


Rab11 GTPase proteins are required for cytokinesis, ciliogenesis, and lumenogenesis. Rab11a is critical for apical delivery of podocalyxin (PODXL) during lumen formation in epithelial cells. SH3BP5 and SH3BP5L are guanine nucleotide exchange factors (GEFs) for Rab11. We show that SH3BP5 and SH3BP5L are required for activation of Rab11a and cyst lumen formation. Using proximity-dependent biotin identification (BioID) interaction proteomics, we have identified SH3BP5 and its paralogue SH3BP5L as new substrates of the poly-ADP-ribose polymerase Tankyrase and the E3 ligase RNF146. We provide data demonstrating that epithelial polarity via cyst lumen formation is governed by Tankyrase, which inhibits Rab11a activation through the suppression of SH3BP5 and SH3BP5L. RNF146 reduces Tankyrase protein abundance and restores Rab11a activation and lumen formation. Thus, Rab11a activation is controlled by a signaling pathway composed of the sequential inhibition of SH3BP5 paralogues by Tankyrase, which is itself suppressed by RNF146.
Introduction
Rab11 proteins belong to the Rab family of the Ras superfamily of small GTPases and are encoded by three distinct genes, known as Rab11a, Rab11b, and Rab25 (Rab11c). These proteins are localized on recycling endosomes and regulate late recycling of cargo through these vesicles (Ullrich et al., 1996; Casanova et al., 1999; Schlierf et al., 2000). Like other GTPases, Rab proteins cycle between GDP and GTP and the GTP bound states. The Rab11 GTP bound form engages downstream effector signaling molecules, including Rab11FIP1, Rab11FIP2, Rab11FIP3, Rab11FIP4, and Rab11FIP5/Rip11 (Hales et al., 2001; Wallace et al., 2002). The nucleotide-binding state of small GTPases is regulated by guanine nucleotide exchange factors (GEFs), which facilitate GDP to GTP exchange and GTPase activating proteins, which promote GTP hydrolysis (Bourne et al., 1991). Rab11 proteins are targeted to vesicle membranes by geranylgeranylation of two cysteine residues located in their C terminus, which is catalyzed by geranylgeranyltransferase II (Pfeffer and Aivazian, 2004). Rab11 proteins facilitate several biological processes, including cytokinesis, neurite formation, lumenogenesis, and ciliogenesis (Wilson et al., 2005; Shirane and Nakayama, 2006; Bryant et al., 2010; Knödler et al., 2010; Westlake et al., 2011).
Rab11a initiates a signaling cascade during lumenogenesis and primary ciliogenesis, where Rab11a-GTP binds the Rab8 GEF Rabin8 and stimulates its GEF activity toward Rab8 (Bryant et al., 2010; Knödler et al., 2010). Rab11a facilitates the apical transport of podocalyxin (PODXL) from the basal membrane to the apical membrane during lumen formation (Bryant et al., 2010); however, the identities of the proteins that regulate Rab11a activation during these processes have not been determined.
SH3BP5 and its paralogue SH3BP5L have been identified as GEFs for Rab11 (Sakaguchi et al., 2015; Jenkins et al., 2018; Goto-Ito et al., 2019). We sought to elucidate the upstream signaling events that regulate SH3BP5 and SH3BP5L, and hence Rab11, activity during lumenogenesis. We show that SH3BP5 and SH3BP5L are required for Rab11a activation during lumen formation in an MDCK cyst model. We characterized SH3BP5 and SH3BP5L as novel substrates of the poly-ADP-ribose (PAR) polymerases, Tankyrase-1 and 2 (TNKS/TNKS2), as well as the E3-ligase RNF146. We show that Tankyrase suppressed SH3BP5 and SH3BP5L function, which resulted in diminished Rab11a activity and impaired lumen formation in epithelial cells. Finally, we demonstrated that RNF146 destabilized Tankyrase protein levels, thereby enabling the SH3BP5 and SH3BP5L GEFs to activate Rab11a during lumenogenesis.
Results
SH3BP5 and SH3BP5L are Rab11a GEFs that mediate nucleotide exchange through a novel GEF domain
We characterized the interactions between SH3BP5 or SH3BP5L and Rab11a or Rab11a mutants reported to stabilize the GDP- or GTP-bound forms. GST-SH3BP5 or GST-SH3BP5L were coexpressed with either GFP-Rab11a, GFP-Rab11a-S25N (SN—a GDP-locked mutant) or GFP-Rab11a-Q70L (QL—a GTP-locked mutant defective for hydrolysis) in HEK293T cells (Ullrich et al., 1996). Rab11a immune complexes were immunoblotted for GST-SH3BP5 or GST-SH3BP5L. Both SH3BP5 and SH3BP5L showed enhanced binding to Rab11a-GDP (SN) relative to the WT or GTP-bound form (QL; Fig. 1 A), consistent with the behavior of other GEFs. To evaluate the GEF activity of SH3BP5 and SH3BP5L, we developed a real-time nucleotide exchange assay using real-time nuclear magnetic resonance (NMR) to analyze the kinetics of Rab11a nucleotide exchange. 15N-labeled Rab11a was monitored by collecting 1H-15N heteronuclear single quantum coherence (HSQC) spectra during the time course of nucleotide exchange, and two specific pairs of peaks that are specific to Rab11a-GDP or Rab11a-GTP were identified and used to monitor the exchange reaction and determine exchange rates (Fig. 1 B). Recombinant SH3BP5 or SH3BP5L were added to determine whether they accelerate nucleotide exchange. Both proteins enhanced the nucleotide exchange rate approximately fourfold when added at a GEF:GTPase molar ratio of 1:6,000 (Fig. 1 C), indicating that SH3BP5 and SH3BP5L are potent GEFs for Rab11a. These data provide independent evidence for the exchange activity of SH3BP5 and SH3BP5L toward Rab11a.

SH3BP5 and SH3BP5L are Rab11a GEFs that mediate exchange through a novel GEF domain. (A) HEK293T cells were coexpressed with GFP-Rab11a WT, S25N (SN), Q70L (QL), and GST-SH3BP5 (left) or GST-SH3BP5L (right) and lysates were immunoprecipitated with GFP antibody. Immune complexes were subjected to Western blot analysis and probed with GST and GFP antibodies. (B) Overlay of 1H-15N-HSQC spectra of 15N-Rab11a in its GDP (black) and GTP (red) bound forms. The two pairs of nucleotide-specific peaks monitored during nucleotide exchange are highlighted in black boxes. (C) Comparison of nucleotide exchange rates of Rab11a between its intrinsic activity and with addition of recombinant full-length SH3BP5 or SH3BP5L. The GEF:GTPase molar ratio is 1:6,000. Data represent mean ± SD of three independent experiments. (D) Protein domain maps of SH3BP5 and SH3BP5L. Green boxes represent the conserved four α-helices predicted by the PSIPRED server. (E) Left: GST-tagged full-length and deletions of SH3BP5 and SH3BP5L coexpressed with GFP-Rab11a S25N in HEK293T cells. Lysates were immunoprecipitated with GFP antibody and immune complexes were analyzed by Western blotting and probed with GST and GFP antibodies. Right: Illustration of each expression construct with helices highlighted as red boxes. (F) Nucleotide exchange rates of Rab11a in the presence of recombinant full-length SH3BP5 and SH3BP5L versus their respective truncated minimal two-helix catalytic domain constructs. Data represent mean ± SD of three independent experiments. Molecular weights (kD) for blots are indicated on the left of each panel.
SH3BP5 and SH3BP5L are Rab11a GEFs that mediate exchange through a novel GEF domain. (A) HEK293T cells were coexpressed with GFP-Rab11a WT, S25N (SN), Q70L (QL), and GST-SH3BP5 (left) or GST-SH3BP5L (right) and lysates were immunoprecipitated with GFP antibody. Immune complexes were subjected to Western blot analysis and probed with GST and GFP antibodies. (B) Overlay of 1H-15N-HSQC spectra of 15N-Rab11a in its GDP (black) and GTP (red) bound forms. The two pairs of nucleotide-specific peaks monitored during nucleotide exchange are highlighted in black boxes. (C) Comparison of nucleotide exchange rates of Rab11a between its intrinsic activity and with addition of recombinant full-length SH3BP5 or SH3BP5L. The GEF:GTPase molar ratio is 1:6,000. Data represent mean ± SD of three independent experiments. (D) Protein domain maps of SH3BP5 and SH3BP5L. Green boxes represent the conserved four α-helices predicted by the PSIPRED server. (E) Left: GST-tagged full-length and deletions of SH3BP5 and SH3BP5L coexpressed with GFP-Rab11a S25N in HEK293T cells. Lysates were immunoprecipitated with GFP antibody and immune complexes were analyzed by Western blotting and probed with GST and GFP antibodies. Right: Illustration of each expression construct with helices highlighted as red boxes. (F) Nucleotide exchange rates of Rab11a in the presence of recombinant full-length SH3BP5 and SH3BP5L versus their respective truncated minimal two-helix catalytic domain constructs. Data represent mean ± SD of three independent experiments. Molecular weights (kD) for blots are indicated on the left of each panel.
