


Ligand binding triggers clathrin-mediated and, at high ligand concentrations, clathrin-independent endocytosis of EGFR. Clathrin-mediated endocytosis (CME) of EGFR is also induced by stimuli activating p38 MAPK. Mechanisms of both ligand- and p38-induced endocytosis are not fully understood, and how these pathways intermingle when concurrently activated remains unknown. Here we dissect the mechanisms of p38-induced endocytosis using a pH-sensitive model of endogenous EGFR, which is extracellularly tagged with a fluorogen-activating protein, and propose a unifying model of the crosstalk between multiple EGFR endocytosis pathways. We found that a new locus of p38-dependent phosphorylation in EGFR is essential for the receptor dileucine motif interaction with the σ2 subunit of clathrin adaptor AP2 and concomitant receptor internalization. p38-dependent endocytosis of EGFR induced by cytokines was additive to CME induced by picomolar EGF concentrations but constrained to internalizing ligand-free EGFRs due to Grb2 recruitment by ligand-activated EGFRs. Nanomolar EGF concentrations rerouted EGFR from CME to clathrin-independent endocytosis, primarily by diminishing p38-dependent endocytosis.
Introduction
EGF receptor (EGFR), the archetypal receptor tyrosine kinase, plays important roles in regulation of cell motility, metabolism, proliferation, survival, and differentiation (Sibilia et al., 2000). Mutations and overexpression of EGFR are associated with tumorigenesis and metastatic processes. Ligand binding to the extracellular domain of EGFR leads to receptor dimerization, activation of the tyrosine kinase, and phosphorylation of tyrosine residues in the cytoplasmic domain, which serve as docking sites for downstream signaling proteins containing Src homology 2 (SH2) and phosphotyrosine-binding domains (Lemmon and Schlessinger, 2010). Ligand binding also results in rapid endocytosis and subsequent lysosomal degradation of EGFR (Sorkin and Goh, 2009). Endocytic trafficking of EGFR is proposed to play a key role in the spatiotemporal regulation of EGFR signaling, and dysregulation of this trafficking is associated with cell transformation (Mellman and Yarden, 2013; Sigismund et al., 2012; Tomas et al., 2014; von Zastrow and Sorkin, 2021). However, the molecular mechanisms of EGFR endocytosis are not well-defined, which hinders development of experimental approaches for defining the impact of dysregulated endocytosis on signal transduction processes.
EGFR ligand concentrations within 20–200 pM (the range of KD values measured for high-affinity EGF binding) induce EGFR internalization via a clathrin-mediated endocytosis (CME) pathway (Sorkin and Goh, 2009). These ligands are present in picomolar concentrations (≤170–340 pM or 1–2 ng/ml) in most mammalian tissues and tumors (Fisher and Lakshmanan, 1990; Hirata and Orth, 1979; Ishikawa et al., 2005; Oka and Orth, 1983; Rich et al., 2017). Therefore, CME has been proposed to be the predominant pathway of EGFR internalization in vivo (Pinilla-Macua et al., 2017). High concentrations of EGFR ligands (10–100 ng/ml) are found in fluids such as saliva, milk, and urine (Connolly and Rose, 1988; Dvorak, 2010; Murdoch-Kinch et al., 2011) and may be generated locally, for example, during wound healing (Fisher and Lakshmanan, 1990; Schultz et al., 1991). At these concentrations, substantial clathrin-independent endocytosis (CIE) occurs in some, though not all, types of cultured cells tested (Boucrot et al., 2015; Caldieri et al., 2017; Orth et al., 2006; Sigismund et al., 2013; Sigismund et al., 2005). The underlying molecular mechanisms responsible for this concentration-dependent rerouting of EGFR endocytosis are not understood. CME of EGFR is proposed to be controlled by two redundant mechanisms: (1) ubiquitination of the receptor by the E3 ubiquitin ligase Cbl, which interacts with the receptor directly as well as through the SH2 adaptor protein Grb2, with subsequent binding of the ubiquitinated receptor to ubiquitin adaptors in clathrin-coated pits; and (2) direct interaction of the receptor internalization motifs with the clathrin adaptor complex AP2 (Fortian et al., 2015; Goh et al., 2010). The balance of the ubiquitination- and AP2-dependent mechanisms in EGFR endocytosis varies among cell types and in different studies (Ahmad et al., 2014; Sorkin et al., 1996; Wang et al., 2015). Notably, Cbl-dependent ubiquitination of EGFR has been also implicated in the CIE of EGFR (Sigismund et al., 2013). However, the molecular mechanisms of EGFR recruitment into endocytic carriers generated at the plasma membrane during ubiquitination-dependent and other types of CIE processes are unknown.
Activation of p38 MAPKs (hereafter referred to p38) by stressors, such as inflammatory cytokines, UV light, thermic or osmotic shocks, antibiotic anisomycin, and chemotherapeutics, such as cisplatin, cause robust EGFR internalization in a ligand-independent manner (Frey et al., 2006; Vergarajauregui et al., 2006; Zwang and Yarden, 2006). The p38-induced internalization of EGFR is via clathrin-coated pits and has been proposed to require AP2 (Grandal et al., 2012; Vergarajauregui et al., 2006). Furthermore, it has been demonstrated that p38-induced internalization is also triggered by EGF and requires phosphorylation of a cluster of serine and threonine residues (1015–1018) of EGFR (Tanaka et al., 2018). However, molecular mechanisms by which p38 activation leads to AP2-mediated endocytosis of EGFR have not been defined. Moreover, how p38 and ligand-induced internalization processes interact in either the normal tissue microenvironment or in tumors, where EGFR ligands, inflammatory cytokines, and other stress signals may coexist, remains sparsely studied.
EGFR is the most studied experimental model of stimulated endocytosis, in large part owing to the availability of fluorescent and radiolabeled EGF. In this study, we wanted to analyze both ligand-dependent and -independent internalization, and therefore took advantage of an experimental approach for a quantitative analysis of EGFR endocytosis we recently developed, which is completely independent of the ligand (Larsen et al., 2019; Perez Verdaguer et al., 2019). Central to this approach is the use of CRISPR/Cas9 gene-edited HeLa cells expressing endogenous EGFR that is extracellularly tagged with a fluorogen-activating protein (FAP; hereafter referred as HeLa/FAP-EGFR cells). Binding of cell-impermeant malachite green (MG) fluorogen derivatives to FAP-EGFR allows specific labeling of cell-surface EGFR and monitoring of its endocytic trafficking. This approach was combined with biochemical endocytosis assays, mutagenesis of EGFR and AP2, RNAi, and mass spectrometry to reveal a key role for a novel site of p38-dependent phosphorylation in EGFR, serine 1006, that facilitates ligand-free EGFR endocytosis through the interaction of the receptor dileucine-motif with the σ2 subunit of AP2. These new mechanistic insights and FAP-based high-throughput analysis of the kinetics of various EGFR endocytic pathways allowed us to propose a model that explains the relative contributions of p38- and ligand-induced pathways in EGFR endocytic trafficking and the mechanistic crosstalk between these pathways.
Results
p38 activation leads to robust clathrin- and AP2-dependent endocytosis of FAP-EGFR
To analyze the mechanisms of p38-induced EGFR endocytosis and its relationships with the ligand-induced internalization of the receptor, we used HeLa cells expressing an endogenous FAP-EGFR fusion protein (HeLa/FAP-EGFR cells; Larsen et al., 2019). MG dyes become fluorescent upon binding to FAP with excitation at ∼640 nm and emission at ∼670–700 nm (Perkins and Bruchez, 2020). Two cell-impermeant MG derivatives were used to label cell surface FAP-EGFR: (1) pH-insensitive MG-B-Tau; and (2) pH-sensitive MG-Bis-SA to quantify FAP-EGFR endocytosis using a fluorescence excitation ratiometric imaging (FERI) assay developed in our recent studies (Larsen et al., 2019; Perez Verdaguer et al., 2019). Briefly, MG-Bis-SA is a tandem dye in which MG is linked to a pH-sensitive Cy3. Due to obligate intramolecular fluorescence resonance energy transfer (FRET) between Cy3 (donor) and MG (acceptor), MG-Bis-SA can emit at ∼670–700 nm both when directly excited at 640 nm and due to a FRET transition from the innate Cy3 when excited at ∼560 nm (561-nm laser line in our setup). The Cy3-MG FRET is enhanced (by several-fold) at the lower pH found in endolysosomal compartments compared with neutral pH (extracellular media). Therefore, the ratio between the FRET and directly excited signal (henceforth referred to as the 561/640 ratio) is proportional to the fraction of endosomal (internalized) relative to total MG-Bis-SA bound to FAP-EGFR.
We have previously demonstrated strong EGF-induced endocytosis of FAP-EGFR in HeLa/FAP-EGFR cells labeled with both MG dyes (Larsen et al., 2019). To test whether activation of p38 also results in FAP-EGFR endocytosis, HeLa/FAP-EGFR cells were treated with TNFα, a cytokine capable of p38 activation, or anisomycin, an antibiotic known to activate p38. TNFα caused strong but transient p38 activation that peaked at 10–15 min (Fig. 1 A), whereas anisomycin induced sustained p38 activation (Fig. 1 B). The FERI internalization assay in cells stimulated with TNFα or EGF demonstrated that the amount of endocytosed FAP-EGFR is maximal after ∼10–15 min of stimulation at 37°C (Fig. S1 A). Therefore, 15-min stimulation with EGF, TNFα, or anisomycin is used for most experiments in the present study. Fluorescence microscopy of MG-Bis-SA–labeled FAP-EGFR in cells treated with TNFα, anisomycin, or EGF for 15 min at 37°C shows dramatic increase of the vesicular far-red fluorescence excited at 561 nm when compared with unstimulated cells (Fig. S1 B). A dose–response analysis of the effects of TNFα and anisomycin indicates that the 561/640 ratio plateaued at ∼3–10 ng/ml TNFα and ∼100 nM anisomycin (Fig. 1, C and D), concentrations used in subsequent experiments.

p38 activation triggers clathrin- and AP2-dependent FAP-EGFR internalization in HeLa/FAP-EGFR cells. (A and B) Cells were incubated with 3 ng/ml TNFα (A) or 100 nM anisomycin (B) for indicated times and lysed. Cell lysates were resolved by electrophoresis and probed by Western blotting with antibodies to phosphorylated p38 (p-p38), pS1046/47 EGFR, and Grb2 (loading control). (C and D) FAP-EGFR internalization was measured using FERI assay in cells treated with TNFα (0.48–100 ng/ml) or anisomycin (0.001–100 µM) for 15 min. Mean values of the 561/640 ratio with SEM of duplicates are plotted against the log of concentrations. TNFα or anisomycin concentrations used in subsequent experiments are indicated by the bar in C and the arrow in D. (E) Cells were pretreated with DMSO (veh) for 90 min, 10 µM SB202190 for 30 min (SB), or 100 nM BIRB796 for 90 min (BIRB), and further incubated with 3 ng/ml TNFα or 100 nM anisomycin for 15 min in the presence of inhibitors. Cell lysates were resolved by electrophoresis and immunoblotted with antibodies to active p38 (p-p38), phosphorylated MAPKAPK2 (pMAPKAPK2), and Grb2 (loading control). (F) FERI internalization assay was performed in cells pretreated with DMSO (vehicle) or 100 nM BIRB796 for 90 min and then incubated with 3 ng/ml TNFα (blue) or 100 nM anisomycin (red). Mean values of the 561/640 ratio with SEM of duplicates are plotted. P values against vehicle were calculated using the unpaired Student’s t test. (G) Cells were pretreated with DMSO (vehicle) or 100 nM BIRB796 for 90 min and labeled with MG-B-Tau, and then left untreated or treated with 10 ng/ml TNFα or 100 nM anisomycin for 15 min. 3D images were acquired through the 640-nm channel. Representative confocal sections through the middle of the cell are shown. Scale bars, 10 µm. (H) Cells were reverse-transfected with nontargeting siRNA (NT) or siRNAs targeting CHC, Grb2, c-Cbl/Cbl-b (Cbl), or the μ2 subunit of AP2. FERI assay was performed 4 d later, after TNFα or anisomycin stimulation. Values of the 561/640 ratio were normalized by this value in NT-transfected cells. Mean values with SEM from three independent experiments are shown. P values against NT were calculated using the unpaired Student’s t test. The efficiency of protein depletion is shown in Fig. S2.
p38 activation triggers clathrin- and AP2-dependent FAP-EGFR internalization in HeLa/FAP-EGFR cells. (A and B) Cells were incubated with 3 ng/ml TNFα (A) or 100 nM anisomycin (B) for indicated times and lysed. Cell lysates were resolved by electrophoresis and probed by Western blotting with antibodies to phosphorylated p38 (p-p38), pS1046/47 EGFR, and Grb2 (loading control). (C and D) FAP-EGFR internalization was measured using FERI assay in cells treated with TNFα (0.48–100 ng/ml) or anisomycin (0.001–100 µM) for 15 min. Mean values of the 561/640 ratio with SEM of duplicates are plotted against the log of concentrations. TNFα or anisomycin concentrations used in subsequent experiments are indicated by the bar in C and the arrow in D. (E) Cells were pretreated with DMSO (veh) for 90 min, 10 µM SB202190 for 30 min (SB), or 100 nM BIRB796 for 90 min (BIRB), and further incubated with 3 ng/ml TNFα or 100 nM anisomycin for 15 min in the presence of inhibitors. Cell lysates were resolved by electrophoresis and immunoblotted with antibodies to active p38 (p-p38), phosphorylated MAPKAPK2 (pMAPKAPK2), and Grb2 (loading control). (F) FERI internalization assay was performed in cells pretreated with DMSO (vehicle) or 100 nM BIRB796 for 90 min and then incubated with 3 ng/ml TNFα (blue) or 100 nM anisomycin (red). Mean values of the 561/640 ratio with SEM of duplicates are plotted. P values against vehicle were calculated using the unpaired Student’s t test. (G) Cells were pretreated with DMSO (vehicle) or 100 nM BIRB796 for 90 min and labeled with MG-B-Tau, and then left untreated or treated with 10 ng/ml TNFα or 100 nM anisomycin for 15 min. 3D images were acquired through the 640-nm channel. Representative confocal sections through the middle of the cell are shown. Scale bars, 10 µm. (H) Cells were reverse-transfected with nontargeting siRNA (NT) or siRNAs targeting CHC, Grb2, c-Cbl/Cbl-b (Cbl), or the μ2 subunit of AP2. FERI assay was performed 4 d later, after TNFα or anisomycin stimulation. Values of the 561/640 ratio were normalized by this value in NT-transfected cells. Mean values with SEM from three independent experiments are shown. P values against NT were calculated using the unpaired Student’s t test. The efficiency of protein depletion is shown in Fig. S2.

