M1 plate containing 4% agar, air-dried, incubated overnight under continuous light, and then continued to be incubated for a week in the dark. The plates were then moved to incubate at −20°C for 2 d followed by culturing at room temperature under continuous light for at least 10 d. The arl3- and bbs8-double null mutants were screened by detecting the loss of ARL3 and BBS8 proteins through immunoblotting of whole cell extracts with both ARL3 and BBS8 antibodies as described below.
Total RNA and genomic DNA manipulations
Genomic DNA of Chlamydomonas cells was extracted and purified using a Wizard Genomic DNA Purification Kit (Promega) following the kit’s protocol. To characterize the arl3-282 cell at the genomic level, 20 ng of genomic DNA was applied as PCR template to amplify ARL3 genomic sequence. The PCR reactions were performed at 95°C for 5 min followed by 30 cycles of 95°C for 20 s, 61°C for 20 s, and 72°C for 5 min with the primer pair gARL3-FOR and gARL3-REV as listed in Table 3. Total RNA of Chlamydomonas cells was extracted and purified according to our protocol reported previously (Dong et al., 2017a). Five micrograms of RNA were reverse-transcribed at 42°C for 1 h using M-MLV Reverse Transcriptase (Promega) and oligo(T)18 primers (Takara). ARL3 cDNA and cDNAs encoding the six BBS proteins BBS1, BBS2, BBS4, BBS5, BBS7, and BBS8 were amplified by PCR using primer pairs cARL3-FOR1 and cARL3-REV1, cBBS1-FOR and cBBS1-REV, cBBS2-FOR and cBBS2-REV, cBBS4-FOR and cBBS4-REV, cBBS5-FOR and cBBS5-REV, cBBS7-FOR and cBBS7-REV, and cBBS8-FOR and cBBS8-REV, respectively, as listed in Table 3. The PCR reactions were performed at 95°C for 5 min followed by 30 cycles of 95°C for 20 s, 61°C for 20 s, and 72°C for 4 min.
Ciliary length measurement
Ciliary length measurement was carried out according to our previous report (Fan et al., 2010). In brief, Chlamydomonas cells growing in M1 medium to a concentration of ∼107 cells were fixed with 1% polyglutaraldehyde, placed onto the surface of glass slides and covered by cover glass. Cells were viewed with an Olympus IX83 inverted fluorescent microscopy equipped with a 100×/1.49 NA TIRF oil immersion objective len (Olympus). Phase contrast images were then taken with a back-illuminated scientific CMOS camera (Prime 95B, Photometrics). Ciliary length was measured using ImageJ (version 1.42g; National Institutes of Health). The data were processed with GraphPad Prism 8.30 (GraphPad Software). For each strain, a total of 20 cilia were measured.
Isolation of cilia and cell bodies
Isolation of cilia and cell bodies was performed according to our protocol reported previously (Fan et al., 2010). In brief, Chlamydomonas cells were grown in 10 liters of TAP medium to a final density of 108 cells/ml, collected by centrifugation at 1,000 × g for 15 min, and suspended in 150 ml of TAP (pH 7.4). Cells were incubated for 2 h under strong light with bubbling before 0.5 M acetic acid was added to adjust the pH value to 4.5 for cell deciliation. Afterwards, 0.5 M KOH was added to adjust the pH value to 7.4. Cell body pellets and cilia in the supernatant were collected separately after centrifugation at 600 × g at 4°C for 5 min. To avoid the possible cell body contamination, cilia were repeatedly washed with HMDEKN buffer (30 mM Hepes [pH 7.4], 5 mM MgSO4, 1 mM DTT, 0.5 mM EGTA, 25 mM KCl, and 125 mM NaCl) by centrifugation at 12,000 × g for 10 min until the green color disappeared completely. All the experiments were done at 4°C.
Preparation of ciliary fractions
Ciliary fractions were prepared according to our protocol reported previously (Liu et al., 2021). In brief, cell body-depleted cilia were dissolved in HMDEKN buffer supplemented with protein inhibitors (PI; 1 mM PMSF, 50 μg/ml soy-bean trypsin inhibitor, 1 μg/ml pepstatin A, 2 μg/ml aprotinin, and 1 μg/ml leupeptin) and frozen in liquid nitrogen. After three cycles of frozen-and-thaw, the solution was centrifugated at 12,000 × g at 4°C for 15 min and the ciliary matrix fraction was collected as the supernatant. The pellets were then dissolved in HMEDKN (see above) buffer containing 0.5% nonidet P-40 (NP-40) and stayed on ice for 15 min. After centrifugation at 12,000 × g at 4°C for 10 min, the supernatant and pellet were collected as membrane and axonemal fractions, respectively.