Secondary structure prediction (PSIPRED) of the N termini of SH3BP5 and SH3BP5L suggested the presence of highly helical regions, comprising four separate α-helices that may be structurally similar to known α-helical Rab GEF domains, like Sec2p and Rabin8/GRAB, which form dimeric coiled-coil domains (Fig. 1 D; Dong et al., 2007; Guo et al., 2013; Buchan et al., 2013); however, there is not sufficient conservation of sequence to conclude that they are homologous. To elucidate which helical structures of SH3BP5/SH3BP5L are required for binding, deletion analysis was performed. A series of SH3BP5 and SH3BP5L constructs with deleted helices were expressed and tested for their ability to bind to GFP-Rab11a-S25N as described above. These deletion studies showed that helix 1 and helix 4 in SH3BP5 and SH3BP5L were both required for interaction with Rab11a-GDP (S25N; Fig. 1 E). The minimal binding unit contained helix 1 and helix 4 (1–280Δ100-207 for SH3BP5; 1–299Δ115-221 for SH3BP5L), and these recombinant truncations were also sufficient to mediate nucleotide exchange for Rab11a. Notably, the truncated version of SH3BPL exhibited enhanced exchange activity compared with full-length protein (compared at the same molar ratio), suggesting that helices 2 and 3 may have a negative regulatory function regulating GEF activity (Fig. 1 F), whereas the full-length and truncated versions of SH3BP5 exhibited similar activity. While this manuscript was in preparation, a crystal structure of SH3BP5 in complex with Rab11a was reported, which confirmed that these two helices form the interface with Rab11a (Jenkins et al., 2018; Goto-Ito et al., 2019). These results substantiate that SH3BP5 and SH3BP5L are Rab11a GEFs and mediate catalysis through a novel two-helix GEF domain.
SH3BP5 and SH3BP5L are required for Rab11a activation during lumen formation in MDCK cysts
Rab11a is required for the transport of PODXL from the basal membrane to the apical membrane during lumen formation (Bryant et al., 2010). We therefore queried whether the Rab11a GEFs SH3BP5 and SH3BP5L may play a role in promoting lumenogenesis. We first tested whether SH3BP5 or SH3BP5L could stimulate Rab11a activation by creating MDCKII cells stably expressing RFP fusion proteins (Tag-RFP-T) of SH3BP5 and SH3BP5L. Both proteins showed cytoplasmic and vesicular localization when grown in 2D cell culture (Fig. 2 A). SH3BP5 and SH3BPL increased Rab11a activation in MDCKII cells, assessed by estimating Rab11a-GTP levels using a Rab11a-GTP pull-down assay based on the Rab11-binding domain from the effector protein Rab11FIP3 (FIP3RBD), which specifically captures the GTP-bound form of Rab11a (Fig. 2, B and C). Overexpressed SH3BP5 and SH3BP5L localized below the apical membrane (labeled by PODXL) in all cysts (Fig. 2 D). Expression of either SH3BP5 or SH3BP5L increased the percentage of cysts with a single well-formed lumen in MDCK cells cultured in Matrigel and increased Rab11a staining below the lumen compared with control cells (Fig. 2, D–F). We also observed that 79.4% ± 9.1% of RFPT-SH3BP5 and 70.6% ± 9.9% of RFPT-SH3BP5L colocalized with Rab11a in cysts below the formed lumen (Fig. 2 G).

SH3BP5 and SH3BP5L are required for Rab11a activation during lumen formation in MDCK cysts. (A) 2D confocal images of MDCKII cells expressing Tag-RFP-T-SH3BP5 and Tag-RFP-T-SH3BP5L. 63× magnification and scale bar represents 10 µm. (B) Rab11a-GTP analysis of Tag-RFP-T-SH3BP5/SH3BP5L expression cells. 500 µg of lysate was immunoprecipitated with 20 µg GST-FIP3RBD, and immune complexes were analyzed by Western blot with Rab11a antibody. Lysates were examined for SH3BP5, SH3BP5L, and Rab11a expression. (C) Quantification of Rab11a-GTP pulldowns by ImageJ. Values were normalized to pMX control Rab11a-GTP levels. Data represent mean ± SD of three independent experiments. (D) Representative confocal images of 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (E) Quantification of single lumen cysts normalized to pMX control single lumen rate. Data represent the mean ± SD of three independent experiments. *, P > 0.05. pMX n = 397, RFPT 3BP5 n = 395, RFPT 3BP5L n = 367. (F) Representative confocal images of 4-d cysts that were incubated with Rab11a antibody. Colocalization channel was built using Coloc tool in Imaris. Scale bar = 10 µm; 63× magnification. (G) Quantification of colocalization of Tag-RFP-T-SH3BP5/SH3BP5L with Rab11a. Individual colocalization values and mean ± SD of 32 cysts expressing RFPT-SH3BP5 and 26 cysts expressing RFPT-SH3BP5L pooled from three independent experiments were plotted using GraphPad Prism 8. (H) Western blot analysis of MDCKII CRISPR generated single-knockout (KO) and double-knockout (dKO) cell lines for SH3BP5 and SH3BP5L. Lysates were examined for SH3BP5 and SH3BP5L protein expression, while α-tubulin serves as a loading control. (I) Rab11a-GTP pulldown analysis of SH3BP5/SH3BP5L double- and single-knockout MDCKII cells. Experiment was performed as in Fig. 2 B. (J) Quantification of Rab11a-GTP levels normalized to pLCKO control cells. Data represent mean ± SD of three independent experiments. (K) Representative confocal image of SH3BP5/SH3BP5L double-knockout MDCKII cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (L) Quantification of cysts with single lumens between SH3BP5/SH3BP5L double-knockout and single-knockout MDCKII cells. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 359, cl14 n = 356, cl26 n = 314, SH3BP5 KO n = 315, SH3BP5L KO n = 314. Molecular weights (kD) for blots are indicated on the left of each panel.
SH3BP5 and SH3BP5L are required for Rab11a activation during lumen formation in MDCK cysts. (A) 2D confocal images of MDCKII cells expressing Tag-RFP-T-SH3BP5 and Tag-RFP-T-SH3BP5L. 63× magnification and scale bar represents 10 µm. (B) Rab11a-GTP analysis of Tag-RFP-T-SH3BP5/SH3BP5L expression cells. 500 µg of lysate was immunoprecipitated with 20 µg GST-FIP3RBD, and immune complexes were analyzed by Western blot with Rab11a antibody. Lysates were examined for SH3BP5, SH3BP5L, and Rab11a expression. (C) Quantification of Rab11a-GTP pulldowns by ImageJ. Values were normalized to pMX control Rab11a-GTP levels. Data represent mean ± SD of three independent experiments. (D) Representative confocal images of 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (E) Quantification of single lumen cysts normalized to pMX control single lumen rate. Data represent the mean ± SD of three independent experiments. *, P > 0.05. pMX n = 397, RFPT 3BP5 n = 395, RFPT 3BP5L n = 367. (F) Representative confocal images of 4-d cysts that were incubated with Rab11a antibody. Colocalization channel was built using Coloc tool in Imaris. Scale bar = 10 µm; 63× magnification. (G) Quantification of colocalization of Tag-RFP-T-SH3BP5/SH3BP5L with Rab11a. Individual colocalization values and mean ± SD of 32 cysts expressing RFPT-SH3BP5 and 26 cysts expressing RFPT-SH3BP5L pooled from three independent experiments were plotted using GraphPad Prism 8. (H) Western blot analysis of MDCKII CRISPR generated single-knockout (KO) and double-knockout (dKO) cell lines for SH3BP5 and SH3BP5L. Lysates were examined for SH3BP5 and SH3BP5L protein expression, while α-tubulin serves as a loading control. (I) Rab11a-GTP pulldown analysis of SH3BP5/SH3BP5L double- and single-knockout MDCKII cells. Experiment was performed as in Fig. 2 B. (J) Quantification of Rab11a-GTP levels normalized to pLCKO control cells. Data represent mean ± SD of three independent experiments. (K) Representative confocal image of SH3BP5/SH3BP5L double-knockout MDCKII cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (L) Quantification of cysts with single lumens between SH3BP5/SH3BP5L double-knockout and single-knockout MDCKII cells. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 359, cl14 n = 356, cl26 n = 314, SH3BP5 KO n = 315, SH3BP5L KO n = 314. Molecular weights (kD) for blots are indicated on the left of each panel.