EGF and TNFα stimulated endocytosis of FAP-EGFR demonstrated using FERI assay and single-cell imaging. (A) Time course of FAP-EGFR internalization. HeLa/FAP-EGFR cells were labeled with MG-Bis-SA and further incubated in starvation medium (Untreated) or stimulated with either EGF (0.5, 5, or 50 ng/ml) or TNFα (10 ng/ml) for indicated times. The 561/640 ratio was measured using the FERI internalization assay. In each experiment, the background ratio at time “zero” was determined by y-intersection of the linear regression slope calculated using ratio values in untreated cells incubated for 5 and 10 min. This ratio value was subtracted from raw ratio values in each experiment, and the resulting background-subtracted ratio values were normalized to the ratio in cells stimulated with 50 ng/ml EGF for 30 min in each time-course experiment. The data in the graph represent mean values with SEMs (n = 3 independent experiments). SEMs are not shown if they are smaller than markers. (B) Examples of single-cell FAP-EGFR ratiometric imaging. Cells labeled with MG-Bis-SA were either untreated or treated with 100 nM anisomycin, 4 ng/ml EGF, or 3 ng/ml TNFα for 15 min at 37°C. 3D stacks of x-y confocal images were acquired from living cells through the 640-nm channel (Ex 640/Em 680; pH-independent) and the FRET channel (Ex 561/Em 680, pH-sensitive). Individual confocal sections through the middle of cells are presented. The 561/640 ratio is presented as pseudocolored image modulated to the intensity of the 640-nm channel. Scale bar, 10 µm. Em, emission; Ex, excitation.
EGF and TNFα stimulated endocytosis of FAP-EGFR demonstrated using FERI assay and single-cell imaging. (A) Time course of FAP-EGFR internalization. HeLa/FAP-EGFR cells were labeled with MG-Bis-SA and further incubated in starvation medium (Untreated) or stimulated with either EGF (0.5, 5, or 50 ng/ml) or TNFα (10 ng/ml) for indicated times. The 561/640 ratio was measured using the FERI internalization assay. In each experiment, the background ratio at time “zero” was determined by y-intersection of the linear regression slope calculated using ratio values in untreated cells incubated for 5 and 10 min. This ratio value was subtracted from raw ratio values in each experiment, and the resulting background-subtracted ratio values were normalized to the ratio in cells stimulated with 50 ng/ml EGF for 30 min in each time-course experiment. The data in the graph represent mean values with SEMs (n = 3 independent experiments). SEMs are not shown if they are smaller than markers. (B) Examples of single-cell FAP-EGFR ratiometric imaging. Cells labeled with MG-Bis-SA were either untreated or treated with 100 nM anisomycin, 4 ng/ml EGF, or 3 ng/ml TNFα for 15 min at 37°C. 3D stacks of x-y confocal images were acquired from living cells through the 640-nm channel (Ex 640/Em 680; pH-independent) and the FRET channel (Ex 561/Em 680, pH-sensitive). Individual confocal sections through the middle of cells are presented. The 561/640 ratio is presented as pseudocolored image modulated to the intensity of the 640-nm channel. Scale bar, 10 µm. Em, emission; Ex, excitation.
To confirm that TNFα- and anisomycin-induced FAP-EGFR endocytosis are both dependent on p38 activity, chemical inhibitors of p38, SB202190 and BIRB796, were used. Both inhibitors efficiently blocked phosphorylation of MAPKAPK2, a substrate of p38 (Ben-Levy et al., 1995; Kuma et al., 2005), although only BIRB796 reduced activating phosphorylation of p38 as well (Fig. 1 E). As shown in Fig. 1, F and G, BIRB796 strongly inhibited FAP-EGFR internalization in cells treated with TNFα and anisomycin. Finally, siRNA knockdown of clathrin heavy chain (CHC) or μ2 subunit of AP2 dramatically inhibited FAP-EGFR endocytosis induced by maximally effective concentrations of TNFα and anisomycin (Fig. 1 H and Fig. S2). By contrast, depletion of Grb2 or Cbl proteins, required for EGF-induced internalization (Jiang et al., 2003; Jiang and Sorkin, 2003), did not affect p38-induced endocytosis (Fig. 1 H and Fig. S2). Together, the data in Fig. 1 and its supplements corroborate that p38 activation results in CME of FAP-EGFR, which requires AP2, and validate HeLa/FAP-EGFR cells as an appropriate model to study the mechanism of this endocytic process.

Efficiencies of siRNAs knockdowns and σ2-GFP assembly into AP2 in cells transfected with σ2 siRNA. (A–D) Typical efficiencies of protein depletion in siRNA experiments in HeLa/FAP-EGFR cells described in Figs. 1, 2, 7, and 9. HeLa/FAP-EGFR cells were transfected with nontargeting (NT) siRNA and Grb2 (A), c-Cbl and Cbl-b (A), CHC (B), μ2 (C), or σ2 (C and D) siRNAs. Cells were lysed 3–4 d after transfections, and lysates were blotted with indicated antibodies. β-Actin and Grb2 are loading controls. (E) HeLa/FAP-EGFR cells were transfected with σ2 siRNA for 2 d and then transfected with WT σ2-GFP plasmid. 2 d later, cells were lysed, and σ2-GFP was immunoprecipitated using the GFP antibody. Lysates (total cell lysates [TCL]) and immunoprecipitates (IP) were probed by Western blotting (WB) with GFP and α-adaptin antibodies to demonstrate the assembly of WT σ2-GFP into AP2 complexes. (F) PAE/EGFR-GFP cells were transfected with μ2 siRNAs as described in Materials and methods. Cells were lysed 5 d after two transfections, and lysates were blotted with α-adaptin and Grb2 (loading control) antibodies.
Efficiencies of siRNAs knockdowns and σ2-GFP assembly into AP2 in cells transfected with σ2 siRNA. (A–D) Typical efficiencies of protein depletion in siRNA experiments in HeLa/FAP-EGFR cells described in Figs. 1, 2, 7, and 9. HeLa/FAP-EGFR cells were transfected with nontargeting (NT) siRNA and Grb2 (A), c-Cbl and Cbl-b (A), CHC (B), μ2 (C), or σ2 (C and D) siRNAs. Cells were lysed 3–4 d after transfections, and lysates were blotted with indicated antibodies. β-Actin and Grb2 are loading controls. (E) HeLa/FAP-EGFR cells were transfected with σ2 siRNA for 2 d and then transfected with WT σ2-GFP plasmid. 2 d later, cells were lysed, and σ2-GFP was immunoprecipitated using the GFP antibody. Lysates (total cell lysates [TCL]) and immunoprecipitates (IP) were probed by Western blotting (WB) with GFP and α-adaptin antibodies to demonstrate the assembly of WT σ2-GFP into AP2 complexes. (F) PAE/EGFR-GFP cells were transfected with μ2 siRNAs as described in Materials and methods. Cells were lysed 5 d after two transfections, and lysates were blotted with α-adaptin and Grb2 (loading control) antibodies.
p38-dependent EGFR internalization requires interaction of EGFR dileucine motif with the σ2 subunit of AP2
The inhibition of p38-induced EGFR endocytosis in AP2-depleted cells may be due to (1) diminished assembly of clathrin-coated pits (Motley et al., 2003) and/or (2) abolished interaction of EGFR with AP2. Previous studies suggested the role of EGFR carboxyl-terminal tyrosine-based (YRAL974-977) and dileucine (LL1010/1011) AP2-binding motifs in the anisomycin-induced endocytosis of the receptor (Grandal et al., 2012; Tomas et al., 2015). Therefore, we explored several approaches to define the precise function of these motifs in p38-dependent endocytosis of EGFR.
Chimeric proteins consisting of extracellular and transmembrane domains of Tac antigen (interleukin-2 receptor α chain) fused to cytoplasmic tails of either murine H2-DMβ containing YxxΘ motif (YTPL; Tac-Y) or murine CD3γ containing dileucine motif (DKQTLL; Tac-LL; Marks et al., 1996) were transiently expressed in HeLa/FAP-EGFR cells to test for their ability to competitively inhibit p38-induced FAP-EGFR endocytosis. As shown in Fig. 2, A and B, expression of Tac-LL but not Tac-Y significantly decreased TNFα-induced internalization. The inhibition of TNFα-induced FAP-EGFR endocytosis by Tac-LL measured using the FERI assay was partial (Fig. 2 B), probably because high expression of Tac-LL, such that it competed effectively for binding of FAP-EGFR to AP2, was limited in the cell population. These results are consistent with the main role of the LL1010/11 motif in the p38-induced EGFR internalization, although they do not rule out involvement of an intermediary protein containing a dileucine motif.

p38-dependent EGFR internalization is mediated by the interaction of the dileucine motif with the σ2 subunit of AP2. (A and B ) HeLa/FAP-EGFR cells were mock-transfected (Ctrl) or transfected with Tac-LL or Tac-Y chimeric constructs, and used for experiments 2 d later. In A, cells were labeled with MG-B-Tau, incubated with 3 ng/ml TNFα for 15 min, and fixed. Tac was immunolabeled using secondary FITC-conjugated antibodies. 3D imaging was performed through the 488-nm (cyan, Tac) and 640-nm (red, MG-B-Tau) channels. Maximum intensity projections of 3D images are shown. Borders of cells expressing Tac chimeras are indicated by white lines. Scale bars, 10 µm. The expression levels of Tac-Y and Tac-LL chimeric proteins were similar. In B, cells were labeled with MG-Bis-SA, treated with 3 ng/ml TNFα for 15 min, and analyzed using the FERI assay. Bar graph represents mean values of the 561/640 ratio with SEM from quadruplicates. P values were calculated using the unpaired Student’s t test against control. (C) PAE cells stably expressing WT, LL1010/11A, or 974A EGFR mutants were treated with 100 nM anisomycin for 15 min and immunolabeled with EGFR (AB_2246311) and EEA1 antibodies. 3D images were acquired through the 488-nm (green, EGFR) and 640-nm (red, EEA1) channels. Individual confocal sections through the middle of z-stacks are shown. Scale bars, 10 µm. (D) Quantification of the fraction of EGFR (WT or mutants as indicated) colocalized with EEA1 endosomes in images exemplified in C. Scatter dot plot represents mean values with SDs (n = 10). P values against untreated WT were determined by multiple-comparison one-way ANOVA. (E–G) HeLa/FAP-EGFR cells were reverse-transfected with nontargeting (siNT) or σ2 siRNA (siRNAσ2). 2 d later, the cells were either not transfected (E), or transfected with WT σ2-GFP or its mutants (A63W, L65S, or V98D; F and G), and assayed after an additional 2 d. The efficiency of σ2 depletion is shown in Fig. S2. In E and F, the cells labeled with MG-B-Tau were treated with TNFα for 15 min and imaged through 640-nm (red, FAP-EGFR) and 488-nm (green, GFP) channels. Maximum intensity projections of 3D images are shown. Scale bars, 10 µm. In G, the cells were labeled with MG-Bis-SA and treated with TNFα for 15 min, and the FERI assay was performed. Bar graph represents mean values of the 561/640 ratio with SEM of quadruplicates. P values against siNT were determined by multiple-comparison one-way ANOVA.
p38-dependent EGFR internalization is mediated by the interaction of the dileucine motif with the σ2 subunit of AP2. (A and B ) HeLa/FAP-EGFR cells were mock-transfected (Ctrl) or transfected with Tac-LL or Tac-Y chimeric constructs, and used for experiments 2 d later. In A, cells were labeled with MG-B-Tau, incubated with 3 ng/ml TNFα for 15 min, and fixed. Tac was immunolabeled using secondary FITC-conjugated antibodies. 3D imaging was performed through the 488-nm (cyan, Tac) and 640-nm (red, MG-B-Tau) channels. Maximum intensity projections of 3D images are shown. Borders of cells expressing Tac chimeras are indicated by white lines. Scale bars, 10 µm. The expression levels of Tac-Y and Tac-LL chimeric proteins were similar. In B, cells were labeled with MG-Bis-SA, treated with 3 ng/ml TNFα for 15 min, and analyzed using the FERI assay. Bar graph represents mean values of the 561/640 ratio with SEM from quadruplicates. P values were calculated using the unpaired Student’s t test against control. (C) PAE cells stably expressing WT, LL1010/11A, or 974A EGFR mutants were treated with 100 nM anisomycin for 15 min and immunolabeled with EGFR (AB_2246311) and EEA1 antibodies. 3D images were acquired through the 488-nm (green, EGFR) and 640-nm (red, EEA1) channels. Individual confocal sections through the middle of z-stacks are shown. Scale bars, 10 µm. (D) Quantification of the fraction of EGFR (WT or mutants as indicated) colocalized with EEA1 endosomes in images exemplified in C. Scatter dot plot represents mean values with SDs (n = 10). P values against untreated WT were determined by multiple-comparison one-way ANOVA. (E–G) HeLa/FAP-EGFR cells were reverse-transfected with nontargeting (siNT) or σ2 siRNA (siRNAσ2). 2 d later, the cells were either not transfected (E), or transfected with WT σ2-GFP or its mutants (A63W, L65S, or V98D; F and G), and assayed after an additional 2 d. The efficiency of σ2 depletion is shown in Fig. S2. In E and F, the cells labeled with MG-B-Tau were treated with TNFα for 15 min and imaged through 640-nm (red, FAP-EGFR) and 488-nm (green, GFP) channels. Maximum intensity projections of 3D images are shown. Scale bars, 10 µm. In G, the cells were labeled with MG-Bis-SA and treated with TNFα for 15 min, and the FERI assay was performed. Bar graph represents mean values of the 561/640 ratio with SEM of quadruplicates. P values against siNT were determined by multiple-comparison one-way ANOVA.
To test for the direct involvement of EGFR AP2-binding motifs, the effects of mutations of LL1010/1011 and YRAL974-977 motifs on p38-dependent internalization of EGFR were examined. We have previously established and thoroughly characterized porcine aortic endothelial (PAE) cells lacking endogenous EGFR and other ErbBs as a model system to express human recombinant EGFR and its mutants, including alanine substitutions of LL1010/1011 (LL1010/11A) and Y974 (Y974A; Carter and Sorkin, 1998; Jiang et al., 2003). Similar to HeLa/FAP-EGFR cells, anisomycin strongly activated p38 in PAE cells (Fig. S3 A) and hence promoted WT EGFR endocytosis leading to receptor accumulation in endosomes containing early endosome resident protein early endosome antigen 1 (EEA1; Fig. 2 C and Fig. S3 B), whereas the response to TNFα was much weaker (Fig. S3 A). Anisomycin effects were sensitive to BIRB796 (Fig. S3, A and B). Therefore, anisomycin treatment was used to test for the effects of EGFR mutations on p38-induced receptor endocytosis in PAE cells. As shown in Fig. 2, C and D, robust endocytosis of the Y974A mutant but not of the LL1010/11A mutant was observed in anisomycin-treated cells.