Sucrose density gradient centrifugation assay
Ciliary samples were analyzed by sucrose density gradient centrifugation according to our protocol reported previously (Sun et al., 2021). Briefly, linear 12 ml of 10–25% sucrose density gradients were prepared in HMDEKN buffer supplemented with PI (see above) and 1% NP-40. Ciliary extracts were frozen and thawed for three cycles using liquid nitrogen and centrifuged at 12,000 × g at 4°C for 10 min. After the non-soluble debris was removed, 700 μl of samples were loaded on the top of the gradients and separated at 38,000 rpm for 14 h in a SW41Ti rotor (Beckman Coulter). After the gradients were fractioned into 24 0.5 ml aliquots, 20 μl of each fraction was loaded into SDS-PAGE gel for electrophoresis and analyzed by immunoblotting as described below. The centrifugation was done at 4°C.
Immunoblotting
Whole cell, cell body, and ciliary samples were mixed with Laemmli SDS sample buffer and boiled for 5 min before centrifugation at 2,500 × g for 5 min. If not otherwise specified, either 20 µg of total protein from each whole cell and cell body sample or 100 µg of total protein from each ciliary sample was loaded in the 12% SDS-PAGE gel for electrophoresis followed by electrotransferring onto a nitrocellulose membrane. After the membrane was blocked with 5% non-fat dry milk in TBS (10 mM Tris, pH 7.5, 166 mM NaCl) plus 0.05% Tween-20, primary antibodies were diluted in the blocking solution and then incubated with the membrane for 2 h at room temperature (RT). After washing three times with TBS plus 0.05% Tween-20, HRP-conjugated secondary antibodies (The Jackson Laboratory) and chemiluminescence were used to detect the primary antibodies using the Mini Chemiluminescent/Fluorescent Imaging and Analysis System (Sagecreation). Primary and secondary antibodies were diluted for immunoblotting with a ratio as shown in Table 1. If needed, ImageJ software (version 1.42g; National Institutes of Health) was used to quantify the target proteins by measuring the immunoblot intensity. The immunoblot intensity was normalized to the intensity of a loading control protein.
Immunoprecipitation
Immunoprecipitation was performed according to our protocol reported previously (Liu et al., 2021). In brief, cell body and ciliary samples isolated from cells expressing HA-YFP, HA-YFP-tagged ARL3, or its variants were resuspended in HMDEKN or DTT-deprived HMEKN buffer supplemented with protein inhibitors (see above). The samples were lysed by adding NP-40 to a final concentration of 1% followed by centrifugation at 14,000 × g, 4°C for 10 min. Afterward, the supernatants were collected for agitating with 5% BSA-pretreated camel anti-YFP antibody-conjugated agarose beads (V-nanoab Biotechnology) for 2 h at 4°C. After continuous washing with HMEKN buffer, HMEK buffer containing 50 mM NaCl, and HMEK without containing NaCl, the beads were collected by centrifugation at 2,500 × g for 2 min, mixed with Laemmli SDS sample buffer, and boiled for 5 min before centrifugation at 2,500 × g for 5 min. The immunoprecipitants in the supernatants were collected for immunoblotting analysis as described above. If needed, immunoprecipitation was performed in the presence of excessive GTPγS (20 mM) or GDP (20 mM).
Liposome flotation assay
The ARL3 cDNA was inserted into the NcoI and NotI sites of pET-28a (Novagen) to result in pET-28a-cARL3. The desiring deletion of N-terminal 15 amino acid residues and the mutations Q70L and T30N were introduced into ARL3 by regular PCR using primer pair cARL3ΔN15-FOR and cARL3-REV1 or site-directed mutagenesis using primer pairs ARL3Q70L-FOR and ARL3Q70L-REV and ARL3T30N-FOR and ARL3T30N-REV, resulting plasmids pET-28a-cARL3Q70L, pET-28a-cARL3T30N, pET-28a-cARL3ΔN15, pET-28a-cARL3ΔN15Q70L, and pET-28a-cARL3ΔN15T30N, respectively. The primer sequences are listed in Table 3. After these plasmids were transformed into the Escherichia coli strain BL21(DE3), the bacterially expressed C-terminal 6×His tagged ARL3 and its variants were purified with Ni Sepharose 6 Fast Flow beads (GE Healthcare) following the kit’s protocol. E. coli total lipid extract was purchased from Avanti (Cat# 100500; Avanti), dissolved in chloroform, and dried under nitrogen gas. Lipid was rehydrated in the lipid reconstitution buffer (30 mM Tris, 150 mM NaCl, 2 mM MgCl2, and 2 mM DTT, pH 7.5) by centrifuging at 1,000 rpm for 1 h at RT. Liposomes were made from the resuspended lipid with an extruder (200 nm pore size, 20 strokes manually; Avanti) at RT. Nucleotide loading to ARL3 was performed by strictly following the protocol reported previously (Gotthardt et al., 2015). In brief, 20 µg of the bacterially purified ARL3 (100 µM) were incubated in the presence of 50 mM EDTA and a five-molar excess of GTPγS or GDP over ARL3 (500 µM) for 2 h. 100 mM MgCl2 was then added to samples and the ARL3 protein was separated from the excess of nucleotides by Zeba Spin desalting columns (Thermal Fisher Scientific). Afterward, 10 µg of each of the pretreated ARL3, 75 μl of liposomes, and lipid reconstitution buffer were mixed to reach a final volume of 150 μl. 100 μl of 75% sucrose solution (in lipid reconstitution buffer) was next added to the liposome-protein mixtures for making 250 μl of 30% sucrose solution, which was loaded as the bottom layer in the centrifuge tube. 200 μl of 25% sucrose solution (in lipid reconstitution buffer) and 50 μl of lipid reconstitution buffer were added sequentially on the top of the bottom layer. The sucrose gradients were centrifuged at 240,000 × g for 2 h in a TLS-55 rotor (Beckman Coulter). After the centrifugation, 50 μl of solution was removed from the top of the sucrose gradients and 10 μl of them was subjected to immunoblotting as described above. For the ARL3 variants, they were used directly for liposome flotation assays without nucleotide preloading. For immunoblotting, 1 µg of ARL3/variant protein was loaded for evaluating their binding/input ratio shown as percentile.
Fixed imaging
Immunofluorescence staining was performed according to our protocol reported previously (Wang et al., 2009). Experiments were performed at RT if not otherwise specified. In brief, Chlamydomonas cells growing in TAP medium were seeded to 0.1% polyethyleneimine-coated coverslips for 8 min. Following permeabilization and fixation with methanol twice each for 10 min, cells were rehydrated with phosphate buffered saline (PBS) and incubated with primary antibodies in blocking buffer (5% BSA, 1% cold water fish gelatin, and 10% goat serum in PBS) for 2 h. After washing 10 times in PBS, cells were incubated with secondary antibodies in blocking buffer for 2 h. After washing additional 10 times in PBS, the coverslips were mounted with SlowFade Antifade reagent (Molecular Probes) and images were captured with an Olympus IX83 inverted fluorescent microscopy equipped with a back-illuminated scientific CMOS camera (Prime 95B, Photometrics), a 100×/1.40 NA oil objective lens (Olympus), and 488and 561-nm lasers from Coherent OBIS Laser Module. All images were acquired and processed with CellSens Dimension (version 2.1; Olympus). The primary antibodies against CEP290, BBS8, IFT46, IFT81, YFP, HA, and the secondary antibodies including Alexa-Fluor594-conjugated goat anti-rabbit, Alexa-Fluor488-conjugated goat anti-mouse, and Alexa-Fluor488-conjugated goat anti-rat (Molecular Probes) were listed in Table 1 with their suggested dilutions.
Live-cell imaging
For live-cell imaging, cells (10 μl) growing in TAP medium were placed onto the larger coverslip (24 × 60-mm No.1.5) and allowed to settle for 2 min at RT. After a ring of petroleum jelly was added around the droplet, a smaller coverslip (22 × 22-mm No.1.5) containing 5 μl of 10 mM HEPES, pH 7.4, and 5 mM EGTA was inverted onto the larger one to generate a sealed observation chamber. TIRF microscopy was applied to visualize the motility of YFP-tagged IFT43, IFT22, IFT38, IFT46, BBS8, and ARL3 and its variants in cilia. YFP-tagged proteins were imaged at 15 frames per second (fps) using an Olympus IX83 inverted fluorescent microscopy equipped with a through-the-objective TIRF system, a 100×/1.49 NA TIRF oil immersion objective lens (Olympus), a back-illuminated scientific CMOS camera (Prime 95B; Photometrics), and 488-nm laser from Coherent OBIS Laser Module as detailed previously (Xue et al., 2020). CellSens Dimension (version 2.1; Olympus) was used to capture videos.
Kymogram analysis
Kymography was generated according to our protocol reported previously (Dong et al., 2017a). In brief, YFP-tagged IFT, BBSome, and ARL3 proteins were imaged with TIRF (15 Hz) for ∼20 s. The videos obtained were processed with CellSens Dimension (version 2.1; Olympus) for generating kymographs. In kymographs, lines drawn along the long axis of the cilium (“the leg”) and the processive movement (“the hypotenuse”) represent the anterograde and retrograde IFT tracks. The angle of the lines was measured (“the included angle”). Comparison of the leg and the hypotenuse was applied for quantifying the frequency of an IFT- or BBS-containing train. Comparison of the hypotenuse and the included angle was applied for measuring the velocity of IFT and BBSome trains. KymographClear was applied to deconvolve retrograde from anterograde trains for clearly showing BBSome diffusion through the TZ (e.g., Fig. 5 E; Mangeol et al., 2016).