To address the requirement of SH3BP5 and SH3BP5L to mediate lumen formation, we generated single- and double-knockout MDCKII cells by CRISPR/Cas9 gene editing (Fig. 2 H). We observed that Rab11a-GTP levels were reduced by 40–50% in either SH3BP5 or SH3BP5L single-knockout cells with a further reduction of Rab11a-GTP levels by 70–75% when both SH3BP5 and SH3BP5L were knocked out in MDCKII cells (Fig. 2, I and J). These data demonstrate that SH3BP5 and SH3BP5L are major contributors to Rab11a nucleotide exchange in epithelial cells and suggests the potential presence of other GEFs that contribute to the basal levels of Rab11a GTP in the absence of both SH3BP5 paralogues (Fig. 2, I and J). We next examined the ramifications of both single and double knockout of SH3BP5 and SH3BP5L on cyst formation in MDCKII cells. Double-knockout cells were impaired in their ability to form single-lumen cysts compared with single-knockout cells (Fig. 2, K and L), which is consistent with decreased levels of Rab11a-GTP observed in double-knockout cells (Fig. 2, I and J). These data show that both SH3BP5 and SH3BP5L contribute to Rab11a activation in epithelial cells, and that this is required for lumenogenesis.
SH3BP5 and SH3BP5L are Tankyrase substrates
To elucidate potential upstream regulators of SH3BP5, we used proximity-dependent biotin identification (BioID) to identify the ensemble of SH3BP5-interacting proteins in HEK293 Flp-In cells using FlagBirA*-tagged SH3BP5 as bait (Fig. S1). 36 high-confidence interactors were identified and 10 SH3BP5 proximity proteins were related to vesicle trafficking including Rab11a (Fig. 3 A and Table S1). Among the list of interactors were members of the PAR polymerase family of Tankyrases, TNKS and TNKS2 (Table S1). Tankyrases regulate protein abundance through poly-ADP-ribosylation, or PARylation, of its substrates, which triggers subsequent ubiquitylation and degradation by the E3 ligase RNF146 (Zhang et al., 2011; Callow et al., 2011). We hypothesized that SH3BP5 could be a novel Tankyrase substrate since it contains a conserved Tankyrase binding motif RxxxxG and was observed in several independent Tankyrase immunoprecipitation mass spectrometry experiments (Guettler et al., 2011; Wang et al., 2015; Li et al., 2017; Huttlin et al., 2015; Bhardwaj et al., 2017). SH3BP5 contains two Tankyrase binding motifs, and its paralogue SH3BP5L contains three putative binding sites (Fig. 3 B).

Generation of HEK293T Flp-In SH3BP5 BioID cells. HEK293T Flp-In cell line validation for expression of SH3BP5. Flag-BirA expression was induced with 1 µg/ml tetracycline and 50 µM biotin for 24 h. Whole-cell lysates were subjected to Western blot analysis and examined for Flag expression, while protein biotinylation was detected using streptavidin-HRP. Molecular weights (kD) for blots are indicated on the left.
Generation of HEK293T Flp-In SH3BP5 BioID cells. HEK293T Flp-In cell line validation for expression of SH3BP5. Flag-BirA expression was induced with 1 µg/ml tetracycline and 50 µM biotin for 24 h. Whole-cell lysates were subjected to Western blot analysis and examined for Flag expression, while protein biotinylation was detected using streptavidin-HRP. Molecular weights (kD) for blots are indicated on the left.

SH3BP5 and SH3BP5L are Tankyrase substrates. (A) Interaction network of significant SH3BP5 interactors identified by BioID. The interaction network was created using Cytoscape and grouped by biological function using the Gene Ontology database. Hexagon size indicates the spectrum sum of each BioID run. Solid black lines indicate known protein interactions imported from the GeneMANIA plugin and dotted lines represent new interactions identified by BioID. (B) Protein domain maps of SH3BP5 and SH3BP5L. Green boxes represent the conserved four α-helices predicted by the PSIPRED server. (C and D) HEK293T lysates coexpressing Flag-SH3BP5 or Flag-SH3BP5L and Myc-TNKS or Myc-TNKS2 were immunoprecipitated with Myc (9E10) antibody and immune complexes were subjected to Western blot analysis and probed for Flag and Myc expression. Whole-cell lysates were examined for Flag and Myc expression and α-tubulin serves as a loading control. (E and F) Lysates from HEK293T coexpressing Flag-SH3BP5 or Flag-SH3BP5L or respective individual or combined TBM deletion mutants (SH3BP5 - Δ1; Δ2; Δ1,2 or SH3BP5L - Δ1; Δ2; Δ3; Δ1,2; Δ1,3; Δ2,3; Δ1,2,3) with Myc-TNKS or Myc-TNKS2. Lysates were immunoprecipitated with Flag antibody and complexes were subjected to Western blot analysis for Myc and Flag expression. Whole-cell lysates were examined for Myc and Flag expression and α-tubulin serves as a loading control. (G and H) HEK293T were coexpressed with Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS or Myc-TNKS2. Lysates were immunoprecipitated with Flag antibody and immune complexes were washed and incubated with 10 µM Biotin-NAD for 1 h. This mixture was subjected to Western analysis and PARylation was detected by streptavidin-HRP. Complexes were also probed for Myc and Flag expression while whole-cell lysates were examined for Myc and Flag and α-tubulin serves as a loading control. * indicates TNKS/TNKS2 bands and → indicates SH3BP5 bands. (I and J) Same as G and H, but cells were treated with and without 5 µM TNKS656 inhibitor for 24 h before lysis. * indicates TNKS/TNKS2 bands and → indicates SH3BP5/SH3BP5L bands. TBM, Tankyrase binding Motif. Molecular weights (kD) for blots are indicated on the left of each panel.
SH3BP5 and SH3BP5L are Tankyrase substrates. (A) Interaction network of significant SH3BP5 interactors identified by BioID. The interaction network was created using Cytoscape and grouped by biological function using the Gene Ontology database. Hexagon size indicates the spectrum sum of each BioID run. Solid black lines indicate known protein interactions imported from the GeneMANIA plugin and dotted lines represent new interactions identified by BioID. (B) Protein domain maps of SH3BP5 and SH3BP5L. Green boxes represent the conserved four α-helices predicted by the PSIPRED server. (C and D) HEK293T lysates coexpressing Flag-SH3BP5 or Flag-SH3BP5L and Myc-TNKS or Myc-TNKS2 were immunoprecipitated with Myc (9E10) antibody and immune complexes were subjected to Western blot analysis and probed for Flag and Myc expression. Whole-cell lysates were examined for Flag and Myc expression and α-tubulin serves as a loading control. (E and F) Lysates from HEK293T coexpressing Flag-SH3BP5 or Flag-SH3BP5L or respective individual or combined TBM deletion mutants (SH3BP5 - Δ1; Δ2; Δ1,2 or SH3BP5L - Δ1; Δ2; Δ3; Δ1,2; Δ1,3; Δ2,3; Δ1,2,3) with Myc-TNKS or Myc-TNKS2. Lysates were immunoprecipitated with Flag antibody and complexes were subjected to Western blot analysis for Myc and Flag expression. Whole-cell lysates were examined for Myc and Flag expression and α-tubulin serves as a loading control. (G and H) HEK293T were coexpressed with Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS or Myc-TNKS2. Lysates were immunoprecipitated with Flag antibody and immune complexes were washed and incubated with 10 µM Biotin-NAD for 1 h. This mixture was subjected to Western analysis and PARylation was detected by streptavidin-HRP. Complexes were also probed for Myc and Flag expression while whole-cell lysates were examined for Myc and Flag and α-tubulin serves as a loading control. * indicates TNKS/TNKS2 bands and → indicates SH3BP5 bands. (I and J) Same as G and H, but cells were treated with and without 5 µM TNKS656 inhibitor for 24 h before lysis. * indicates TNKS/TNKS2 bands and → indicates SH3BP5/SH3BP5L bands. TBM, Tankyrase binding Motif. Molecular weights (kD) for blots are indicated on the left of each panel.