PAE cells as a cell model for studying p38-dependent endocytosis of EGFR-GFP and its mutants: activation of p38, steady-state EGFR-GFP subcellular distribution, and p38-dependent and EGF-stimulated EGFR-GFP endocytosis. (A) PAE cells stably expressing EGFR-GFP were incubated with 10 ng/ml EGF (E), 20 ng/ml TNFα (T), or 100 nM anisomycin (A) for 15 min at 37°C after preincubation with DMSO (vehicle) or BIRB976 for 90 min. Cells were lysed, and lysates were probed by Western blotting (WB) with antibodies to p1046/47, active p38 (p-p38), pY1068, pERK1/2, and Grb2 (loading control). (B) PAE cells stably expressing EGFR-GFP were incubated with 100 nM anisomycin for 15 min at 37°C after preincubation with DMSO (vehicle) or BIRB976 for 90 min. Cells were immunolabeled with the EEA1 antibody to mark early endosomes. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections are shown. Scale bars, 10 µm. (C) PAE cells stably expressing either WT or indicated EGFR-GFP mutants were fixed and immunolabeled with EGFR antibody Mab528 to label cell-surface EGFR-GFP followed by secondary antibodies conjugated to Cy5. 3D images were acquired through 488-nm (GFP, total EGFR-GFP) and 640-nm (Cy5, surface EGFR-GFP) channels. The ratio of Cy5 to GFP fluorescence intensities corresponds to the fraction of cell-surface EGFR-GFP. Bar graph represents mean values with SDs (n = 10–14). (D) PAE cells expressing either WT or indicated EGFR-GFP mutants were stimulated with 1 ng/ml EGF-Rh for 15 min at 37°C. Cells were 3D-imaged through the 488-nm channel (GFP) and the 561-nm channel (EGF-Rh, not shown). Maximum intensity projection images are presented. Scale bars, 10 µm. Ctrl, control.
PAE cells as a cell model for studying p38-dependent endocytosis of EGFR-GFP and its mutants: activation of p38, steady-state EGFR-GFP subcellular distribution, and p38-dependent and EGF-stimulated EGFR-GFP endocytosis. (A) PAE cells stably expressing EGFR-GFP were incubated with 10 ng/ml EGF (E), 20 ng/ml TNFα (T), or 100 nM anisomycin (A) for 15 min at 37°C after preincubation with DMSO (vehicle) or BIRB976 for 90 min. Cells were lysed, and lysates were probed by Western blotting (WB) with antibodies to p1046/47, active p38 (p-p38), pY1068, pERK1/2, and Grb2 (loading control). (B) PAE cells stably expressing EGFR-GFP were incubated with 100 nM anisomycin for 15 min at 37°C after preincubation with DMSO (vehicle) or BIRB976 for 90 min. Cells were immunolabeled with the EEA1 antibody to mark early endosomes. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections are shown. Scale bars, 10 µm. (C) PAE cells stably expressing either WT or indicated EGFR-GFP mutants were fixed and immunolabeled with EGFR antibody Mab528 to label cell-surface EGFR-GFP followed by secondary antibodies conjugated to Cy5. 3D images were acquired through 488-nm (GFP, total EGFR-GFP) and 640-nm (Cy5, surface EGFR-GFP) channels. The ratio of Cy5 to GFP fluorescence intensities corresponds to the fraction of cell-surface EGFR-GFP. Bar graph represents mean values with SDs (n = 10–14). (D) PAE cells expressing either WT or indicated EGFR-GFP mutants were stimulated with 1 ng/ml EGF-Rh for 15 min at 37°C. Cells were 3D-imaged through the 488-nm channel (GFP) and the 561-nm channel (EGF-Rh, not shown). Maximum intensity projection images are presented. Scale bars, 10 µm. Ctrl, control.
Together, the data presented in Fig. 2, A–D, demonstrate that p38-dependent EGFR internalization in both HeLa and PAE cells requires LL1010/1011 but not the YRAL974-977 motif of the receptor. Strikingly, though, mutations of LL1010/1011 were shown to block phosphorylation of R1 cluster residues (Tanaka et al., 2018). These observations create a dilemma: is the role of the LL1010/11 motif to bind AP2 or, alternatively, to promote R1 cluster phosphorylation, with the latter mediating endocytosis through another mechanism, for example via an interaction of a phosphoserine acidic cluster with the μ2 subunit of AP2 (Singh et al., 2018)? To directly demonstrate that p38-dependent EGFR endocytosis requires dileucine-AP2 interaction, we performed AP2 siRNA knockdown-and-rescue experiments in HeLa/FAP-EGFR cells. Dileucine motifs bind to the AP2 core in two consecutive hydrophobic pockets of the σ2 subunit (Kelly et al., 2008). siRNA knockdown of σ2 resulted in down-regulation of the entire AP2 complex due to instability of unassembled AP2 subunits as demonstrated by down-regulation of the α subunit (α-adaptin; Fig. S2, C and D). TNFα-induced FAP-EGFR internalization quantified using the FERI assay was reduced by ∼50% in σ2-depleted cells (Fig. 2 G), equivalent to the effect of the μ2 knockdown (Fig. 1 H). The endocytosis-inhibitory effect of σ2 knockdown was readily observed by fluorescence microscopy imaging of HeLa/FAP-EGFR cells labeled with MG-B-Tau (Fig. 2 E). Transient expression of GFP-tagged siRNA-resistant WT σ2 (σ2-GFP) in σ2-depleted cells resulted in the efficient assembly of WT σ2-GFP into the AP2 complex (Fig. S2 E) and a punctate GFP fluorescence pattern at the basal cell membrane, characteristic of clathrin-coated structures (Fig. 2 F). WT σ2-GFP efficiently restored FAP-EGFR endocytosis induced by TNFα in σ2-depleted cells (Fig. 2, F and G).
In vitro analysis of σ2 interaction with dileucine-containing peptides identified several residues in σ2, whose substitution did not prevent σ2 incorporation into AP2 but dramatically reduced the binding affinity for dileucine peptides either by disrupting the hydrophobic pockets (V98S and L65S) or by filling them with bulky hydrophobic side chains (A63W; Kelly et al., 2008). Hence, HeLa/FAP-EGFR cells, transfected with σ2 siRNA, were further transfected with corresponding GFP-tagged σ2 mutants. Unlike WT σ2-GFP, none of the σ2 mutants rescued FAP-EGFR internalization, even though they shared the punctate localization pattern on the basal cell membrane seen with WT σ2-GFP (Fig. 2, F and G). These data support the notion that the interaction of the LL1010/1011 motif with σ2 is essential for p38-induced EGFR endocytosis.
Phosphorylation of EGFR serine 1006 is necessary for p38-dependent endocytosis
Dileucine motifs are capable of constitutive binding to AP2 and mediate basal cargo endocytosis (Traub and Bonifacino, 2013). Why p38 activation is necessary for such interaction of EGFR is unclear. To elucidate the mechanisms responsible for the LL1010/1011:AP2 interaction and EGFR endocytosis induced by p38 activation, alanine mutagenesis of residues surrounding LL1010/1011 was performed (Fig. 3 A), and mutants were stably expressed in PAE cells. Using EGFR-GFP as template for mutations allowed rapid screening of the steady-state localization of mutants and triaging clones with abnormal receptor distribution. Typically, 70–80% of total cellular WT and mutant EGFR-GFPs were present at the cell surface (Fig. S3 C), and the surface levels of various EGFR-GFP mutants were comparable. Expression of the LL1010/11A EGFR mutant in PAE cells previously showed that this motif is not essential for EGF-induced CME of ligand-bound EGFR in these cells (Huang et al., 2003; Jiang et al., 2003). In fact, all EGFR mutants described below were efficiently internalized upon EGF stimulation (Fig. S3 D).

Serine 1006 and the R1 serine/threonine cluster are necessary for p38-induced, LL1010/1011-mediated EGFR internalization. (A) Schematic representation of EGFR region encompassed by residues 999–1020 including the dileucine motif and surrounding clusters of serines and threonines. Residues 1015–1018 are designated as the R1 cluster per Tanaka et al. (2018). Key mutations and mutant names are indicated. (B) PAE cells stably expressing EGFR-GFP (WT or indicated mutants) were treated with 100 nM anisomycin for 15 min and immunolabeled with the EEA1 antibody. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (C) Quantification of the fraction of EGFR-GFP colocalized with EEA1 endosomes in images exemplified in B. Scatter dot plot represents mean values with SDs (n = 8–15). P values were determined by multiple-comparison one-way ANOVA. P values against “WT plus anisomycin” are shown in black. P values against LL1010/11A mutant are shown in red. (D and E) PAE cells stably expressing EGFR-GFP (WT or mutants as indicated) were treated with anisomycin for 15 min and lysed, and EGFR-GFP was immunoprecipitated. In D, total cell lysates (TCL) and immunoprecipitates (IP) were probed by Western blotting (WB) with antibodies to α-adaptin subunit of AP2 (α-Ad), pS1015, phosphorylated p38 (p-p38), total phosphoserine (pSer), and GFP. The identity of α-adaptin band was confirmed by immunoblotting analysis of cells depleted of AP2 by μ2 siRNA (Fig. S2 F). In E, the amount of AP2 coimmunoprecipitated with EGFR-GFP was normalized by the amount of immunoprecipitated EGFR-GFP, and the values of the AP2/EGFR-GFP ratio were further normalized to this value obtained in immunoprecipitates from anisomycin-treated WT cells in each independent experiment. Bar graph represents mean values with SEM (n = 2–4 independent experiments). P values against WT treated with anisomycin were determined by the unpaired Student’s t test.
Serine 1006 and the R1 serine/threonine cluster are necessary for p38-induced, LL1010/1011-mediated EGFR internalization. (A) Schematic representation of EGFR region encompassed by residues 999–1020 including the dileucine motif and surrounding clusters of serines and threonines. Residues 1015–1018 are designated as the R1 cluster per Tanaka et al. (2018). Key mutations and mutant names are indicated. (B) PAE cells stably expressing EGFR-GFP (WT or indicated mutants) were treated with 100 nM anisomycin for 15 min and immunolabeled with the EEA1 antibody. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (C) Quantification of the fraction of EGFR-GFP colocalized with EEA1 endosomes in images exemplified in B. Scatter dot plot represents mean values with SDs (n = 8–15). P values were determined by multiple-comparison one-way ANOVA. P values against “WT plus anisomycin” are shown in black. P values against LL1010/11A mutant are shown in red. (D and E) PAE cells stably expressing EGFR-GFP (WT or mutants as indicated) were treated with anisomycin for 15 min and lysed, and EGFR-GFP was immunoprecipitated. In D, total cell lysates (TCL) and immunoprecipitates (IP) were probed by Western blotting (WB) with antibodies to α-adaptin subunit of AP2 (α-Ad), pS1015, phosphorylated p38 (p-p38), total phosphoserine (pSer), and GFP. The identity of α-adaptin band was confirmed by immunoblotting analysis of cells depleted of AP2 by μ2 siRNA (Fig. S2 F). In E, the amount of AP2 coimmunoprecipitated with EGFR-GFP was normalized by the amount of immunoprecipitated EGFR-GFP, and the values of the AP2/EGFR-GFP ratio were further normalized to this value obtained in immunoprecipitates from anisomycin-treated WT cells in each independent experiment. Bar graph represents mean values with SEM (n = 2–4 independent experiments). P values against WT treated with anisomycin were determined by the unpaired Student’s t test.
Similar to untagged EGFR (Fig. 2, C and D), mutation of LL1010/1011 essentially eliminated anisomycin-stimulated accumulation of EGFR-GFP in EEA1-containing endosomes (Fig. 3, B and C). Binding of dileucine motifs to AP2 is facilitated by a negatively charged residue at the −4 position (Traub and Bonifacino, 2013). Such acidic residue is not present upstream of the LL1010/11 motif in EGFR (Fig. 3 A). On the other hand, it has been suggested that a negative charge downstream from a dileucine motif may also facilitate AP2 binding (Lindwasser et al., 2008; Pitcher et al., 1999). Thus, phosphorylation of the R1 cluster may potentially play this role in the p38-induced EGFR:AP2 interaction. However, triple mutation STS1015/17/18A in EGFR-GFP substantially reduced but did not fully block anisomycin-induced internalization, as evidenced by considerable colocalization of this mutant with EEA1 compared with that of the LL1010/11A mutant (Fig. 3, B and C). Mutations of other potential phosphorylation sites, such as SS1046/1047 or SS1001/1002, known to be phosphorylated by p38 (Tong et al., 2014), had limited or no effect on p38-dependent EGFR-GFP endocytosis (Fig. 3 C). Surprisingly, triple mutant STS1004/05/06A displayed negligible anisomycin-stimulated endocytosis, and the S1006A mutation alone essentially reduced anisomycin-induced internalization of EGFR-GFP to the level observed for the LL1010/11A mutant (Fig. 3, B and C). Anisomycin triggered the interaction of WT EGFR-GFP with AP2 as measured by AP2 coimmunoprecipitation with EGFR-GFP (Fig. 3, D and E). Coimmunoprecipitation of AP2 with LL1010/11A, STS1004/05/06A and S1006A mutants was essentially abolished, while only partially inhibited by STS1015/17/18A mutations (Fig. 3, D and E). Altogether, the data in Fig. 3 demonstrate a major role for serine 1006 in p38-induced EGFR internalization mediated by the LL1010/1011:AP2 interaction.
Phosphorylation of S1006 could generate a negative charge at the −4 position of the dileucine motif and hence facilitate AP2 binding. Constitutive phosphorylation of S1006 was detected in lung cancer cells (Zhang et al., 2011) but has not been, to our knowledge, reported previously in response to p38 activation or EGF stimulation, despite an abundance of mass spectrometry studies of the EGFR phosphoproteome (Dephoure et al., 2008; Hornbeck et al., 2012; Tong et al., 2014). To determine whether S1006 is phosphorylated by p38, we performed mass-spectrometric analysis of FAP-EGFR immunoprecipitated from HeLa/FAP-EGFR cells treated or not with 10 ng/ml TNFα or 100 nM anisomycin. Trypsin digestion of this region of the EGFR molecule yields long peptides containing numerous serines and threonines. Therefore, FAP-EGFR was first digested by trypsin, and then chymotrypsin digestion was performed. Mass spectrometry analysis revealed a potential phosphorylation site on S1006 on the doubly phosphorylated peptide FSSPSTSRTPL (Table S1) in TNFα- and anisomycin-stimulated cells. Subsequently, parallel reaction monitoring targeting FSSPSTSRTPL peptide with two phosphorylation sites was performed. Peptides with phosphorylated S1006 identified in these experiments were likely also phosphorylated at S1001 or S1002 (known phosphorylation sites; Fig. 4 A).