Protein–protein interaction assay
The cDNA encoding BBS9 was synthesized by Genewiz. The cDNAs encoding BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9 were inserted into the BamHI and Hind III sites of pMal-C2x (Nova Lifetech) to generate pMal-C2x-cBBS1, pMal-C2x-cBBS2, pMal-C2x-cBBS4, pMal-C2x-cBBS5, pMal-C2x-cBBS7, pMal-C2x-cBBS8, and pMal-C2x-cBBS9. After these plasmids and the empty pMal-C2x plasmid were transformed into the E. coli strain BL21(DE3), the bacterially expressed N-terminal MBP-tagged BBS proteins were purified with Dextrin Sepharose High Performance with MBP-tagged protein purification resin (GE Healthcare). One hundred micrograms of MBP and the MBP-tagged BBS proteins were individually mixed with 100 µg of bacterially expressed ARL3Q70L or ARL3T30N (see above) to form a combination of 16 reactions. After incubated for 2 h at RT, the mixtures were purified with Dextrin Sepharose High Performance MBP-tagged protein purification resin (GE Healthcare). 10 micrograms of proteins from elutes were resolved on 12% SDS-PAGE gels and visualized with Coomassie blue staining. Immunoblotting assay was also performed to verify the interaction between BBS proteins and ARL3 variants with α-ARL3.
Statistical analysis
Statistical analysis was done with GraphPad Prism 8.30 (GraphPad Software). For comparisons on velocities and frequencies of the YFP- and HA-YFP–labeled proteins and ciliary length measurement, one-sample unpaired student t-test was used on samples. The data were presented as mean ± SD ns represents non-significance. Data distribution was assumed to be normal, but this was not formally tested.
Online supplemental material
Fig. S1 analyzes the conservation of ARL3 across ciliated species. Fig. S2 characterizes the ARL3 CLiP mutant arl3-282. Fig. S3 characterizes the anti-ARL3 antibody and studies the influence of ARL3 on globe IFT protein content and ciliation. Fig. S4 characterizes the influence of ARL3 on ciliary BBSome content and integrity. Fig. S5 studies the influence of ARL3 nucleotide state on basal body targeting of IFT proteins and the interaction of ARL3 with IFT subcomplexes and the BBSome in cytoplasm. Video 1 shows the distribution of ARL3-HA-YFP in arl3-282; ARL3:HA:YFP-TG cilia. Video 2 shows the distribution of ARL3T30N-HA-YFP in arl3-282; ARL3T30N:HA:YFP-TG cilia. Video 3 shows the distribution of ARL3Q70L-HA-YFP in arl3-282; ARL3Q70L:HA:YFP-TG cilia. Video 4 shows the distribution of ARL3ΔN15Q70L-HA-YFP in arl3-282; ARL3ΔN15Q70L:HA:YFP-TG cilia. Video 5 shows the IFT movement of BBS8-YFP in bbs8; BBS8:YFP-TG cilia. Video 6 shows the IFT movement of BBS8-YFP in arl3; bbs8; BBS8:YFP-TG cilia. Video 7 shows the IFT movement of IFT46-YFP across ift46-1; IFT46:YFP-TG transition zone. Video 8 shows both the IFT movement and diffusion of BBS8-YFP across bbs8; BBS8:YFP-TG transition zone. Video 9 shows the IFT movement but not diffusion of BBS8-YFP across arl3; bbs8; BBS8:YFP-TG transition.
Data availability
Data supporting the findings of this study were contained within this paper and the supplemental files. Protocols and materials are available to the scientific community upon request.
Acknowledgments
Research reported in this publication was supported by National Natural Science Foundation of China (32070698 to Z.-C. Fan and 32100541 to B. Xue) and China Postdoctoral Science Foundation (2021M702457 to Y.-X. Liu and 2021M692403 to B. Xue). The founders have no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare no competing financial interests.
Author contributions: Y.-X. Liu performed genetic and biochemical experiments, acquired data at the microscope, performed image processing, analyzed the data, and interpreted the data. W.-Y. Sun performed experiments and acquired data at the microscope. B. Xue prepared the samples. R.-K. Zhang prepared the samples. W.-J. Li prepared the samples. X. Xie analyzed the data. Z.-C. Fan designed the experiments, performed image processing, analyzed the data, interpreted the data, and wrote the manuscript.