To validate the interaction between Tankyrase and SH3BP5 observed by BioID, HEK293T cells were cotransfected with Flag-SH3BP5 and Myc-TNKS or Myc-TNKS2 expression vectors. Cell lysates were immunoprecipitated with anti-Myc agarose beads and probed for Flag-SH3BP5 by immunoblot analysis. SH3BP5 was detected in immunoprecipitates of both TNKS and TNKS2 (Fig. 3 C). We observed that the abundance of TNKS but not TNKS2 protein, increased dramatically upon coexpression of SH3BP5, suggesting a unique regulatory function of SH3BP5 proteins in controlling the steady levels of TNKS (Fig. 3 C). Similar results were observed for the interaction between SH3BP5L and TNKS or TNKS2 (Fig. 3 D). Each of the Tankysase binding motifs were deleted alone or in combination in SH3BP5/SH3BP5L, and these constructs were tested for interaction with TNKS or TNKS2. Whereas both Tankyrase binding motifs had to be deleted in SH3BP5 to abolish Tankyrase binding, loss of either of the two motifs in the N terminus of SH3BP5L, together with deletion of the third motif, was sufficient to uncouple SH3BP5L from TNKS/TNKS2 (Fig. 3, E and F). Loss of TNKS interaction with SH3BP5/SH3BP5L decreased the abundance of TNKS protein, suggesting an unanticipated regulatory function of SH3BP5 and SH3BP5L in controlling TNKS protein levels, but not TNKS2 (Fig. 3, E and F).
SH3BP5 and SH3BP5L were next tested for their ability to serve as Tankyrase substrates using an in vitro PARylation assay. Flag-SH3BP5 or Flag-SH3BP5L were immunoprecipitated from HEK293T cells coexpressing Myc-TNKS or Myc-TNKS2. Biotin-tagged NAD+, which is used as a source for PAR that can be transferred to target proteins by Tankyrases, was incubated with anti-Flag immune complexes that were then subjected to Western blot analysis. Biotin-NAD was detected by streptavidin-HRP as an indicator for PARylation. Both SH3BP5 and SH3BP5L proteins exhibited strong modification by PARylation, and Tankyrase auto-PARylation was also observed (Fig. 3, G and H). To ensure that the PAR modification was specific to the PAR polymerase catalytic activity of Tankyrase, a Tankyrase-specific inhibitor (TNKS656) was added to HEK293T cells for 24 h after transfection. Comparison of treated and untreated samples showed that the inhibitor abolished the PARylation signal (Fig. 3, I and J). These results demonstrated SH3BP5 and SH3BP5L are bona fide Tankyrase binders and substrates in transfected cellular model systems.
SH3BP5 and SH3BP5L are RNF146 substrates
The established mechanism of PARylation-dependent ubiquitylation (PARdU) involves recognition of iso-ADP-ribose on Tankyrase substrates by the WWE domain of RNF146 (Wang et al., 2012). This interaction induces a conformational change in the RING domain of RNF146 that facilitates the transfer of ubiquitin molecules from the E2 enzyme to the PARylated target (DaRosa et al., 2015).
We identified SH3BP5 as a potential RNF146-interacting protein using BioID- and FlagBirA*-tagged RNF146 as bait (Fig. S2 and Table S2). To validate the interaction between SH3BP5 and RNF146, HEK293T cells were cotransfected with the following combination of expression constructs: Flag-SH3BP5 with Myc-TNKS and HA-RNF146. A similar transfection combination was performed with Flag-SH3BP5L, Myc-TNKS, and HA-RNF146. Flag-SH3BP5 was immunoprecipitated using Flag-agarose beads and subjected to immunoblotting for HA-RNF146. SH3BP5 bound to RNF146 only in the presence of either TNKS. A point mutation within the RNF146 WWE domain (R163A) has been shown to disrupt the interaction between RNF146 and PARylated protein (Zhang et al., 2011). SH3BP5 did not bind to RNF146 when the WWE domain was mutated (R163A or RA), demonstrating the requirement for a functional WWE domain to mediate the interaction (Fig. 4 A). The protein abundance of SH3BP5 was decreased in the presence of TNKS and RNF146, suggesting that all three proteins form a degradation complex. SH3BP5 and Tankyrase protein levels were unaffected when expressed with the RNF146 WWE domain mutant (RA), demonstrating the importance of PAR recognition by RNF146 to trigger proteolysis. Similar findings were observed with SH3BP5L (Fig. 4 B).
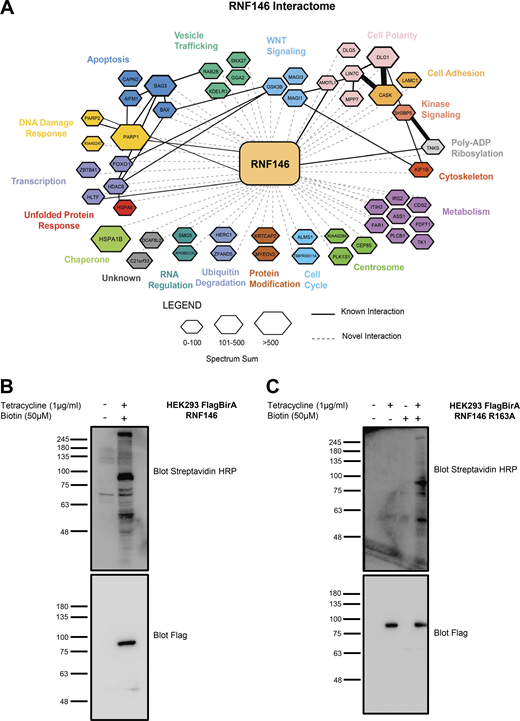
Elucidation of RNF146 interactome by BioID. (A) Representation of all significant RNF146 interactors from each BioID sample. The interaction network was created using Cytoscape and grouped by biological function using the Gene Ontology database (http://www.geneontology.org). The hexagon size indicates the spectral sum of peptides obtained from each BioID run. Solid black lines indicate known protein interactions imported from the GeneMANIA plugin and dotted lines represent new interactions identified by BioID. To identify PARylated proteins, which bound to RNF146 through the WWE domain, we generated a WWE domain loss-of-function RNF146 mutant (R163A) that is unable to bind to PAR. To identify proteins whose steady-state levels are regulated by RNF146-mediated ubiquitylation, we examined RNF146 binding proteins whose abundance increased in the presence the proteasome inhibitor MG132. Therefore, four experimental conditions were interrogated by BioID: WT RNF146 in the absence or presence of MG132 compared with the WWE mutant RNF146-R163A in the absence or presence of MG132. 51 significant proteins were identified that satisfied statistical testing (SAINT cutoff of 0.92) in at least one of the four samples (Table S2). (B and C) HEK293T Flp-In cell line validation for expression of Flag-BirA RNF146 and RNF146 R163A. Flag-BirA expression was induced with 1 µg/ml tetracycline and 50 µM biotin for 24 h. Whole-cell lysates were subjected to Western blot analysis and examined for Flag expression, while protein biotinylation was detected using streptavidin-HRP. Molecular weights (kD) for blots are indicated on the left of each panel.
Elucidation of RNF146 interactome by BioID. (A) Representation of all significant RNF146 interactors from each BioID sample. The interaction network was created using Cytoscape and grouped by biological function using the Gene Ontology database (http://www.geneontology.org). The hexagon size indicates the spectral sum of peptides obtained from each BioID run. Solid black lines indicate known protein interactions imported from the GeneMANIA plugin and dotted lines represent new interactions identified by BioID. To identify PARylated proteins, which bound to RNF146 through the WWE domain, we generated a WWE domain loss-of-function RNF146 mutant (R163A) that is unable to bind to PAR. To identify proteins whose steady-state levels are regulated by RNF146-mediated ubiquitylation, we examined RNF146 binding proteins whose abundance increased in the presence the proteasome inhibitor MG132. Therefore, four experimental conditions were interrogated by BioID: WT RNF146 in the absence or presence of MG132 compared with the WWE mutant RNF146-R163A in the absence or presence of MG132. 51 significant proteins were identified that satisfied statistical testing (SAINT cutoff of 0.92) in at least one of the four samples (Table S2). (B and C) HEK293T Flp-In cell line validation for expression of Flag-BirA RNF146 and RNF146 R163A. Flag-BirA expression was induced with 1 µg/ml tetracycline and 50 µM biotin for 24 h. Whole-cell lysates were subjected to Western blot analysis and examined for Flag expression, while protein biotinylation was detected using streptavidin-HRP. Molecular weights (kD) for blots are indicated on the left of each panel.