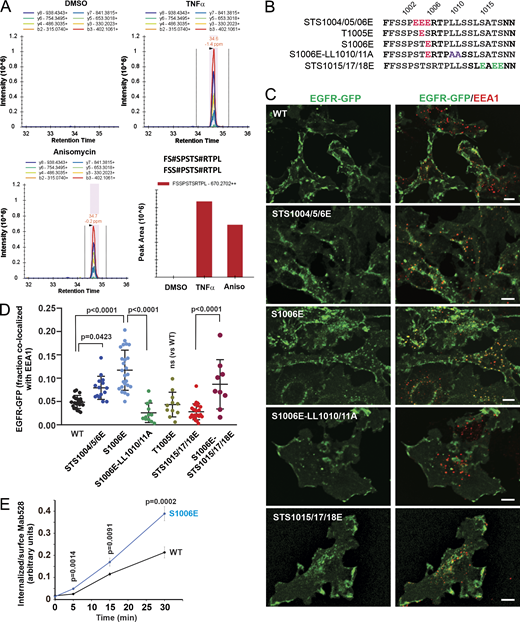
Mass spectrometry and phosphomimetic mutations demonstrate the importance of serine 1006 phosphorylation for p38-induced EGFR endocytosis. (A) HeLa/FAP-EGFR cells were untreated or treated with 10 ng/ml TNFα or 100 nM anisomycin (Aniso) for 15 min, and lysed. EGFR was immunoprecipitated, and immunoprecipitates were resolved by SDS-PAGE. Mass spectrometry analysis of corresponding gel bands was performed. Peaks corresponding to the peptides containing phosphoS1006 are shown. These peptides were also phosphorylated at S1001 or S1002. See Table S1. (B) Main phosphomimetic mutations with mutant names indicated. (C) PAE cells stably expressing EGFR-GFP (WT or mutants as indicated) were immunostained with the EEA1 antibody. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (D) Quantification of the fraction of EGFR-GFP (WT and mutants as indicated) colocalized with EEA1 endosomes in images exemplified in C and Fig. S4. Scatter dot plot represents mean values with SDs (n = 8–21). P values were determined by multiple-comparison one-way ANOVA. (E) The antibody-uptake endocytosis assay was performed in PAE cells expressing WT or the S1006E EGFR-GFP mutant. The cells were preincubated with the EGFR antibody Mab528 for 10 min at RT, and then incubated at 37°C for indicated times. Cell-surface and internalized Mab528 was labeled with secondary antibodies conjugated with, respectively, Cy5 and Cy3, as described in Materials and methods. 3D images were acquired through 640-nm (surface Mab528) and 561-nm (internalized Mab528) channels. The ratio of internalized/surface EGFR (Cy3/Cy5) was calculated, and mean values with SEM (n = 13–16) were plotted against time. P values were determined by the unpaired Student’s t test.
Mass spectrometry and phosphomimetic mutations demonstrate the importance of serine 1006 phosphorylation for p38-induced EGFR endocytosis. (A) HeLa/FAP-EGFR cells were untreated or treated with 10 ng/ml TNFα or 100 nM anisomycin (Aniso) for 15 min, and lysed. EGFR was immunoprecipitated, and immunoprecipitates were resolved by SDS-PAGE. Mass spectrometry analysis of corresponding gel bands was performed. Peaks corresponding to the peptides containing phosphoS1006 are shown. These peptides were also phosphorylated at S1001 or S1002. See Table S1. (B) Main phosphomimetic mutations with mutant names indicated. (C) PAE cells stably expressing EGFR-GFP (WT or mutants as indicated) were immunostained with the EEA1 antibody. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximum intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (D) Quantification of the fraction of EGFR-GFP (WT and mutants as indicated) colocalized with EEA1 endosomes in images exemplified in C and Fig. S4. Scatter dot plot represents mean values with SDs (n = 8–21). P values were determined by multiple-comparison one-way ANOVA. (E) The antibody-uptake endocytosis assay was performed in PAE cells expressing WT or the S1006E EGFR-GFP mutant. The cells were preincubated with the EGFR antibody Mab528 for 10 min at RT, and then incubated at 37°C for indicated times. Cell-surface and internalized Mab528 was labeled with secondary antibodies conjugated with, respectively, Cy5 and Cy3, as described in Materials and methods. 3D images were acquired through 640-nm (surface Mab528) and 561-nm (internalized Mab528) channels. The ratio of internalized/surface EGFR (Cy3/Cy5) was calculated, and mean values with SEM (n = 13–16) were plotted against time. P values were determined by the unpaired Student’s t test.
The requirement of S1006 for EGFR:AP-2 interaction (Fig. 3) and the detection of pS1006 (Fig. 4 A) in cells with activated p38 prompted us to test for the importance of a negative charge at this position. Accordingly, we compared the ability of phosphomimetic mutations of S1006 and other serine/threonine residues known to be phosphorylated by p38 to enable EGFR internalization in unstimulated PAE cells. Triple STS1015/17/18E and STS15/17/18D mutants as well as a single mutant S1015D stably expressed in PAE cells were almost exclusively localized to the cell surface and showed minimal accumulation in endosomes (Fig. 4, B–D; and Fig. S4). By contrast, the S1006E mutant and, to a lesser extent, the STS1004/05/06E mutant were clearly detected in intracellular vesicles and displayed strong colocalization with EEA1 (Fig. 4, B–D). Constitutive internalization of the S1006E mutant was negated by LL1010/11A mutations (Fig. 4, B–D). Furthermore, increased constitutive endocytosis of the S1006E mutant as compared with WT EGFR-GFP was demonstrated by measuring internalization rates of Mab528, an antibody that binds to the extracellular domain of EGFR but does not activate the receptor (Gill et al., 1984; Fig. 4 E). Therefore, a negative charge at the position 1006 is sufficient to facilitate dileucine motif–mediated internalization of unstimulated EGFR.

Effect of phosphomimetic mutations on EGFR-GFP localization. (A) PAE cells stably expressing EGFR-GFP mutants S1006E, T1005E, S1006E-STS1015/17/18E, S1015D, or STS1015/17/18D were fixed and immunolabeled with the EEA1 antibody to mark early endosomes. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximal intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (B) Quantification of the fraction of EGFR-GFP (WT and indicated mutants) colocalized with EEA1 endosomes. Scatter dot plot represents mean values with SDs (n = 5–7 images, each depicting multiple cells). P values were determined by multiple-comparison one-way ANOVA.
Effect of phosphomimetic mutations on EGFR-GFP localization. (A) PAE cells stably expressing EGFR-GFP mutants S1006E, T1005E, S1006E-STS1015/17/18E, S1015D, or STS1015/17/18D were fixed and immunolabeled with the EEA1 antibody to mark early endosomes. 3D images were acquired through the 488-nm (green, EGFR-GFP) and 640-nm (red, EEA1) channels. Maximal intensity projections of three consecutive confocal sections are shown. Scale bars, 10 µm. (B) Quantification of the fraction of EGFR-GFP (WT and indicated mutants) colocalized with EEA1 endosomes. Scatter dot plot represents mean values with SDs (n = 5–7 images, each depicting multiple cells). P values were determined by multiple-comparison one-way ANOVA.
Having established a key role for pS1006 in p38-induced endocytosis of EGFR mediated by the LL1010/11 motif, we next examined the relationship between the LL1010/11 motif and phosphorylation of the R1 cluster. The LL1010/11A mutations completely abolished the phosphorylation detected by the p1015 antibody, but not S1046/47 phosphorylation, similar to the mutations of the R1 cluster itself (Fig. 3 D and Fig. S5 A). Interestingly, LL1010/11A mutations dramatically decreased the phosphorylation signal detected by the antibody to total phosphoserine (anti-pSer), suggesting that phosphorylation of the R1 cluster is the main contributor to p38-induced phosphorylation of EGFR (Fig. 3 D). The blockade of the R1 cluster phosphorylation by LL1010/1011A mutations could be due to the direct impairment of p38-mediated phosphorylation (presumably if LL is part of the p38 recognition site on the receptor) or the consequence of abolished interaction of this di-leucine motif with AP2. The latter is ruled out by showing that the siRNA knockdown of μ2 did not affect S1015 phosphorylation (Fig. S5 B). Therefore, given that LL1010/11A mutations do not inhibit p38 kinase activity and S1046/47 phosphorylation (Fig. 3 D and Fig. S5), it is much more likely that residues LL1010/11 are part of the p38 substrate–recognition site that is involved specifically in phosphorylation of residues 1015–1018.

p38-induced phosphorylation of EGFR Ser1015 is dependent on LL1010/1011 and independent on AP2. (A) PAE cells expressing WT EGFR-GFP or the LL1010/11A mutant were incubated with vehicle (Ctrl), 10 ng/ml EGF (E), or 100 nM anisomycin (A) for 15 min at 37°C. Cells were lysed, and lysates were probed by Western blotting (WB) with antibodies to pS1046/47, pS1015, pY1068, activated p38 (p-p38), pERK1/2, and α-actinin (loading control). (B) HeLa/FAP-EGFR cells were transfected with nontargeting (NT) or μ2 siRNAs. 4 d later, the cells were stimulated with vehicle (Ctrl), 4 ng/ml EGF (E), 10 ng/ml TNFα (T), or 100 nM anisomycin (A). Cells were lysed, and lysates were immunoblotted with antibodies to pS1015, α-adaptin, and Grb2 (loading control).
p38-induced phosphorylation of EGFR Ser1015 is dependent on LL1010/1011 and independent on AP2. (A) PAE cells expressing WT EGFR-GFP or the LL1010/11A mutant were incubated with vehicle (Ctrl), 10 ng/ml EGF (E), or 100 nM anisomycin (A) for 15 min at 37°C. Cells were lysed, and lysates were probed by Western blotting (WB) with antibodies to pS1046/47, pS1015, pY1068, activated p38 (p-p38), pERK1/2, and α-actinin (loading control). (B) HeLa/FAP-EGFR cells were transfected with nontargeting (NT) or μ2 siRNAs. 4 d later, the cells were stimulated with vehicle (Ctrl), 4 ng/ml EGF (E), 10 ng/ml TNFα (T), or 100 nM anisomycin (A). Cells were lysed, and lysates were immunoblotted with antibodies to pS1015, α-adaptin, and Grb2 (loading control).
Together, these observations made using alanine substitutions, phosphomimetic mutations, and mass spectrometry suggest that p38 phosphorylation of S1006 may serve as a “classical” negative charge at the −4 position from the dileucine motif and that it is essential for the interaction of this motif with AP2 and subsequent EGFR endocytosis upon activation of p38. Phosphorylation of the R1 cluster is necessary for maximally efficient p38-induced AP2 binding and endocytosis.
EGF promotes p38-dependent internalization of ligand-free EGFR into endosomes containing EGF-bound EGFR
p38-induced internalization of ligand-free EGFR has been demonstrated to occur upon EGF stimulation (Tanaka et al., 2018). We confirmed that EGF at physiological concentration (1 ng/ml) strongly activates p38 in HeLa/FAP-EGFR cells (Fig. 5 A). To determine the contribution of p38-dependent internalization in the overall EGF-induced FAP-EGFR internalization, the dependence of FAP-EGFR endocytosis on EGF concentration in the absence or presence of BIRB796 was examined using the FERI assay. As shown in Fig. 5 B, the p38-dependent component of FAP-EGFR endocytosis (endocytosis inhibited by BIRB796) peaked at ∼1 ng/ml EGF when ∼40–50% of internalized FAP-EGFR was dependent on p38 activity (48.4 ± 12.8% at 0.78 ng/ml EGF and 40.9 ± 7.8% at 1.56 ng/ml EGF; mean ± SDs from three experiments exemplified in Fig. 5 B). Increasing EGF concentration gradually reduced and ultimately abolished the p38-dependent component of EGF–induced FAP-EGFR internalization. BIRB796 did not significantly alter the internalization of 1 ng/ml 125I-EGF (Fig. 5 C), suggesting that EGF-induced p38 activity caused endocytosis of ligand-free FAP-EGFRs without significantly affecting the internalization of EGF:FAP-EGFR complexes. Together, the data in Fig. 5, B and C, indicate that after 15-min stimulation with 1 ng/ml EGF, about half of internalized FAP-EGFRs are ligand-free receptors.

EGF-induced p38-independent and p38-dependent internalization target ligand-occupied and free EGFRs to the same endosomes. (A) HeLa/FAP-EGFR cells were treated with 1 ng/ml EGF for indicated times at 37°C and lysed. Lysates were probed by Western blotting with pY1068, pS1046/47, phospho-p38 (p-p38), phospho-ERK1/2 (pERK), and α-actinin antibodies (loading control). (B) HeLa/FAP-EGFR cells were incubated with DMSO (vehicle, black) or 100 nM BIRB796 (red) for 90 min and then treated with 0.48–100 ng/ml EGF for 15 min. FAP-EGFR internalization was measured using the FERI assay. Mean values with SEM of duplicates are plotted against log of EGF concentrations. Mean values of the 561/640 ratio in cells treated with BIRB796 were subtracted from those values in vehicle-treated cells to estimate the relative contribution of the p38-dependent internalization (dashed violet). This experiment is representative of three independent experiments. (C) HeLa/FAP-EGFR cells were pretreated with DMSO (vehicle, black) or 100 nM BIRB796 (red) for 90 min, and then incubated with 1 ng/ml 125I-EGF for indicated times at 37°C. Surface-bound and internalized 125I-EGF was measured, and the ratio of the amounts of internalized and surface ligand is plotted against time. The data are normalized to the maximum value of the internalized/surface 125I-EGF ratio at the 10-min time point. Mean values with SDs from two independent experiments are presented. The difference between internalization rates in vehicle- and BIRB796-treated cells is not statistically significant. (D–F) HeLa/FAP-EGFR cells were pretreated for 90 min with DMSO (vehicle) or 100 nM BIRB796, labeled with MG-B-Tau, and stimulated with 0.5 ng/ml or 20 ng/ml EGF-Rh for 15 min. Cells were fixed, and 3D imaging through 640-nm (green, EGFR) and 561-nm (red, EGF-Rh) channels was performed. In D, single confocal sections are shown. Scale bars, 10 µm. In E, the ratio of EGF-Rh and MG-B-Tau fluorescence (EGF-Rh/FAP-EGFR) in individual (ind.) endosomes was calculated in 3D images generated as in D. Median and quartiles are shown on the violin graph; n is >5,000 endosomes. In F, the interpretation of the data in E is proposed. Stimulation with 0.5 ng/ml EGF results in internalization of EGF:EGFR dimers and monomeric ligand-free receptors (in a p38-dependent manner) to the same endosomes. Inhibition of p38 results in endocytosis of only EGF:EGFR complexes but not ligand-free receptors, which leads to an apparent increase of the EGF/EGFR ratio per endosome. When cells are stimulated with the saturating concentration of EGF (20 ng/ml), p38-dependent internalization of free EGFR is negligible, and therefore, BIRB796 does not change the EGF:EGFR ratio in endosomes. A considerable fraction of EGF:EGFR dimers with 1:2 stoichiometry may exist in cells treated with 0.5 ng/ml EGFR (Macdonald and Pike, 2008).
EGF-induced p38-independent and p38-dependent internalization target ligand-occupied and free EGFRs to the same endosomes. (A) HeLa/FAP-EGFR cells were treated with 1 ng/ml EGF for indicated times at 37°C and lysed. Lysates were probed by Western blotting with pY1068, pS1046/47, phospho-p38 (p-p38), phospho-ERK1/2 (pERK), and α-actinin antibodies (loading control). (B) HeLa/FAP-EGFR cells were incubated with DMSO (vehicle, black) or 100 nM BIRB796 (red) for 90 min and then treated with 0.48–100 ng/ml EGF for 15 min. FAP-EGFR internalization was measured using the FERI assay. Mean values with SEM of duplicates are plotted against log of EGF concentrations. Mean values of the 561/640 ratio in cells treated with BIRB796 were subtracted from those values in vehicle-treated cells to estimate the relative contribution of the p38-dependent internalization (dashed violet). This experiment is representative of three independent experiments. (C) HeLa/FAP-EGFR cells were pretreated with DMSO (vehicle, black) or 100 nM BIRB796 (red) for 90 min, and then incubated with 1 ng/ml 125I-EGF for indicated times at 37°C. Surface-bound and internalized 125I-EGF was measured, and the ratio of the amounts of internalized and surface ligand is plotted against time. The data are normalized to the maximum value of the internalized/surface 125I-EGF ratio at the 10-min time point. Mean values with SDs from two independent experiments are presented. The difference between internalization rates in vehicle- and BIRB796-treated cells is not statistically significant. (D–F) HeLa/FAP-EGFR cells were pretreated for 90 min with DMSO (vehicle) or 100 nM BIRB796, labeled with MG-B-Tau, and stimulated with 0.5 ng/ml or 20 ng/ml EGF-Rh for 15 min. Cells were fixed, and 3D imaging through 640-nm (green, EGFR) and 561-nm (red, EGF-Rh) channels was performed. In D, single confocal sections are shown. Scale bars, 10 µm. In E, the ratio of EGF-Rh and MG-B-Tau fluorescence (EGF-Rh/FAP-EGFR) in individual (ind.) endosomes was calculated in 3D images generated as in D. Median and quartiles are shown on the violin graph; n is >5,000 endosomes. In F, the interpretation of the data in E is proposed. Stimulation with 0.5 ng/ml EGF results in internalization of EGF:EGFR dimers and monomeric ligand-free receptors (in a p38-dependent manner) to the same endosomes. Inhibition of p38 results in endocytosis of only EGF:EGFR complexes but not ligand-free receptors, which leads to an apparent increase of the EGF/EGFR ratio per endosome. When cells are stimulated with the saturating concentration of EGF (20 ng/ml), p38-dependent internalization of free EGFR is negligible, and therefore, BIRB796 does not change the EGF:EGFR ratio in endosomes. A considerable fraction of EGF:EGFR dimers with 1:2 stoichiometry may exist in cells treated with 0.5 ng/ml EGFR (Macdonald and Pike, 2008).
To examine the localization of internalized free and ligand-bound FAP-EGFR in EGF-stimulated cells, FAP-EGFR was labeled with MG-B-Tau, and 3D imaging of cells stimulated with low (0.5 ng/ml) or high (20 ng/ml) concentrations of EGF-rhodamine conjugate (EGF-Rh) was performed. Virtually complete colocalization of EGF-Rh and FAP-EGFR in endosomes after 15-min EGF stimulation was seen regardless of BIRB796 pretreatment (Fig. 5 D), indicating that free and ligand-bound FAP-EGFRs are accumulated in the same endosomes. The mean ratio of EGF-Rh and FAP-EGFR fluorescence intensities (EGF-Rh/FAP-EGFR) in individual endosomes of cells stimulated with 0.5 ng/ml EGF-Rh was less than half of that seen in cells stimulated with 20 ng/ml EGF-Rh (Fig. 5 E). BIRB796 significantly (∼1.8-fold) increased the mean EGF-Rh/FAP-EGFR ratio in cells treated with 0.5 ng/ml but not in cells treated with 20 ng/ml EGF-Rh. These data indicate that the fraction of ligand-free receptors in endosomes in cells stimulated with 0.5 ng/ml EGF-Rh was substantially reduced when p38-dependent endocytosis was inhibited, and that this fraction was negligible in the presence of high EGF concentration (20 ng/ml EGF-Rh). These results are summarized in Fig. 5 F.
Activation of p38 by TNFα concurrently with EGF stimulation increases EGFR internalization only at low EGF concentrations
Fig. 5 demonstrated that EGF-induced p38-dependent endocytosis of free EGFRs is minimal in the presence of moderate/high EGF concentrations. Therefore, we tested whether a robust activation of p38 by TNFα can expand the capacity of the p38-dependent endocytic pathway in HeLa/FAP-EGFR cells stimulated with the range of EGF concentrations used in this study. Essentially, the goal of the experiments was to recapitulate the complex tissue microenvironment in vivo, where cells are exposed to multiple stimuli and stress factors under both physiological and pathological conditions. TNFα (3 ng/ml) substantially increased FAP-EGFR internalization in the presence of low EGF concentrations as measured using the FERI assay (Fig. 6 A). However, TNFα-dependent increase in receptor internalization was inversely proportional to the EGF concentration, gradually decreasing from 70–90% of the overall uptake at EGF concentrations of 0.1–0.5 ng/ml to <50% at EGF concentrations >1–2 ng/ml (Fig. 6 A). Importantly, TNFα did not affect the internalization rate of 125I-EGF used in low (0.2 ng/ml) or moderate concentrations (6 ng/ml; Fig. 6, B and C). These data indicate that TNFα-induced p38 activity predominantly promotes endocytosis of ligand-free but does not significantly affect endocytosis of ligand-bound FAP-EGFR.

Simultaneous cell stimulation with EGF and TNFα enhances ligand-free EGFR internalization only at low concentrations of EGF and targets ligand-bound and free EGFRs to the same endosomes. (A) HeLa/FAP-EGFR cells were treated with 0.48–100 ng/ml EGF in the absence (black) or presence (green) of 3 ng/ml TNFα for 15 min, and the FERI internalization assay was performed. Mean values of the 561/640 ratio with SEM of duplicates are plotted against log of EGF concentration. Values obtained in the absence of TNFα were subtracted from those in the presence of TNFα to estimate the contribution of TNFα-induced internalization of FAP-EGFR (dotted blue). This experiment is representative of several independent experiments. (B and C) HeLa/FAP-EGFR cells were incubated with 0.2 ng/ml (B) or 6 ng/ml (C) of 125I-EGF in the absence or presence of 3 ng/ml TNFα for 10 min at 37°C. Surface-bound and internalized 125I-EGF were measured, and the ratio of internalized and surface 125I-EGF is plotted against time. The data were normalized to the value of the internalized/surface 125I-EGF ratio at the 10-min point in the absence of TNFα. Mean values with SDs from three independent experiments are presented. Differences between internalization rates in control and TNFα-stimulated cells are not statistically significant. (D and E) HeLa/FAP-EGFR cells were labeled with MG-B-Tau and treated with 0.2 ng/ml or 10 ng/ml EGF-Rh for 15 min in the absence or presence of 3 ng/ml TNFα. After fixation, 3D images were acquired through 640-nm (green, FAP-EGFR) and 561-nm (red, EGF-Rh) channels. In D, single confocal sections of cells treated with 0.2 ng/ml EGF-Rh are shown. Insets are higher magnification images of regions marked by white rectangles in which the 561-nm channel image (red, EGF-Rh) was shifted by 3 pixels as indicated by the white arrow for better visualization of EGF-Rh and FAP-EGFR colocalization in individual (ind.) endosomes. Scale bars, 10 µm. In E, the ratio of EGF-Rh and MG-B-Tau fluorescence (EGF-Rh/FAP-EGFR) in individual endosomes calculated in images generated as in D. Median and quartiles are shown on the violin graph; n > 5,000 endosomes.
Simultaneous cell stimulation with EGF and TNFα enhances ligand-free EGFR internalization only at low concentrations of EGF and targets ligand-bound and free EGFRs to the same endosomes. (A) HeLa/FAP-EGFR cells were treated with 0.48–100 ng/ml EGF in the absence (black) or presence (green) of 3 ng/ml TNFα for 15 min, and the FERI internalization assay was performed. Mean values of the 561/640 ratio with SEM of duplicates are plotted against log of EGF concentration. Values obtained in the absence of TNFα were subtracted from those in the presence of TNFα to estimate the contribution of TNFα-induced internalization of FAP-EGFR (dotted blue). This experiment is representative of several independent experiments. (B and C) HeLa/FAP-EGFR cells were incubated with 0.2 ng/ml (B) or 6 ng/ml (C) of 125I-EGF in the absence or presence of 3 ng/ml TNFα for 10 min at 37°C. Surface-bound and internalized 125I-EGF were measured, and the ratio of internalized and surface 125I-EGF is plotted against time. The data were normalized to the value of the internalized/surface 125I-EGF ratio at the 10-min point in the absence of TNFα. Mean values with SDs from three independent experiments are presented. Differences between internalization rates in control and TNFα-stimulated cells are not statistically significant. (D and E) HeLa/FAP-EGFR cells were labeled with MG-B-Tau and treated with 0.2 ng/ml or 10 ng/ml EGF-Rh for 15 min in the absence or presence of 3 ng/ml TNFα. After fixation, 3D images were acquired through 640-nm (green, FAP-EGFR) and 561-nm (red, EGF-Rh) channels. In D, single confocal sections of cells treated with 0.2 ng/ml EGF-Rh are shown. Insets are higher magnification images of regions marked by white rectangles in which the 561-nm channel image (red, EGF-Rh) was shifted by 3 pixels as indicated by the white arrow for better visualization of EGF-Rh and FAP-EGFR colocalization in individual (ind.) endosomes. Scale bars, 10 µm. In E, the ratio of EGF-Rh and MG-B-Tau fluorescence (EGF-Rh/FAP-EGFR) in individual endosomes calculated in images generated as in D. Median and quartiles are shown on the violin graph; n > 5,000 endosomes.
Imaging of FAP-EGFR labeled with MG-B-Tau in cells stimulated with 0.2 ng/ml EGF-Rh with or without TNFα showed strong colocalization of EGF-Rh and FAP-EGFR in endosomes regardless of the presence of TNFα (Fig. 6 D), thus demonstrating accumulation of both ligand-bound and free FAP-EGFRs in the same endosomes. TNFα substantially (two times) decreased the mean value of the EGF-Rh/FAP-EGFR fluorescence intensity ratio in endosomes of cells stimulated with 0.2 ng/ml EGF-Rh, indicative of an increased fraction of ligand-free FAP-EGFR in these endosomes. By contrast, the ligand–receptor ratio was not significantly changed by costimulation with TNFα in cells treated with 10 ng/ml EGF-Rh (Fig. 6 E), which is consistent with the minimal contribution of p38-dependent endocytosis under these conditions (Fig. 6 A). Therefore, the robust activation of p38 by TNFα does not overcome the predominance of EGF-induced, p38-independent endocytosis that takes place in the presence of moderate and receptor-saturating concentrations of EGF.
p38 induces internalization of ligand-bound EGFR in Grb2-depleted cells
The data presented in Figs. 5 and 6 raised two questions: (1) why p38 activation does not promote endocytosis of ligand-bound EGFRs, and (2) why the contribution of p38-induced EGFR endocytosis in the overall internalization of EGFR decreases even with moderate increases in the concentration of EGF. To address the first question, we examined whether EGF-induced post-translational modifications and interactions of ligand-bound EGFR interfere with p38-dependent internalization of these receptor complexes. It has been demonstrated that the kinase-dead monomeric EGFR mutant can be internalized in a p38-dependent manner (Tanaka et al., 2018). To further explore this observation, we found that inhibition of the EGFR kinase activity by erlotinib, which does not prevent ligand-induced receptor dimerization but blocks ligand-induced CME of the receptor, allowed for TNFα-induced endocytosis of EGF-Rh–occupied EGFR (Fig. 7 A). These data suggested that the receptor kinase activity is important for preventing p38-dependent endocytosis of ligand-bound receptors.

p38 activation by TNFα rescues inhibited EGFR internalization in Grb2-depleted HeLa/FAP-EGFR cells. (A) After pretreatment with DMSO (vehicle) or BIRB796 for 90 min, the cells were simultaneously labeled with MG-B-Tau and treated with DMSO (vehicle) or erlotinib (1 µM) for 10 min. The cells were then stimulated with 1 ng/ml EGF-Rh alone or together with 10 ng/ml TNFα for 15 min. Live-cell imaging was performed through 640-nm (green, FAP-EGFR) and 561-nm channels (red, EGF-Rh). Scale bars, 10 µm. (B) The cells were treated with 0.5 or 10 ng/ml EGF in the presence of 10 ng/ml TNFα for 15 min at 37°C. Cell lysates were immunoprecipitated with EGFR Mab528, pS1015 or pY1068 antibodies. Both total cell lysates (TCL) and immunoprecipitates (IP) were probed by immunoblotting with pY1068, pS1015, and EGFR (AB_631420) antibodies. (C and D) 3 d after transfection with nontargeting (NT) or Grb2 siRNAs, cells were stimulated with 1 or 10 ng/ml EGF (C) or EGF-Rh (D) in the absence or presence of 10 ng/ml TNFα. In C, FAP-EGFR internalization was examined using the FERI assay. For each treatment variant, the value of the 561/640 ratio was normalized to that ratio in NT-transfected cells treated with 10 ng/ml EGF. In D, FAP-EGFR was labeled with MG-B-Tau. 3D images were acquired through 640-nm (green, FAP-EGFR) and 561-nm (red, EGF-Rh) channels. The amount of EGF-Rh in FAP-EGFR–containing endosomes per cell was quantified. These values were normalized by maximum values obtained in NT cells treated with 10 ng/ml EGF-Rh in each experiment. Graph bars in C and D represent mean values with SEMs (n = 3 independent experiments). P values of Grb2 against NT were calculated using the unpaired Student’s t test. a.l.u.f.i., arbitrary linear unit of fluorescence intensity; WB, Western blot.
p38 activation by TNFα rescues inhibited EGFR internalization in Grb2-depleted HeLa/FAP-EGFR cells. (A) After pretreatment with DMSO (vehicle) or BIRB796 for 90 min, the cells were simultaneously labeled with MG-B-Tau and treated with DMSO (vehicle) or erlotinib (1 µM) for 10 min. The cells were then stimulated with 1 ng/ml EGF-Rh alone or together with 10 ng/ml TNFα for 15 min. Live-cell imaging was performed through 640-nm (green, FAP-EGFR) and 561-nm channels (red, EGF-Rh). Scale bars, 10 µm. (B) The cells were treated with 0.5 or 10 ng/ml EGF in the presence of 10 ng/ml TNFα for 15 min at 37°C. Cell lysates were immunoprecipitated with EGFR Mab528, pS1015 or pY1068 antibodies. Both total cell lysates (TCL) and immunoprecipitates (IP) were probed by immunoblotting with pY1068, pS1015, and EGFR (AB_631420) antibodies. (C and D) 3 d after transfection with nontargeting (NT) or Grb2 siRNAs, cells were stimulated with 1 or 10 ng/ml EGF (C) or EGF-Rh (D) in the absence or presence of 10 ng/ml TNFα. In C, FAP-EGFR internalization was examined using the FERI assay. For each treatment variant, the value of the 561/640 ratio was normalized to that ratio in NT-transfected cells treated with 10 ng/ml EGF. In D, FAP-EGFR was labeled with MG-B-Tau. 3D images were acquired through 640-nm (green, FAP-EGFR) and 561-nm (red, EGF-Rh) channels. The amount of EGF-Rh in FAP-EGFR–containing endosomes per cell was quantified. These values were normalized by maximum values obtained in NT cells treated with 10 ng/ml EGF-Rh in each experiment. Graph bars in C and D represent mean values with SEMs (n = 3 independent experiments). P values of Grb2 against NT were calculated using the unpaired Student’s t test. a.l.u.f.i., arbitrary linear unit of fluorescence intensity; WB, Western blot.