SH3BP5 and SH3BP5L are RNF146 substrates. (A and B) Lysates from HEK293T coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 or R163A mutant (RA) were immunoprecipitated with Flag antibody and subjected to Western blot analysis. Immune complexes and lysates were probed with HA, Myc, and Flag antibodies, while α-tubulin serves as a loading control. (C and D) HEK293T cells coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 WT, ΔRING (ΔR), and R163A (RA) were treated with 10 µM MG132 4 h before lysis. Lysates were immunoprecipitated with Flag antibody and immune complexes were subjected to Western blot analysis and examined for ubiquitin lysine 48 linkages. Complexes and lysates were also examined for HA, Flag, and Myc expression while α-tubulin serves as a loading control. (E and F) HEK293T cells coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 were treated with and without 10 µM MG132 for 4 h before lysis. Cell lysates were examined for HA, Flag, and Myc expression, while α-tubulin serves as a loading control. Molecular weights (kD) for blots are indicated on the left of each panel.
SH3BP5 and SH3BP5L are RNF146 substrates. (A and B) Lysates from HEK293T coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 or R163A mutant (RA) were immunoprecipitated with Flag antibody and subjected to Western blot analysis. Immune complexes and lysates were probed with HA, Myc, and Flag antibodies, while α-tubulin serves as a loading control. (C and D) HEK293T cells coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 WT, ΔRING (ΔR), and R163A (RA) were treated with 10 µM MG132 4 h before lysis. Lysates were immunoprecipitated with Flag antibody and immune complexes were subjected to Western blot analysis and examined for ubiquitin lysine 48 linkages. Complexes and lysates were also examined for HA, Flag, and Myc expression while α-tubulin serves as a loading control. (E and F) HEK293T cells coexpressing Flag-SH3BP5 or Flag-SH3BP5L with Myc-TNKS and HA-RNF146 were treated with and without 10 µM MG132 for 4 h before lysis. Cell lysates were examined for HA, Flag, and Myc expression, while α-tubulin serves as a loading control. Molecular weights (kD) for blots are indicated on the left of each panel.
To determine whether attenuation of SH3BP5 protein abundance was due to ubiquitin-mediated proteolysis, cells were transiently treated with 10 µM MG132 to block proteasome-mediated degradation and stabilize ubiquitin-conjugated proteins. We have previously shown that RNF146 stimulates lysine 48 ubiquitylation of Tankyrase substrates (Levaot et al., 2011). Flag-SH3BP5 was immunoprecipitated from each condition, subjected to Western blotting, and probed with an antibody specific for polyubiquitin chains linked through lysine 48. Expression of RNF146 strongly enhanced lysine 48–linked ubiquitylation of SH3BP5 in the presence of TNKS, whereas there was no lysine 48 ubiquitylation detected in the absence of TNKS expression (Fig. 4 C). Deletion of the RNF146 catalytic RING domain (ΔR) impaired SH3BP5 ubiquitylation, demonstrating the specific role of RNF146 as an SH3BP5-specific E3 ubiquitin ligase. Importantly, the RNF146 RA mutant was unable to ubiquitylate SH3BP5, indicating the requirement of PAR recognition by RNF146 to execute ubiquitylation of its substrate (Fig. 4 C). Similar to what we observed with SH3BP5, SH3BP5L underwent lysine 48–linked ubiquitylation in the presence of Tankyrase and RNF146 coexpression, but notably less than SH3BP5 (Fig. 4 D). These results demonstrated that RNF146 binds to and ubiquitylates SH3BP5 and SH3BP5L in a Tankyrase- and ADP-ribose–dependent manner.
To determine whether SH3BP5 was degraded by the proteasome, the abundance of SH3BP5 in the presence of TNKS and RNF146 was assayed in the presence of MG132. We observed that both SH3BP5 and SH3BP5L proteins were stabilized in the presence of MG132 (Fig. 4, E and F). TNKS and RNF146 levels also stabilized, suggesting that all these proteins reside in the same destruction complex. These results show that SH3BP5 and SH3BP5L are novel PARdU-regulated proteins whose steady-state levels are controlled by the proteasome.
RNF146 controls Tankyrase stability and blocks Tankyrase inhibition of SH3BP5 and SH3BP5L GEF activity
We established SH3BP5 and SH3BP5L as GEFs required for Rab11a activation during lumenogenesis and characterized them as Tankyrase and RNF146 substrates. Based on these observations, we hypothesized that Tankyrase and RNF146 are upstream regulators of Rab11a activation. To examine the role of Tankyrase on SH3BP5 function during epithelial lumenogenesis, we generated TNKS and TNKS2 double-knockout MDCKII cells using CRISPR/Cas9 gene editing. In the absence of Tankyrase, SH3BP5 protein levels were elevated, demonstrating that SH3BP5 is a bona fide substrate of Tankyrase proteins (Fig. 5 A). In distinction, knockout of both TNKS/TNKS2 had little effect on SH3BP5L steady-state protein abundance, suggesting that endogenous SH3BP5L is not regulated by Tankyrases in epithelial cells. (Fig. 5 A). Consistent with elevated SH3BP5 protein expression, TNKS/TNKS2 double-knockout cells exhibited increased Rab11a-GTP activation, which correlated with increased single lumen formation, similar to cells stably expressing SH3BP5 (Fig. 2 E and Fig. 5, B–E). To assess the role of the catalytic activity of Tankyrase in controlling lumenogenesis, we treated SH3BP5 and SH3BP5L single-knockout cells or SH3BP5/SH3BP5L double-knockout cells with the Tankyrase inhibitor TNKS656. The inhibitor reduced the defect in single lumenogenesis of SH3BP5/SH3BP5L double-knockout cells, suggesting that Tankyrases may regulate other protein targets involved in epithelial polarity that are required for lumen formation, such as Angiomotin (Fig. 5 F; Campbell et al., 2016). TNKS656 had little effect on single lumen formation in Rab11a-depleted cells, which indicated that Tankyrase inhibition of lumen formation is predominantly through Rab11a (Fig. 5 G). These data demonstrate that Tankyrases negatively regulate lumenogenesis through PAR-dependent repression of SH3BP5 paralogues and Rab11a activation. Tankyrases may control epithelial morphogenesis through the control of other proteins that operate independently of SH3PB5 proteins.

RNF146 controls Tankyrase stability and blocks Tankyrase inhibition of SH3BP5 and SH3BP5L GEF activity. (A) TNKS/TNKS2 double-knockout (dKO) MDCK cells were analyzed by Western blot for SH3BP5, SH3BP5L, and TNKS/TNKS2 protein expression. α-Tubulin serves as a loading control. (B) Rab11a-GTP loading analysis of TNKS/TNKS2 double-knockout MDCK cells. 500 µg of lysate was immunoprecipitated with 20 µg GST-FIP3RBD, and immune complexes were probed with Rab11a antibody. Total Rab11a expression was examined from lysates. (C) Quantification of Rab11a-GTP levels by ImageJ and values were normalized to pLCKO control. Data represent mean ± SD of three independent experiments. (D) Representative confocal images of TNKS/TNKS2 double-knockout 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (E) Proportion of cysts with single lumens normalized to pLCKO control. Data represent the mean ± SD of three independent experiments. *, P < 0.05. pLCKO n = 308, dKO n = 336. (F) Proportion of cysts with single lumens of 3BP5/3BP5L knockout (KO) lines without and with 4 d treatment of 1 µM TNKS656. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 359, dKO cl14 n = 356, dKO cl26 n = 314, SH3BP5 KO n = 315, SH3BP5L KO n = 314, pLCKO + TNKS656 n = 424, dKO cl14 + TNKS656 n = 309, dKO cl26 + TNKS656 n = 333, SH3BP5 KO + TNKS656 n = 310, SH3BP5L KO + TNKS656 n = 333. (G) Proportion of cysts with single lumens in MDCK cells knocked down with two Rab11a shRNAs without and with 1 µM TNKS656 treatment. Data represent the mean ± SD of three independent experiments. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. shSCM n = 340, sh44 n = 300, sh91 n = 316, shSCM + TNKS656 n = 323, sh44 + TNKS656 n = 300, sh91 + TNKS656 n = 327. (H) Western blot analysis of RNF146 knockout MDCK cells. Lysates from clones derived from two independent gRNAs were examined for TNKS/TNKS2, SH3BP5, SH3BP5L, and RNF146 expression. α-Tubulin serves as a loading control and arrow indicates correct SH3BP5 band. (I) Rab11a-GTP loading analysis of RNF146 knockout MDCK cells as performed in Fig. 5 B. (J) Quantification of Rab11a-GTP levels normalized to pLCKO control cells. Data represent mean ± SD of three independent experiments. (K) Representative confocal images of RNF146 knockout 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (L) Proportion of cysts with single lumens normalized to pLCKO control. Data represent the mean ± SD of three independent experiments. **, P < 0.01. pLCKO n = 383, KO sg2 n = 312, KO sg3 n = 334. (M) Proportion of single lumens of RNF146 knockout 4-d cysts without and with 1 µM TNKS656 treatment. Cysts were treated for 4 d with inhibitor. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 309, KO sg2 n = 321, KO sg3 n = 301, pLCKO + TNKS656 n = 357, KO sg2 + TNKS656 n = 318, KO sg3 + TNKS656 n = 321. Molecular weights (kD) for blots are indicated on the left of each panel.