One possible mechanism could be interference of the receptor tyrosine phosphorylation with the serine-threonine phosphorylation of EGFR by p38. However, reciprocal immunoprecipitations of EGFR from EGF/TNFα costimulated cells using antibodies to pY1068 and pS1015 demonstrated coexistence of both phosphorylated residues on the same receptor molecule, thus arguing against such a mechanism (Fig. 7 B). On the other hand, autophosphorylation of tyrosines 1068 and 1086 creates binding sites for Grb2, which is critical for the CME of ligand-bound EGFR (Jiang et al., 2003; Stang et al., 2004). Therefore, we tested the role of Grb2 in the crosstalk between p38-independent and p38-dependent internalization pathways. As shown in Fig. 1 H, Grb2 siRNA knockdown does not affect TNFα/p38-induced endocytosis in HeLa/FAP-EGFR cells. In cells stimulated with 1 ng/ml EGF, Grb2 knockdown partially but significantly inhibited the endocytic uptake of FAP-EGFR; the residual endocytosis is largely a combination of p38-dependent endocytosis of free receptors, redundant CME mechanisms, and possibly, minimal CIE (Fig. 7 C). TNFα stimulation of cells treated with 1 ng/ml EGF increased FAP-EGFR endocytosis in control cells (nontargeting siRNA), and most importantly, restored FAP-EGFR endocytosis in Grb2-depleted cells to the level observed in control cells. Similar trends were observed in cells treated with 10 ng/ml EGF, although Grb2 depletion and rescue effects were less pronounced (Fig. 7 C).
The rescue of EGFR endocytosis in Grb2-depleted cells by strong activation of p38 suggests that in the absence of Grb2, ligand-bound active receptors may be internalized in a p38-dependent manner. To test this possibility, the amount of internalized FAP-EGFR ligand (EGF-Rh) was measured in cells transfected with nontargeting or Grb2 siRNAs, and costimulated with TNFα. Grb2 knockdown decreased internalization of 1 ng/ml EGF-Rh by ∼40% (Fig. 7 D). At this EGF-Rh concentration, consistent with the FERI assay (Fig. 7 C), TNFα rescued EGF-Rh internalization in Grb2-depleted cells (Fig. 7 D), demonstrating that ligand-bound receptors can be internalized in a p38-dependent manner in the absence of Grb2. At 10 ng/ml EGF-Rh, Grb2 knockdown did not significantly affect the amount of internalized EGF-Rh with or without TNFα (Fig. 7 D).
High levels of EGFR activation result in CIE and counteract TNFα stimulation of p38-dependent CME without affecting p38 signaling
To address the second question, i.e., why the contribution of p38-induced EGFR endocytosis in the overall internalization of EGFR is decreasing with increasing EGF concentrations, we first examined whether elevated EGFR signaling may affect p38 activity. As shown in Fig. 8 A, 6 ng/ml EGF did not inhibit EGF- (as compared with 0.2 ng/ml EGF) or TNFα-induced p38 autophosphorylation and p38-mediated phosphorylation of EGFR Ser1046/47 and MAPKAPK2. These data indicate that the apparent inhibition of p38-dependent EGFR endocytosis by moderate/high EGF concentrations is not due to inhibition of the p38 activity and its downstream signaling. Vice versa, p38 activation by TNFα did not interfere with EGF-stimulated tyrosine phosphorylation of Y1068 and ubiquitination of EGFR, its coimmunoprecipitation with Cbl, and ERK1/2 activation (Fig. 8 A).

Moderate/high EGF concentrations minimize p38-dependent down-regulation of cell-surface FAP-EGFR without affecting p38 signaling. (A) HeLa/FAP-EGFR cells were incubated with 0, 0.2, or 6 ng/ml EGF for 10 min, in the absence or presence of 3 ng/ml TNFα. Cells were solubilized, and EGFR was immunoprecipitated from cell lysates. Aliquots of lysates (total cell lysates [TCL]) and immunoprecipitates (IP) were immunoblotted with pY1068, pS1046/47, pERK, p-p38, pMAPKAPK2, ubiquitin (Ub), Cbl, EGFR, and Grb2 (loading control) antibodies. (B–D) Cells were left untreated or treated with 0.5, 5, or 50 ng/ml EGF without (black lines) or with 10 ng/ml TNFα (green lines), or with TNFα alone (dashed blue) for 5–30 min. Cell-surface FAP-EGFR was then labeled by incubation with MG-B-Tau at 4°C, and MG-B-Tau fluorescence intensity was measured. All values of fluorescence intensity in treated cells were normalized to that value measured in untreated cells in each experiment. Values from untreated cells were used as 0 time points. The data represent mean values with SEM from four independent experiments plotted against time. WB, Western blot.
Moderate/high EGF concentrations minimize p38-dependent down-regulation of cell-surface FAP-EGFR without affecting p38 signaling. (A) HeLa/FAP-EGFR cells were incubated with 0, 0.2, or 6 ng/ml EGF for 10 min, in the absence or presence of 3 ng/ml TNFα. Cells were solubilized, and EGFR was immunoprecipitated from cell lysates. Aliquots of lysates (total cell lysates [TCL]) and immunoprecipitates (IP) were immunoblotted with pY1068, pS1046/47, pERK, p-p38, pMAPKAPK2, ubiquitin (Ub), Cbl, EGFR, and Grb2 (loading control) antibodies. (B–D) Cells were left untreated or treated with 0.5, 5, or 50 ng/ml EGF without (black lines) or with 10 ng/ml TNFα (green lines), or with TNFα alone (dashed blue) for 5–30 min. Cell-surface FAP-EGFR was then labeled by incubation with MG-B-Tau at 4°C, and MG-B-Tau fluorescence intensity was measured. All values of fluorescence intensity in treated cells were normalized to that value measured in untreated cells in each experiment. Values from untreated cells were used as 0 time points. The data represent mean values with SEM from four independent experiments plotted against time. WB, Western blot.
An alternative possibility is that an increased ligand occupancy of surface EGFR in the presence of high EGF concentrations decreases the pool of free EGFRs at the cell surface, thus abolishing p38-dependent internalization of these receptors. Therefore, surface levels of FAP-EGFR in cells stimulated with the range of EGF concentrations without and with TNFα were measured by labeling cell-surface FAP-EGFR with MG-B-Tau at 4°C. Consistently with the FERI assay (Fig. 6 A), TNFα significantly decreased surface EGFR levels only in the presence of a low EGF concentration (0.5 ng/ml; Fig. 8, B–D). However, a considerable pool of free FAP-EGFR (∼30%) remained at the surface of cells treated with 5 ng/ml EGF (Fig. 8 C) as compared with cells treated with saturating EGF concentration (50 ng/ml), conditions of maximal receptor down-regulation (Fig. 8 D). These data suggested that while the availability of ligand-free EGFRs may, to some extent, limit the contribution of p38-dependent endocytosis in the overall EGFR internalization (in particular, at saturating concentrations of EGF), additional mechanisms exist that minimize p38-dependent endocytosis of EGFR in the presence of moderate EGF concentrations.
To further examine these mechanisms, we analyzed the clathrin requirement for EGFR endocytic pathways in HeLa/FAP-EGFR cells. To this end, FAP-EGFR endocytosis was measured in cells stimulated with a range of concentrations of EGF, TNFα, or both ligands together in cells transfected with nontargeting or CHC-targeting siRNAs. As shown in Fig. 9 A, the CME of EGFR reached a plateau while CIE became substantial in HeLa/FAP-EGFR cells stimulated with 2–3 ng/ml EGF alone. Such concentration saturability suggested an apparent limited capacity of the clathrin-mediated EGF-stimulated pathway of EGFR endocytosis. TNFα increased the amount of FAP-EGFR internalized via the CME pathway to its highest total capacity without EGF (Fig. 9 B) and in the presence of low EGF concentrations (Fig. 9 A). This capacity of TNFα-triggered CME could not be increased by costimulation with increased EGF concentrations but was significantly higher than the maximal capacity of EGF-induced CME (Fig. 9, A and B). Furthermore, the fraction of FAP-EGFRs internalized through a TNFα-dependent CME started to decrease at EGF concentrations as low as 1 ng/ml with the complete loss of this uptake component in the presence of >10 ng/ml EGF (Fig. 9 A). Moreover, high EGF concentrations reduced ligand-dependent CME of EGFR below its maximal capacity (Fig. 9 A). Together, the data in Fig. 9, A and B, show that only robust p38 activation allows CME of EGFR to proceed at its maximum apparent capacity in HeLa/FAP-EGFR cells, and that moderate/high EGF concentrations reroute EGFR to the CIE pathway and decrease the relative contribution of CME to the total EGFR internalization, mainly at the expense of the p38-dependent CME.

Clathrin-dependence of EGF- and TNFα-induced EGFR internalization and a working model of the relationships between various pathways of EGFR endocytosis. (A and B) HeLa/FAP-EGFR cells were transfected with nontargeting (NT; solid lines) or CHC siRNAs (dashed lines). 3 d later, FERI assay was performed after 15-min treatment with (A) 0.48–100 ng/ml EGF in the absence (black) or presence of 3 ng/ml TNFα (green); (B) 0.48–100 ng/ml TNFα in the absence (blue) or presence of 3 ng/ml EGF (purple). Mean values of 561/640 ratio obtained in cells depleted of CHC (internalization through CIE) were subtracted from those values in NT-transfected cells (total internalization) to estimate the amount of internalization via CME. Mean values with SEM from duplicates are shown in the graphs on the right. Experiments in A and B are representative of several independent experiments. (C) Working model of the relationships between distinct pathways of EGFR endocytosis in the presence of EGF and TNFα. The distribution of ligand-bound (red and blue) and free (green) EGFRs after 15-min stimulation is shown. Blue arrows, CME of ligand-bound EGFR; green arrows, CME of ligand-free EGFR; gray arrow, CIE of EGFR. The widths of arrows roughly correspond to the relative contribution of specific endocytic pathways in the total internalization of EGFR. Receptors internalized through a p38-dependent pathway or CIE are indicated by green or gray dotted lines, respectively. See Discussion for the description of the model. The model does not account for heterodimerization of EGFR with ErbB2 given a relatively low level of ErbB2 expression in HeLa cells and an impaired internalization of heterodimers.
Clathrin-dependence of EGF- and TNFα-induced EGFR internalization and a working model of the relationships between various pathways of EGFR endocytosis. (A and B) HeLa/FAP-EGFR cells were transfected with nontargeting (NT; solid lines) or CHC siRNAs (dashed lines). 3 d later, FERI assay was performed after 15-min treatment with (A) 0.48–100 ng/ml EGF in the absence (black) or presence of 3 ng/ml TNFα (green); (B) 0.48–100 ng/ml TNFα in the absence (blue) or presence of 3 ng/ml EGF (purple). Mean values of 561/640 ratio obtained in cells depleted of CHC (internalization through CIE) were subtracted from those values in NT-transfected cells (total internalization) to estimate the amount of internalization via CME. Mean values with SEM from duplicates are shown in the graphs on the right. Experiments in A and B are representative of several independent experiments. (C) Working model of the relationships between distinct pathways of EGFR endocytosis in the presence of EGF and TNFα. The distribution of ligand-bound (red and blue) and free (green) EGFRs after 15-min stimulation is shown. Blue arrows, CME of ligand-bound EGFR; green arrows, CME of ligand-free EGFR; gray arrow, CIE of EGFR. The widths of arrows roughly correspond to the relative contribution of specific endocytic pathways in the total internalization of EGFR. Receptors internalized through a p38-dependent pathway or CIE are indicated by green or gray dotted lines, respectively. See Discussion for the description of the model. The model does not account for heterodimerization of EGFR with ErbB2 given a relatively low level of ErbB2 expression in HeLa cells and an impaired internalization of heterodimers.
Discussion
In the present study, we uncover new insights into the mechanism of p38-dependent endocytosis of EGFR and analyze the contribution of this endocytic pathway to the internalization of EGFR in cells stimulated with EGF alone or together with p38-activating stimuli. Such quantitative analysis was made possible by the development of new methodologies for studying EGFR endocytosis, which do not rely on the use of labeled EGF but are designed to directly measure the internalization rates of the receptor itself, using the endogenous FAP-EGFR fusion protein (Larsen et al., 2019). Endocytic rates of ligand-free EGFR have been previously measured using labeled antibodies (Burke et al., 2001; Burke and Wiley, 1999). However, FAP-based methods provided an opportunity to combine a multiplex, high-throughput measurement with a single-cell high-resolution fluorescence microscopy imaging approach, and circumvent possible caveats associated with the antibody binding to the receptor.
Our analysis addressed two key questions about the mechanism of p38-dependent EGFR endocytosis that were raised in previous studies: (1) is internalization mediated by the LL1010/1011 motif, and if so, is the role of this motif to interact with AP2 or promote phosphorylation of the R1 cluster?; and (2) how does p38 activity facilitate these interactions and receptor internalization? To address the first question, we showed that mutations of LL1010/1011 abolished p38-dependent EGFR:AP2 interaction and internalization of EGFR, and that this internalization was inhibited by overexpressed model cargo containing a dileucine motif. Furthermore, using an siRNA knockdown and rescue approach, we demonstrated that p38-dependent FAP-EGFR endocytosis requires amino acid residues in the σ2 subunit of AP2 that are essential for AP2 interaction with dileucine motifs. Together, these data strongly suggest that the LL1010/1011 motif is crucial for p38-induced EGFR endocytosis because it binds to AP2.
Clues to answer the second question were proposed in the previous study, which demonstrated the importance of phosphorylation of the R1 cluster (residues 1015–1018) for p38-dependent EGFR endocytosis (Tanaka et al., 2018). However, the observation that mutations of residues 1015–1018 only partially inhibited p38-dependent endocytosis of EGFR as compared with the inhibitory effect of LL1010/11A mutations (Fig. 3) motivated new mutagenesis-based analysis. The −4 position of dileucine motifs is most commonly occupied by acidic residues, which bind to the hydrophilic patch with an overall positive charge in σ2. EGFR does not have an acidic residue at the −4 position or in close proximity. Instead, it has a cluster of serines and threonines (residues 1001–1006). Surprisingly, alanine substitution of S1006 virtually abolished p38-dependent endocytosis, phosphomimetic S1006E mutation resulted in a constitutive endocytosis via the dileucine motif, and p38-dependent phosphorylation of S1006 was detected by mass spectrometry. These data cumulatively imply that p38-inducible phosphorylation of S1006 may fulfill the requirement for a negative charge at the −4 position for interaction of the LL1010/1011 motif with AP2. Whereas dileucine-containing peptides, which were cocrystalized with AP2, included phosphoserine in the −5 position of dileucines, this phosphorylated residue was shown, in contrast to a negative change in the −4 position, to not be engaged in the interaction with σ2 or other AP2 subunits (Kelly et al., 2008). Therefore, phosphorylation of Ser1006 is, to our knowledge, the first example of a stimuli-generated negative charge at the −4 position that enables the interaction of a dileucine motif with AP2.
The precise role of the major p38-induced phosphorylation site of EGFR, the R1 cluster, remains unclear. Phosphomimetic mutations of these residues did not increase EGFR endocytosis, which indicates that phosphorylation of residues 1015–1018 may not be involved in direct electrostatic interactions with AP2. Alternatively, such interactions may occur, but they may not be sufficient to facilitate LL:AP2 interactions without the negative charge at S1006. Serine phosphorylation downstream (+positions) from the dileucine motif of CD4 was shown to be necessary for the maximally strong interaction of CD4 with AP2 and efficient CD4 endocytosis (Pitcher et al., 1999). Interestingly, phosphomimetic mutations of those serines did not result in CD4 endocytosis, equivalent to mutations of the R1 cluster in EGFR. Therefore, it is reasonable to consider that R1 phosphorylation promotes structural changes in this region of the EGFR molecule, allowing a linear conformation of the dileucine-containing motif, thus favoring its binding to AP2.