RNF146 controls Tankyrase stability and blocks Tankyrase inhibition of SH3BP5 and SH3BP5L GEF activity. (A) TNKS/TNKS2 double-knockout (dKO) MDCK cells were analyzed by Western blot for SH3BP5, SH3BP5L, and TNKS/TNKS2 protein expression. α-Tubulin serves as a loading control. (B) Rab11a-GTP loading analysis of TNKS/TNKS2 double-knockout MDCK cells. 500 µg of lysate was immunoprecipitated with 20 µg GST-FIP3RBD, and immune complexes were probed with Rab11a antibody. Total Rab11a expression was examined from lysates. (C) Quantification of Rab11a-GTP levels by ImageJ and values were normalized to pLCKO control. Data represent mean ± SD of three independent experiments. (D) Representative confocal images of TNKS/TNKS2 double-knockout 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (E) Proportion of cysts with single lumens normalized to pLCKO control. Data represent the mean ± SD of three independent experiments. *, P < 0.05. pLCKO n = 308, dKO n = 336. (F) Proportion of cysts with single lumens of 3BP5/3BP5L knockout (KO) lines without and with 4 d treatment of 1 µM TNKS656. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 359, dKO cl14 n = 356, dKO cl26 n = 314, SH3BP5 KO n = 315, SH3BP5L KO n = 314, pLCKO + TNKS656 n = 424, dKO cl14 + TNKS656 n = 309, dKO cl26 + TNKS656 n = 333, SH3BP5 KO + TNKS656 n = 310, SH3BP5L KO + TNKS656 n = 333. (G) Proportion of cysts with single lumens in MDCK cells knocked down with two Rab11a shRNAs without and with 1 µM TNKS656 treatment. Data represent the mean ± SD of three independent experiments. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. shSCM n = 340, sh44 n = 300, sh91 n = 316, shSCM + TNKS656 n = 323, sh44 + TNKS656 n = 300, sh91 + TNKS656 n = 327. (H) Western blot analysis of RNF146 knockout MDCK cells. Lysates from clones derived from two independent gRNAs were examined for TNKS/TNKS2, SH3BP5, SH3BP5L, and RNF146 expression. α-Tubulin serves as a loading control and arrow indicates correct SH3BP5 band. (I) Rab11a-GTP loading analysis of RNF146 knockout MDCK cells as performed in Fig. 5 B. (J) Quantification of Rab11a-GTP levels normalized to pLCKO control cells. Data represent mean ± SD of three independent experiments. (K) Representative confocal images of RNF146 knockout 4-d cysts that were incubated with PODXL and β-catenin antibodies. Scale bar = 10 µm; 63× magnification. (L) Proportion of cysts with single lumens normalized to pLCKO control. Data represent the mean ± SD of three independent experiments. **, P < 0.01. pLCKO n = 383, KO sg2 n = 312, KO sg3 n = 334. (M) Proportion of single lumens of RNF146 knockout 4-d cysts without and with 1 µM TNKS656 treatment. Cysts were treated for 4 d with inhibitor. Data represent the mean ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. pLCKO n = 309, KO sg2 n = 321, KO sg3 n = 301, pLCKO + TNKS656 n = 357, KO sg2 + TNKS656 n = 318, KO sg3 + TNKS656 n = 321. Molecular weights (kD) for blots are indicated on the left of each panel.
Since Tankyrases regulated endogenous SH3BP5 stability in MDCK cells, we next interrogated the role of RNF146 during lumenogenesis using RNF146 knockout MDCKII cells. Loss of RNF146 resulted in the stabilization of both TNKS and TNKS2 proteins (Fig. 5 H). These data confirm that Tankyrase proteins are subject to negative regulation by RNF146. We note that RNF146 deletion decreased levels of Rab11a-GTP, which correlated with impaired single lumen formation (Fig. 5, I–L). Importantly, the defect in lumen formation was rescued with TNKS656 treatment, suggesting that Tankyrase abundance and PARylation are the key negative regulators of this process (Fig. 5 M). These genetic experiments suggest that other ubiquitin-modifying enzymes are principally involved in SH3BP5 and SH3BP5L protein turnover. These data support a model whereby Tankyrase proteins are negatively regulated by RNF146 in epithelial cells. Tankyrase levels, in turn, negatively regulate SH3BP5 activation of Rab11a.
Discussion
We have used proteomics and functional genetics to demonstrate that Rab11a activity is negatively regulated by Tankyrases and positively regulated by RNF146 through their modulation of the abundance of the Rab11 GEFs SH3BP5 and SH3BP5L. Thus, we propose a model whereby the regulation of Rab11a-dependent lumenogenesis is controlled by a concatenation of regulatory interactions between RNF146, Tankyrases, SH3BP5, SH3BP5L, and Rab11a. Tankyrases interact with and PARylate SH3BP5 and SH3BP5L, which impairs their ability to activate Rab11a, leading to a defect in lumen formation. RNF146 positively regulates Rab11a-mediated lumen formation through the suppression of Tankyrase by proteasome-mediated degradation (Fig. 6).

Tankyrase and RNF146 have opposing roles during lumen formation. Tankyrase interaction and modification of exchange factors SH3BP5 and SH3BP5L inhibits Rab11a activation, resulting in a multilumen phenotype in MDCKII cysts. RNF146 downregulates Tankyrase protein abundance, which leads to increased Rab11a GDP to GTP exchange mediated by SH3BP5 and SH3BP5L and correct single lumen formation.
Tankyrase and RNF146 have opposing roles during lumen formation. Tankyrase interaction and modification of exchange factors SH3BP5 and SH3BP5L inhibits Rab11a activation, resulting in a multilumen phenotype in MDCKII cysts. RNF146 downregulates Tankyrase protein abundance, which leads to increased Rab11a GDP to GTP exchange mediated by SH3BP5 and SH3BP5L and correct single lumen formation.
Our work also demonstrates cell-type specificity that governs Tankyrase and RNF146 substrate recognition. TNKS/TNKS2 deletion confirmed that endogenous SH3BP5 protein is a substrate of Tankyrases. These data suggest the possibility that PARylation of SH3BP5 paralogues may inhibit their GEF activities and promote SH3BP5 degradation. It is possible that PARylation of these GEFs might directly impair their catalytic activity. Tankyrase has recently been shown to modify Asp and Glu residues, several of which are involved in direct contact with Rab11a where the addition of PAR could introduce a disruptive steric clash (Jenkins et al., 2018; Eisemann et al., 2019). Tankyrases form oligomeric structures through the SAM domain, and bound substrates can become part of this large protein complex (De Rycker et al., 2003). Tankyrase oligomers result in large vesicular aggregates in cells and contribute to PARylation activity (DaRosa et al., 2016; Mariotti et al., 2016; Riccio et al., 2016). Tankyrase and the transactive response DNA binding protein 43 (TDP-43) have recently been shown to assemble into phase-separated foci during stress (McGurk et al., 2018). Tankyrase may therefore negatively regulate SH3BP5 and SH3BP5L, and hence Rab11a, activation by sequestering both SH3BP5 proteins and Rab11a into insoluble protein complexes.