Advances in defining the mechanism of p38-dependent CME of EGFR and the availability of a high-throughput FAP-based internalization assay enabled better understanding of the reciprocal relationships between multiple pathways of EGFR endocytosis. In Fig. 9 C, we propose a working model of these relationships in cells, such as HeLa, with a moderate level of EGFR expression.
As depicted in Fig. 9 C, a, in cells stimulated with low EGF concentrations (≤170 pM or ≤1 ng/ml), ∼50–60% of EGFRs in endosomes are ligand-bound EGFR dimers, which are internalized by CME using primarily the Grb2-Cbl-ubiquitin–dependent mechanism. The other 40–50% are ligand-free monomeric EGFRs internalized through the CME pathway using the dileucine:AP2 interaction mechanism involving activation of p38 by EGF (Fig. 9 C, a). The fraction of receptors internalized using this mechanism is substantially increased if p38 is robustly activated by stress stimuli like TNFα (Fig. 9 C, b). In the presence of TNFα, with or without low EGF concentrations, the CME pathway reaches its maximal contribution to total EGFR internalization. The fraction of CME-internalized receptors does not increase with increasing EGF or TNFα concentrations (Fig. 9, A and B). This maximal capacity of the CME for EGFR (∼50% of surface EGFRs internalized in 15 min) may correspond to a general cargo-handling capacity of the CME machinery, or may be limited by the retention of a fraction of surface EGFRs from entering clathrin-coated pits (Haslekås et al., 2005).
We further propose, as shown in Fig. 9 C, c, that Grb2 binding to activated EGFR prevents p38-induced internalization of ligand-bound EGFR, and that such internalization can be observed in Grb2-depleted cells, as demonstrated in Fig. 7, C and D. Multiple molecules of Grb2 can bind to a single EGFR (Fortian and Sorkin, 2014), which may sterically hinder the interaction of the EGFR dileucine motif with AP2, thus preventing p38-dependent endocytosis of EGF-activated receptors. Alternatively, rapid CME of ligand-activated EGFR facilitated by Grb2 may minimize the use of the p38/LL-dependent endocytosis because of the lag period in activation of the latter pathway (Tanaka et al., 2018).
In the presence of moderate concentrations of EGF, such as 10–20 ng/ml (Fig. 9 C, d), most receptors are ligand-bound, CME is at its plateau, and internalization through the CIE pathway is substantial. Under these conditions, the effect of TNFα on total EGFR internalization is virtually abolished (Fig. 9 A), suggesting that the p38 component of the EGFR CME is negligible, owing to the lack of free EGFR at the cell surface. An apparent decrease in p38-dependent CME of free EGFRs in the presence of TNFα was observed even with moderate (2–3 ng/ml) EGF concentrations (Fig. 9 A), when free receptors were still available at the cell surface and CIE was minimal. Under these conditions, an increased component of ligand-induced Grb2-dependent CME results in corresponding decrease in the contribution of p38-dependent CME due to the limited total capacity of EGFR CME. Grb2 depletion did not significantly inhibit EGFR endocytosis induced by 10 ng/ml EGF (Fig. 7, C and D), suggesting that Grb2 is not essential for EGFR CIE.
Interestingly, in the presence of high EGF concentrations (>30 ng/ml; Fig. 9 C, e; widely used in studies of EGFR trafficking and signaling), the functional contribution of CME in EGFR internalization is below the maximal capacity of CME as observed in Fig. 9 C, b and d. This observation suggests an “active” role of CIE competing with CME pathways for internalization of EGFR. Early studies by Wiley and co-workers proposed that at high concentrations of EGF, the bulk of receptors are internalized through a slow (presumably CIE) pathway with the kinetics comparable to a constitutive, basal internalization that does not require EGFR kinase activity (Lund et al., 1990). The clathrin independent nature of EGFR internalization induced by high EGF concentrations was later demonstrated by its insensitivity to the clathrin knockdown (Sigismund et al., 2005). Several mechanisms of the EGFR CIE have been proposed (Barbieri et al., 2000; Boucrot et al., 2015; Caldieri et al., 2017; Orth et al., 2006; Sigismund et al., 2013). Most of these mechanisms are known to require EGFR signaling, which, for example, leads to actin cytoskeleton rearrangement, membrane ruffling, and micropinocytosis. Such signaling processes are, however, triggered only by moderate or high EGF concentrations (Wiley, 2018). We therefore propose a modification to the model of the limited capacity of CME, in which we suggest that CIE is not simply an overflow constitutive pathway when CME is saturated, but that EGF stimulation of CIE is part of the mechanism limiting the number of receptors that are internalized through the CME pathway. Multiple CIE mechanisms of EGFR internalization have been observed in HeLa cells. Such redundancy together with the lack of inhibitors that specifically interfere with the CIE of EGFR precluded an efficient blockade of this component of internalization, which would be necessary for an additional test of our model.
Picomolar concentrations of EGFR ligands, at which p38-dependent internalization is substantial and may therefore influence signal transduction processes, are ubiquitously present in mammalian tissues. p38-dependent internalization of ligand-free receptors may result in rapid decrease of the plasma membrane EGFR level in cells expressing low levels of EGFR, and thus considerably reduce ligand binding and downstream signaling (Singhirunnusorn et al., 2007). On the other hand, our observation of internalization of free and ligand-bound EGFRs into the same endosomes suggests an increased stoichiometry of the receptor versus ligand in endosomes under conditions depicted in Fig. 9 C, a and b. This may lead to a shift of binding equilibrium toward ligand-receptor binding, which may extend the activity of receptors in endosomes (Tomas et al., 2015). Under such scenarios, and given the tendency of receptors internalized using the p38-dependent mechanisms to be recycled rather than sorted to lysosomes (Tanaka et al., 2018; Tomas et al., 2015), p38-dependent endocytosis may explain a relatively prolonged signaling in cells treated with low as compared with high EGF concentrations (Sigismund et al., 2008). It should be noted that EGF and cytokines do not induce p38-dependent endocytosis of EGFR in all cell types. Future comparative analysis of multiple types of cells with and without p38-dependent endocytosis may help to clarify the physiological role of this endocytic process in regulating EGFR signaling.
Materials and methods
Reagents
Recombinant human EGF was purchased from BD Biosciences. EGF-Rh was from Molecular Probes (Invitrogen). TNFα was from Miltenyi Biotec. Anisomycin (stored as 1 mM stock solution in DMSO at 4°C), BIRB796 (1 mM stock solution in DMSO stored at −20°C), SB202190 (stored as 10 mM stock in DMSO at 4°C), and erlotinib (10 mM stock solution stored at −20°C) were from Sigma-Aldrich.
Mouse monoclonal antibody to pTyr1068 of EGFR (pY1068; AB_331792) and to pThr180/Tyr182 of p38 (p-p38; AB_331296); and rabbit polyclonal antibodies to EGFR (AB_2246311), pSer1046/47 EGFR (pS1046/47; AB_331129), pThr202/Ser204 of ERK1/2 (pERK; AB_331768), pThr334 of MAPKAPK2 (pMAPKAPK2; AB_490936), and α-actinin (AB_2223798) were from Cell Signaling Technology. Rabbit polyclonal antibodies to CHC (AB_2083165) and mouse monoclonal antibody to GFP (AB_303395) were from Abcam. Rabbit polyclonal antibodies to EGFR (AB_631420), Grb2 (AB_631602), and Cbl (AB_2259627), and mouse monoclonal antibody to ubiquitin (AB_2762364) were from Santa Cruz Biotechnology. Monoclonal IgG2a antibody to α-adaptin (α subunit of AP2; AB_2056321) was from Thermo Fisher Scientific. Mouse monoclonal antibody to EEA1 (AB_397829) was from BD Biosciences. Rabbit polyclonal antibody to phosphoserine (pSer; AB_390205) was from Millipore. Mouse monoclonal antibody Mab528 to EGFR (AB_10978829) was from American Type Culture Collection. Rabbit polyclonal antibody to pSer1015 of EGFR (pS1015) was described previously (Tanaka et al., 2018). Mouse monoclonal antibody to Tac (Marks et al., 1996) was a gift of Dr. M. Marks (University of Pennsylvania, Philadelpha, PA). Secondary goat anti-mouse (AB_10956166), goat anti-mouse IgG2a (AB_2783643), and goat anti-rabbit (AB_621842) IRDye antibodies were from Li-COR. Secondary donkey anti-mouse conjugated to Cy5 (AB_2340820), Cy3 (AB_2315777), or FITC (AB_2335588) and donkey anti-rabbit conjugated to Alexa Fluor 488 (AB_2313584) were from Jackson ImmunoResearch. MG-B-Tau and MG-Bis-SA were synthesized in Dr. M. Bruchez’s laboratory (Carnegie Mellon University, Pittsburgh, PA).
Expression plasmids and site-directed mutagenesis
EGFR-GFP was described previously (Carter and Sorkin, 1998; Addgene; #32751). Human σ2 was amplified from the σ2-GFP plasmid provided by Dr. L. Traub (University of Pittsburgh, Pittsburgh, PA) using primers from Invitrogen that eliminated the stop codon and incorporated EcoRI and XmaI/SmaI restriction sites at the 5′ and 3′ ends of the ORF, respectively: 5′-CAAGCTTCGAATTCCGCCACCATGATCCGCTTTATCCTC-3′, and 5′-GATCCCGGGCCTCCAGGGACTGTAGCATCAGCAGCTGTTTC-3′.
The amplified product was digested by EcoRI and SmaI, and subcloned into pEGFP-N1. EGFR-GFP and σ2-GFP mutants were generated using the QuickChange site-directed mutagenesis kit (Agilent Technologies). Tac chimeras in pCDM8.1 plasmid, generously provided by Dr. M. Marks, were subcloned into pcDNA3.1 (+) using EcoRI and XbaI restriction sites. All constructs were confirmed by sequencing.
Cell culture
Gene-edited HeLa/FAP-EGFR cells were previously described (Larsen et al., 2019). The identity of HeLa cells was confirmed by short tandem repeats genotyping. They were cultured in DMEM containing 10% FBS and 100 U/ml penicillin/streptomycin (GIBCO BRL). Cells were starved in serum-free DMEM for 16 h before experimental treatments. PAE cells were grown in F-12/10% FBS. These cells were starved in serum-free F-12 for 1–2 h before experimental treatments. All pretreatments with inhibitors were performed in the starvation media at 37°C. All experimental treatments were performed in starvation media supplemented with 0.1% BSA at 37°C. The final concentration of DMSO was <0.5% in all assays. FAP-EGFR labeling with MG dyes was performed in starvation media at 37°C except for FAP-EGFR down-regulation assays as specified below.
DNA plasmid transfections and generation of stable cell lines
HeLa/FAP-EGFR and PAE cells were transfected with DNA plasmids using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s instructions. Experiments were performed 48 h after transfection. To generate cells stably expressing WT or mutants of EGFR-GFP, transfected cells were selected in the presence of 0.4 mg/ml Geneticin (G418).
siRNA transfection and rescue experiments
siRNAs (individual, ON-TARGET plus or siGENOME grade) were purchased from Dharmacon as follows: μ2: 5′-AAGUGGAUGCCUUUCGGGUCA-3′ (Huang et al., 2004); σ2 targeting 3′UTR: 5′-CCGUGUGUGUCCCGAGUAA-3′ (J-011833-09-0005); CHC: D-004001-02 (Huang et al., 2004); Grb2: 5′-CAUGUUUCCCCGCAAUUAUUUUU-3′ (Jiang et al., 2003); Cbl-b: 5′-GGACAGACGAAAUCUCACAUU-3′ (Huang et al., 2006); and c-Cbl: 5′-CCUCUCUUCCAAGCACUGAUU-3′ (Huang et al., 2006).
The specificity of siRNAs (except of siRNA to σ2) was validated previously (Huang et al., 2004; Jiang et al., 2003). HeLa/FAP-EGFR cells were reverse-transfected in 6-well dishes with 50 nM siRNA (final concentration) and 3.5 µl DharmaFECT-1 in 1 ml of complete media without antibiotics. 2 d later, cells were split and plated on 96-well plates, coverslips, and other dishes (for Western blot analysis). The next day, the cells were starved overnight and used for experiments on day 4 after siRNA transfection. In AP2σ-GFP rescue experiments, cells were replated into 12-well dishes 36 h after siRNA transfection and transfected with DNA plasmids 12 h after plating. The next day, the cells were split again and plated onto 96-well plates or coverslips, serum-starved overnight, and used for experiments on day 5 after siRNA transfection. PAE cells were transfected with 50 nM μ2 siRNA 24 h after plating. 2 d later, the cells were replated and 8 h later were again siRNA-transfected. 2 d later, the cells were replated and used for experiments 24 h later.
Immunoprecipitation and Western blotting
Cells were washed in ice-cold Ca2+, Mg2+-free PBS (CMF-PBS) and lysed in TGH lysis buffer (1% Triton X-100, 10% glycerol, 20 mM Hepes, pH 7.2, 150 mM NaCl, 2 mM EDTA, and 2 mM EGTA) supplemented with protease and phosphatase inhibitors (10 µg/ml aprotinin, 10 µg/ml of leupeptin, P8340 Sigma-Aldrich protease cocktail [1:1,000 dilution], 10 µM MG132, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, and 1 mM Na3VO4). Lysates were cleared by centrifugation at 16,000 ×g for 15 min at 4°C and electrophoresed. For immunoprecipitation, the lysis buffer was supplemented with 10 mM N-ethylmaleimide. Aliquots of lysates containing 1–1.5 mg protein were incubated with Mab528 (AB_10978829; 10 µg/sample), GFP (AB_303395; 4 µg/sample), pY1068 (AB_331792; 8 µl/sample) or pS1015 (9 µg/sample) for 3–5 h at 4°C, followed by incubation with 75 µl of protein A–Sepharose beads (Sigma-Aldrich) for 1–3 h at 4°C on nutator, except for pY1068 immunoprecipitation, which was incubated with 40 µl protein G-Sepharose beads (Invitrogen). Immunoprecipitates were washed in TGH and denatured in sample buffer at 95°C. Lysates and immunoprecipitates were resolved by SDS-PAGE (7.5% or 10% acrylamide) followed by transfer to nitrocellulose membrane and Western blotting. Secondary antibodies conjugated to far-red fluorescent dyes (IRDye-680 and -800, 1:10,000) were detected using an Odyssey Li-COR system. Quantifications were performed using Li-COR software.
FAP-EGFR labeling and single-cell imaging
Labeling and imaging of FAP-EGFR in HeLa/FAP-EGFR cells was described previously (Larsen et al., 2019; Perez Verdaguer et al., 2019). Briefly, cells were first incubated with 50 nM MG-B-Tau or 100 nM MG-Bis-SA in DMEM for 5 min at 37°C and then experimentally treated. To stop internalization, cells were washed with ice-cold CMF-PBS. MG-B-Tau–labeled cells were kept in cold CMF-PBS or fixed with freshly prepared 4% PFA for 15 min at RT. Cells labeled with MG-Bis-SA were placed in cold pH 8 buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, and 25 mM Hepes, pH 8) at 4°C. Live-cell imaging was immediately performed on a spinning-disk microscope.