The polarity inversion that PODXL undergoes during MDCK lumen formation is regulated by opposing forces that regulate its association with the ECM-contacting surface and translocation to the apical membrane initiation site (AMIS; Bryant et al., 2014). Rab11a activity is required for trafficking multiple proteins to the AMIS for apical membrane assembly and establishment of apical-basal polarity (Bryant et al., 2010; Roland et al., 2011; Vogel et al., 2015). Our work demonstrates that SH3BP5 and SH3BP5L are GEFs required for Rab11a activation. The state of SH3BP5/SH3BP5L catalytic activity could serve to amplify Rab11a-dependent trafficking of PODXL. In addition, these GEFs may also be required for Rab11a activation during protein recycling, cytokinesis, neurite formation, and ciliogenesis. RhoA activation regulates PODXL association with the ECM, and inhibition of RhoA, recognition of ECM by β1-integrins, and phosphorylation of PODXL by PKCβII trigger its translocation to the AMIS in Rab11a-positive vesicles (Bryant et al., 2014). Tankyrase could also be regulating proteins involved with RhoA signaling and integrin recognition at the ECM. Tankyrase regulation of proteins that operate independently of Rab11a and promote PODXL association at the ECM could explain why Tankyrase inhibition reduced the defect in single lumen formation in SH3BP5/SH3BP5L double-knockout cells (Gálvez-Santisteban et al., 2012; Mrozowska and Fukuda, 2016).
SH3BP5 was originally identified as a negative regulator of Bruton’s tyrosine kinase (Matsushita et al., 1998; Yamadori et al., 1999). SH3BP5 has also been reported to be required for sustained JNK activation during cell stress and localizes to the mitochondria (Wiltshire et al., 2002; Win et al., 2011; Win et al., 2014). Interestingly, we did not identify Bruton’s tyrosine kinase or JNK in our BioID screen, suggesting both cell- and stimuli-specific interactions. Stimuli, such as cell stress, could be important for targeting SH3BP5 as a JNK activator as opposed to a Rab11 GEF. It is also possible that Tankyrase and RNF146 are master regulators of SH3BP5 and coordinate its downstream function based on localization and response to different cell signals and stresses.
We have discovered a unique mechanism regulating Rab11a GTPase activation during epithelial cell polarity involving protein–protein interactions and post-translational modifications involving a complex interplay between RNF146, Tankyrases, and SH3BP5 paralogues. We have defined a new model in which RNF146 and Tankyrase have opposing functions in the regulation of Rab11a through the exchange factors SH3BP5 and SH3BP5L to mediate lumenogenesis.
Materials and methods
Cell lines
Cell lines used in this study were HEK293T, HEK293 T-REx (Invitrogen), and MDCKII (gift from Dr. David Bryant, Beatson Institute, Glasgow, UK). HEK293 cells were grown in DMEM (Wisent) with 10% FBS (Wisent) and 100 U/ml penicillin and 100 µg/ml streptomycin (Wisent). MDCKII were grown in DMEM with 5% FBS. HEK293 T-REx cells were additionally maintained with 5 µg/ml blasticidin (BioShop) and 100 µg/ml zeocin (Invitrogen). Cells were cultured at 37°C in a humidified incubator with 5% CO2.
Plasmids
SH3BP5 cDNA was isolated from HEK293T RNA, and human SH3BP5L cDNA was obtained from OpenFreezer (Mount Sinai Hospital). SH3BP5 and SH3BP5L were subcloned into pEBG using KpnI and NotI restriction sites. Deletions for both proteins in pEBG were created by overlap-extension PCR. N-terminal and C-terminal fragments were cloned with inner primers flanking the deleted region and terminal full-length cloning primers. Full-length constructs were generated by PCR with KpnI and NotI cloning primers by using each fragment as template DNA. α-Helix regions were determined based on the boundaries suggested from secondary structure analysis by the PSIPRED prediction server (http://bioinf.cs.ucl.ac.uk/psipred; Buchan et al., 2013). SH3BP5 was subcloned into pCMV10 (Sigma-Aldrich) by PCR using HindIII and EcoRI restriction sites, and SH3BP5L was subcloned into pCMV10 with EcoRI and XbaI sites. Tankyrase binding domain deletion mutants were generated using QuikChange Lightning XL (Agilent Technologies). Human RNF146 plasmids, pcDNA4-TO HA-RNF146, R163A, and ΔRING were kind gifts from Dr. Feng Cong (Novartis, Cambridge, MA; Zhang et al., 2011; Levaot et al., 2011). Plp-dMyc-TNKS/TNKS2 plasmids were previously described in Guettler et al. (2011) and Levaot et al. (2011). SH3BP5, RNF146, and R163A were cloned into the BioID plasmid pcDNA5 FRT.TO FlagBirA R118G by PCR using AscI and NotI restriction sites. pEGFPC1-Rab11a WT and S25N were kind gifts from Dr. Richard Pagano (Addgene plasmids #12674 and #12678, respectively). pEGFPC1-Rab11a Q70L was generated using QuikChange Lightning XL. Pet28a Rab11a 1–183 was subcloned using NheI/XhoI sites and kept in frame to generate a C-terminal 6xHis Tag. Human SH3BP5 and SH3BP5L were cloned into the bacterial expression vector pGEX-4T1 using EcoRI and NotI restriction sites. To generate Tag-RFP-T constructs, human SH3BP5 and SH3BP5L were subcloned using EcoRI/KpnI sites into pTag-RFP-T-C1 (kind gift from Dr. David Bryant).
Retroviral and lentiviral plasmids
Tag-RFP-T-SH3BP5 and SH3BP5L fusions were subcloned into the retroviral plasmid pMXs-IRES-Puro (Clontech) using the NotI/EcoRI restriction sites and InFusion HD cloning kit (Clontech).
sgRNA sequences targeting dog SH3BP5, SH3BP5L, TNKS, TNKS2, and RNF146 were selected from the CRISPOR design tool (http://www.crispor.tefor.net) by entering the cDNA sequence of specific exons (Haeussler et al., 2016). Guide sequences were cloned into the plasmid pLCKO using BfuAI and NsiI sites (Table 1).
gRNA sequences
| sgRNA | Sequence | RefSeq ID and target exon |
|---|---|---|
| Dog_SH3BP5 sg5 | 5′-AGCGACAGTTCGACTCCGCC-3′ | XM_005634401.3 Exon4 |
| Dog_SH3BP5L sg3 | 5′-GCAAGAAAGCTCAACACGCA-3′ | XM_854895.5 Exon3 |
| Dog_TNKS sg2 | 5′-GACCTCCATCATCACGAGCA-3′ | XM_844295.5 Exon2 |
| Dog_TNKS2 sg3 | 5′-CTGTTCGAGGCGTGCCGCAA-3′ | XM_003639982.4 Exon1 |
| Dog_RNF146 sg2 | 5′-GAATATGCGTGGTATTACGA-3′ | XM_533493.6 Exon3 |
| Dog_RNF146 sg3 | 5′-TCTCGCTAGGTTGACCGTGC-3′ | XM_533493.6 Exon3 |
Two shRNAs against dog Rab11a were obtained from the RNAi consortium. Scrambled control (shSCM) hairpin was cloned into pLKO.1 (Addgene #10878) by annealing oligos into pLKO.1 digested with EcoRI and AgeI (Table 2).
shRNA sequences
| shRNA | Sequence | TRC clone ID | Plasmid | RefSeq ID |
|---|---|---|---|---|
| shSCM | 5′-CCTAAGGTTAAGTCGCCCTCG-3′ | NA | pLKO.1 puro | NA |
| Dog_Rab11a sh91 | 5′-CGAGCTATAACATCAGCATAT-3′ | TRCN0000303291 | pLKO.1 puro | NM_001003276.3 |
| Dog_Rab11a sh44 | 5′-CAGAGATATACCGCATTGTTT-3′ | TRCN0000100344 | pLKO.1 puro | NM_001003276.3 |
Antibodies and reagents
The following antibodies were used for immunoblotting: Flag (clone M2, 1:2,000; F1804; Sigma-Aldrich), Myc (clone 9E10, 1:250; sc40; Santa Cruz Biotechnology), HA (1:1,000; #3724; Cell Signaling Technologies), α-tubulin (1:1,000; sc69969; Santa Cruz Biotechnology), streptavidin-HRP (1:2,000; #3999; Cell Signaling Technologies), ubiquitin Lys48 (clone Apu2, 1:1,000; Millipore), GAPDH (1:1,000; #2118; Cell Signaling Technologies), GFP (1:2,000; ab290; Abcam), GST (1:1,000; sc138; Santa Cruz Biotechnology), SH3BP5 (1:300; HPA036445; Sigma-Aldrich), SH3BP5L (1:500; NBP2-38385; Novus Biologics), Tankyrase-1/2 (1:500; sc365897; Santa Cruz Biotechnology), RNF146 (1:1,000; SAB1408054; Sigma-Aldrich), and Rab11a (1:500; 71–5300; Thermo Fisher Scientific). For immunofluorescence: β-Catenin (1:500; #9562; Cell Signaling Technologies), podocalyxin (clone 3F2, 1:1,000; MABS1327; Millipore), Rab11a (1:250; 71–5300; Thermo Fisher Scientific), Alexa Fluor 488 (1:500; A28175/A11008; Thermo Fisher Scientific), and Alexa Fluor 647 (1:500; A27040; Thermo Fisher Scientific). TNKS656 inhibitor was a kind gift from Dr. Feng Cong. Biotin-NAD+ was purchased from Trevigen (cat# 4670–500-01). MG132 was purchased from Enzo Life Sciences (cat# BML-PI102-0005). D-Biotin (BB0078) was purchased from BioBasic. Tetracycline (cat# 87128) was purchased from Sigma-Aldrich.