Spinning-disk confocal microscopy
Images were acquired with a spinning-disk Marianas system based on a Zeiss Axio Observer Z1 inverted fluorescence microscope, equipped with CSU-W1 Yokogawa spinning disk; 405-, 436-, 488-, 515-, 561-, and 640-nm lasers; a 63× Plan Apo PH NA 1.4 oil immersion objective; an Evolve EM-CCD camera (Photometrics), a piezo stage controller, a spherical aberration correction module, and a temperature- and CO2-controlled chamber, all controlled by Slidebook6 software (Intelligent Imaging Innovation) as described previously (Larsen et al., 2019; Perez Verdaguer et al., 2019). Typically, a z-stack of 10 or 20 x-y confocal images was acquired for PAE or HeLa/FAP-EGFR cells, respectively, at 400-nm z-steps. Image acquisition settings were identical for all variants in each experiment.
Immunofluorescence staining
Cells grown on glass coverslips were fixed in freshly prepared 4% PFA for 15 min, washed with 3 mM glycine in CMF-PBS, permeabilized in 0.1% Triton X-100 CMF-PBS for 15 min, and blocked for 30 min in 0.05% Triton X-100 and 0.1% BSA in CMF-PBS, all at RT. Cells were then incubated for 1 h at RT with primary antibodies (EEA1 at 1:500, Tac at 1:50, or EGFR AB_2246311 at 1:500) and secondary antibodies (1:500) both in the blocking buffer. Finally, samples were washed and mounted in ProLong Gold antifade reagent (Invitrogen). To label cell-surface EGFR-GFP, PFA-fixed cells were stained with 1 µg/ml Mab528 for 1 h at RT in CMF-PBS/0.5% BSA followed by secondary Cy5-conjugated antibodies (1:500) and mounted as above.
Antibody-uptake endocytosis assay
To measure EGFR-GFP internalization rates, PAE/EGFR-GFP cells were incubated with 1 µg/ml Mab528 in starvation media for 10 min at RT, and then incubated in the same media at 37°C for indicated times. The cells were fixed with freshly prepared 4% PFA for 15 min at RT and washed in PBS. To maximally occupy surface-bound Mab528 and label surface EGFR-GFP, the cells were incubated with excessive concentration of Cy5-conjugated secondary antibody (1:50) for 1 h at RT in CMF-PBS/0.5% BSA. After washing, cells were fixed again, permeabilized with 0.1% Triton X-100 in CMF-PBS for 15 min at RT, and incubated with the same secondary antibody conjugated with Cy3 (1:500) in CMF-PBS/0.5% BSA for 45 min at RT to label internalized Mab528. Finally, samples were washed and mounted in ProLong Gold. 3D images were acquired through 488- (EGFR-GFP), 561- (Cy3), and 640-nm (Cy5) channels.
Image analysis
To quantify the amount of EGFR-GFP or untagged EGFR (WT and mutants) localized in early endosomes (EEA1-positive), a colocalization analysis was performed. 3D images of immunolabeled untagged EGFR (secondary Alexa Fluor 488–conjugated antibody; 488-nm channel) or EGFR-GFP–expressing cells, colabeled with EEA1 antibodies (secondary Cy5-conjugated antibody; 640-nm channel), were deconvolved using a constrained iterative algorithm of SlideBook6. A segment mask was then generated to select EEA1 endosomes detected through the 640-nm channel (EEA1 mask). Another segment mask was generated with the minimum threshold to include total cellular EGFR or EGFR-GFP fluorescence detected through the 488-nm channel (EGFR mask). For both masks, identical threshold parameters were used for all experimental variables. Objects smaller than 10 voxels were eliminated. A “Colocalization” mask was then generated to select voxels overlapping in EEA1 and EGFR masks. The sum fluorescence intensity of the 488-nm channel in the Colocalization mask was divided by the sum fluorescence intensity of the 488-nm channel in the EGFR mask in each image to calculate the fraction of total cellular EGFR/EGFR-GFP located in EEA1-containing endosomes.
To calculate the ratio of EGF-Rh and FAP-EGFR fluorescence in individual endosomes of cells labeled with MG-B-Tau and incubated with EGF-Rh, a segment mask was generated using background-subtracted 3D images through the 640-nm channel (MG-B-Tau:FAP-EGFR) with the low threshold that allows selection of the FAP-EGFR puncta and a minimal quantity of non–vesicular-shaped fluorescent objects. Under these experimental conditions, intracellular fluorescence puncta detectable by our spinning-disk confocal system represent endosomes. Identical threshold parameters were used for all experimental variables. Objects smaller than 10 voxels were eliminated. Non–vesicular-shaped objects at the cell periphery characteristic of the plasma membrane FAP-EGFR localization were manually removed from the mask. For each individual object (endosomes) of the final mask, the sum fluorescence intensity of the 561-nm channel (EGF-Rh) was divided by the sum fluorescence intensity of the 640-nm channel (FAP-EGFR) to obtain the value of EGF-Rh/FAP-EGFR ratio.
To estimate the total amount of internalized EGF-Rh in cells labeled with MG-B-Tau, a segment mask was created to select FAP-EGFR endosomes using the 640-nm channel (FAP-EGFR) as described above. The sum fluorescence intensity through the 561-nm channel (EGF-Rh) of the mask was normalized by the number of cells in the image. The values obtained in cells stimulated with 10 ng/ml TNFα with a similar pattern of predominant localization of FAP-EGFR in endosomes but not exposed to EGF-Rh were used to determine the mean intensity of the background autofluorescence detected through the 561-nm channel. These values were subtracted from the values of total 561-nm channel fluorescence intensity per cell obtained in EGF-Rh–treated cells to determine the specific EGF-Rh fluorescence intensity. Finally, values of specific 561-nm channel fluorescence per cell were normalized by those values obtained in cells treated with 10 ng/ml EGF.
To quantify the fraction of EGFR-GFP located at the cell surface at steady-state, 3D images of cells that were stained with Mab528 without membrane permeabilization were deconvoluted using a No Neighbors algorithm of SlideBook. Segment masks were then generated to select Cy5 fluorescence (Mab528) detected through the 640-nm channel (Surface mask) and GFP fluorescence detected through the 488-nm channel (Total mask). The sum fluorescence intensity of GFP (488-nm channel) in the Surface mask was divided by the sum fluorescence of GFP in the Total mask to determine the fraction of cell-surface EGFR-GFP.
To quantify the relative amounts of surface and internalized EGFR-GFP in antibody-uptake endocytosis assays, a segment mask was generated from background-subtracted 3D images using the minimal intensity threshold of the 640-nm channel to select Mab528 labeled Cy5-conjugated secondary antibody before cell permeabilization (Surface mask). Another segment mask was generated using the minimal intensity threshold of the 561-nm channel to select the vesicular fluorescence of Mab528 labeled with Cy3-conjugated secondary antibody after permeabilization (Internalized mask). Voxels in the Surface and Internalized masks that were not overlapping with GFP fluorescence (488-nm channel; EGFR-GFP) were eliminated from these masks. The sum fluorescence intensity of the 561-nm channel in the Internalized mask was then divided by the sum fluorescence intensity of the 640-nm channel in the Surface mask in each image to obtain the “Cy3/Cy5” ratio as the surrogate measure of EGFR-GFP internalization.
Image analyses were performed using SlideBook6 software. Quantifications of colocalization and antibody uptake rates in PAE cells were done per a field of view that typically displayed 5–10 cells. No gamma corrections were made in all images.
FAP-EGFR FERI internalization assay
The FERI internalization assay was performed in optical 96-well plates (Greiner bio-one μCLEAR) as described in Larsen et al. (2019) and Perez Verdaguer et al. (2019). Briefly, MG-Bis-SA was added to the final concentration of 100 nM in DMEM. Two or more wells were kept unlabeled to calculate the background fluorescence. After 5 min at 37°C, labeling medium was replaced by treatment medium. Endocytosis was stopped by placing plates on ice. The cells were washed with ice-cold CMF-PBS, placed in ice-cold pH 8 buffer, and imaged immediately at RT on a Nikon ECLIPSE Ti-E microscopy system with an A1 resonant scanner and a 40× 1.15 NA water immersion objective to capture and stitch four image fields of every well using a dual 561-nm/640-nm laser excitation and a single emission (700/75-nm) bypass filter. The Nikon NIS Elements HCA JOBS module automatically calculated the mean intensities of four fields. The ratio of background-subtracted mean fluorescence intensities emitted upon 561-nm and 640-nm excitation (561/640 ratio) was calculated in each image. The mean value of this ratio obtained in wells that were not treated with EGF, TNFα, or anisomycin (basal internalization) was subtracted from the values obtained in the rest of the wells.
FAP-EGFR down-regulation
HeLa/FAP-EGFR cells were grown in optical 96-well plates. Cells were treated with EGF and/or TNFα at 37°C, washed with cold CMF-PBS, and incubated with 100 nM MG-B-Tau in CMF-PBS for 10 min on ice. At least two wells were kept unlabeled to calculate background. Finally, the cells were washed, placed in cold CMF-PBS, and immediately imaged on a Nikon ECLIPSE Ti-E microscope as described above except single 640-nm excitation and 700/75-nm emission were used. Background-subtracted mean fluorescence intensity from four fields in each well was normalized by this intensity measured in wells that were not treated with EGF and/or TNFα.
125I-EGF internalization assay
Mouse receptor-grade EGF was 125I-conjugated, and the internalization assays were performed as described (Sorkin and Duex, 2010). Briefly, confluent HeLa/FAP-EGFR cells in 6-well plates were incubated with 125I-EGF for 0–10 min at 37°C in the absence or presence of unlabeled EGF (50–100-fold excess to estimate the background signal). When indicated, cells were pretreated with DMSO or BIRB796 for 1.5 h, or I125-EGF was applied in the presence of 10 ng/ml TNFα. Endocytosis was stopped by washing with ice-cold DMEM, and surface-bound I125-EGF was stripped by 5 min incubation with ice-cold 0.2 M sodium acetate buffer (pH 2.8). Cells were then lysed in 1 N NaOH for 1 h at 37°C to determine the amount of internalized I125-EGF. After background subtraction, the ratio of internalized and surface 125I-EGF was calculated, normalized to that value at a 10-min time point in control cells, and plotted against time.
Mass spectrometry
HeLa cells/FAP-EGFR grown in 100-mm dishes were left untreated or stimulated with 10 ng/ml TNFα or 100 nM anisomycin for 15 min at 37°C. FAP-EGFR was immunoprecipitated as described above. The TGH lysis buffer was additionally supplemented with 2 mM dithiothreitol and phosphor-STOP (Roche; 04906845001), and immunoprecipitation was performed with 90 µg Mab528 and 300 µl protein A-Sepharose beads per two 100-mm dishes in each treatment. Immunoprecipitates were resolved by 10% acrylamide SDS-PAGE, gels were Coomassie-stained, and FAP-EGFR bands were cut out. The gel bands were washed at RT shaking for 10–20 min in 1 ml of destaining buffer (50% acetonitrile and 50% 100 mM Tris-HCl, pH 8.5, in ultrapure water) several times (until the Coomassie dye was no longer visible in the gel). The gel pieces were dehydrated in 500 µl of acetonitrile for 10 min at RT. Acetonitrile was removed and gel pieces swelled in 300 µl digestion buffer (100 mM Tris-HCl, pH 8.5, and 2 mM CaCl2) with 300 ng trypsin. After digestion for 16-h shaking at 37°C, 300 ng chymotrypsin was added and shaken at 37°C for an additional 16 h. Then, peptides were extracted from gels as described previously (Paulo, 2016) and dried down in a vacuum concentrator, stage-tipped, and dried down again. Prior to liquid chromatography–tandem mass spectrometry analysis, the dried peptides were dissolved in 5% formic acid and 5% ACN and analyzed by a Q Exactive mass spectrometer (Thermo Fisher Scientific). The same samples were then analyzed using a parallel reaction monitoring–based targeted mass spectrometry method (Gallien et al., 2012) targeting to peptide FSSPSTSRTPL with two phosphorylation events (mol wt, 670.2702 D). Mass spectra were processed using a Comet-based pipeline (Eng et al., 2013). Spectra were converted to mzXML using a modified version of ReAdW.EXE. Database searching only included human EGFR sequence (downloaded July 5, 2020). This database was concatenated with one database composed of EGFR protein sequences in the reverse order plus common contaminants. Searches were performed using a 3D precursor ion tolerance for total protein–level analysis. The fragment ion tolerance was set to 0.02 D. Oxidation of methionine residues (+15.995 D) and phosphorylation on serine, tyrosine, and threonine (+79.966331 D) were set as variable modifications. Peptides were filtered by precursor accuracy ± 10 ppm, and Xcorr > 2. The area under the curve of the peptide (FSS#PSTS#RTPL,2+) in three samples was calculated from the MS2 spectrum using Skyline (MacLean et al., 2010).
Statistics
All statistical analyses were performed using GraphPad Prism software (GraphPad). For comparisons of two groups, the unpaired Student’s t test was used. For multiple comparison analyses, a one-way ANOVA followed by Tukey’s or Fisher’s least significant difference, multiple-comparison test was used. Differences were considered significant when the P value was <0.05, with the specific P values shown in the figures and detailed within each figure legend. Because P values are not informative and have no practical importance for comparing large datasets, they are not shown for comparing datasets obtained in the analyses of individual endosomes (Figs. 5 and 6).
Online supplemental material
Fig. S1 shows endocytosis of FAP-EGFR stimulated with EGF and TNFα using the FERI assay and single-cell imaging to set up experimental conditions for subsequent experiments. Fig. S2 shows efficiencies of siRNA knockdowns and σ2-GFP assembly into AP2 in cells transfected with σ2 siRNA. Fig. S3 provides characterization of PAE cells as a cell model for studying p38-dependent endocytosis of EGFR-GFP and its mutants. Fig. S4 shows cellular localization of EGFR-GFP with phosphomimetic mutations in addition to mutations described in Fig. 4. Fig. S5 shows that p38-induced phosphorylation of EGFR at Ser1015 is dependent on L1010/L1011 and independent on AP2. Table S1 contains detailed information about the phosphopeptides identified by mass spectrometry.
Acknowledgments
We are grateful to Drs. M. Bruchez, M. Marks, and L. Traub for generous gifts of reagents.
M. Perez Verdaguer is supported by the postdoctoral fellowship award from the National Cancer Center. A. Sorkin is supported by National Institutes of Health grants CA089151 and GM124186.
The authors declare no competing financial interests.
Author contributions: M. Perez Verdaguer: conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, and writing—review and editing. T. Zhang: investigation and writing—review and editing. J.A. Paulo: instrumentation. S. Gygi: instrumentation and methodology. S.C. Watkins: instrumentation, software, and writing—review and editing. H. Sakurai contributed resources and conceptualization. A. Sorkin contributed conceptualization, formal analysis, supervision, funding acquisition, visualization, validation, project administration, and writing—review and editing.