Retrovirus and lentivirus generation and transduction
Viruses were generated by transfecting 5 × 105 HEK293T cells in six-well plates with 50 ng envelope, 450 ng packaging plasmids, along with 500 ng viral plasmids using X-tremegene 9 (Roche). VSV-G and psPAX2 were used as envelope and packaging plasmids for lentiviral particles, while VSV-G and gag-pol were used for retroviral particles. After 24 h of transfection, the media was replaced with target cell media. Viral supernatants were collected 48 h after transfection and cleared through 0.45-µm filters. Target cells were transduced with viral supernatants along with 8 µg/ml polybrene for 24 h. Antibiotic selection was started 24 h after infection for a further 48 h in fresh media. For MDCKII cells, 5 µg/ml puromycin (BioShop), 500 µg/ml hygromycin (Wisent), and 5 µg/ml blasticidin (BioShop) were used for selection.
MDCKII stable expression lines
Stable MDCKII cells expressing Tag-RFP-T-SH3BP5 and Tag-RFP-T-SH3BP5L were generated by retroviral infection. Surviving cells that had gone through selection with puromycin were sorted by FACS for RFP expression. Cells were single-cell cloned in 96-well plates and uniform expressing clones were selected. These clones were expanded and used for experiments.
Knockout lines created by CRISPR
MDCKII-Cas9 cells were generated by infection of Lenti-Cas9-2A lentiviral particles, expressed from the Lenti-Cas9-2A plasmid with 8 µg/ml polybrene (Hart et al., 2015). 24 h after infection, viral media was replaced with fresh culture media. Cells were placed under selection with 5 µg/ml blasticidin for 48 h. Surviving cells were checked for Flag-Cas9 expression by immunoblot analysis and then seeded into 96-well plates to achieve single-cell clones. Clones were expanded and again tested for Cas9 expression. Clonal lines that maintained Cas9 were subsequently used for gene knockout studies.
sgRNA viruses were used to create SH3BP5, SH3BP5L, and RNF146 knockout lines by infecting MDCKII-Cas9 cells with 8 µg/ml polybrene for 24 h. To generate SH3BP5/SH3BP5L and TNKS/TNKS2 double-knockout lines, equal volumes of gRNA lentiviruses for each gene were used following the same parameters. Cells were placed under selection with 5 µg/ml puromycin and 5 µg/ml blasticidin for 48 h. Surviving cells were seeded into 96-well plates to achieve single-cell clones. Clones were analyzed by Western blotting and clones exhibiting complete deletion of protein were expanded and used for further study.
Western blotting and immunoprecipitation
For immunoprecipitation experiments, 8 × 105 HEK293T cells in six-well plates were transfected for 24 h using LipoD293 (Froggabio). Cells were lysed directly on ice for 10 min using Triton X-100 lysis buffer (1% Triton X-100, 25 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA) with 1× Halt protease and phosphatase cocktails (Thermo Fisher Scientific). For lysine 48 ubiquitylation and protein degradation experiments, cells were treated with 10 µM MG132 for 4 h before lysis. Lysates were cleared by centrifugation at 13,300 rpm for 10 min at 4°C and incubated with 12 µl of a 50% slurry solution of either Flag M2 agarose (Sigma-Aldrich) or Myc agarose (Sigma-Aldrich) for 1 h at 4°C. For Rab11a interaction studies, lysates were incubated with either 1 µg of GFP antibody plus 15 µl of Protein G agarose slurry (BioShop) or 15 µl glutathione agarose (Thermo Fisher Scientific) for 1 h. Beads were washed three times with lysis buffer (without inhibitor cocktails) before boiling with 2× Laemmli buffer for 5 min at 100°C. For immunoblotting, cells were lysed in Triton X-100 buffer containing 1% Triton X-100, 25 mM Tris (pH 7.50), 100 mM NaCl, and 1 mM EDTA, with 1× Halt protease and phosphatase cocktails (Thermo Fisher Scientific). Lysates were cleared by centrifugation at 13,300 rpm for 10 min at 4°C, quantified using the 660-nm protein assay kit (Thermo Fisher Scientific), and boiled with 1× Laemmli buffer for 5 min at 100°C.
Western blotting was performed by separating samples by SDS-PAGE, transferring to PVDF membrane (Millipore), blocking with 5% skim milk (BioShop) in 1× PBS + 0.1% Tween 20 (PBST; BioShop), and incubating with indicated antibodies overnight at 4°C. Membranes were washed three times with PBST and incubated with HRP-conjugated mouse and rabbit antibodies (BioShop) for 1 h in 5% milk. For Rab11a pulldown experiments, a conformation specific secondary antibody (CST #3678) was used to remove denatured rabbit IgG signals. Membranes were washed three times with PBST and developed using ECL or ECL prime (GE Healthcare) and SuperSignal West Pico PLUS or SuperSignal West Femto (Thermo Fisher Scientific) HRP substrates. Membranes were imaged using a MicroChemi 2.0 chemiluminescent imager (DNR Bio-Imaging Systems).
In vitro PARylation assay
HEK293T were transfected in six-well plates for 24–48 h with LipoD293. For Tankyrase inhibition, cells were treated 24 h after transfection with 1 µM TNKS656 for another 24 h. Cells were lysed in PAR lysis buffer (50 mM Hepes [pH 7.5], 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 5 mM EDTA, 1× Halt protease/phosphatase cocktails, and 5 mM DTT) and cleared lysates were immunoprecipitated with 12 µl of a 50% slurry solution of Flag M2 agarose beads for an hour at 4°C. The beads were washed three times with lysis buffer and then washed three times with in vitro assay buffer (50 mM Hepes [pH 7.5], 100 mM NaCl, 0.01% Triton X-100, 2 mM Tris[2-carboxyethyl]phosphine). Beads were incubated in 50 µl of assay buffer plus 10 µM Biotin-NAD+ and shaken at 25°C for 1 h. The reaction was stopped with Laemmli buffer, boiled for 5 min at 100°C, and subjected to SDS-PAGE and immunoblot analysis. To detect PAR chains, blocked membranes were incubated with streptavidin-HRP for 1 h at RT followed by three washes with PBST. Membranes were then imaged using ECL prime or SuperSignal West Femto HRP substrates.
Cyst culture
8-well chamber slides (#354108; BD Falcon) were coated with 10 µl of 100% Matrigel (#356230; Corning). 300 µl of a 1.5 × 104 cells/ml suspension with 2% Matrigel was plated on top of solidified Matrigel. Cysts were grown for 4 d before immunofluorescence analysis. In experiments using TNKS656 inhibitor, the compound was added during plating and kept for 48 h before being replenished for another 48 h.
Immunofluorescence
Cysts grown for 4 d were fixed with 4% PFA at RT for 25 min. Cysts were washed twice with 1× PBS with magnesium and calcium and blocked in 1× PBS (Mg2+ and Ca2+), 1% BSA (BioShop), 5% goat serum (ab7481; Abcam), and 0.025% saponin or 0.05% saponin (for Rab11a staining; S4521; Sigma-Aldrich) for an hour. Cysts were incubated with primary antibodies overnight at RT. Chamber slides were washed three times with 1× PBS (Mg2+ and Ca2+) and incubated with Alexa Fluor 488 and -647 secondary antibody conjugates for 1 h at RT. Slides were washed three times with 1× PBS and incubated with DAPI (Molecular Probes) for 10 min at RT. Slides were washed twice with 1× PBS and mounted with ProLong Gold Antifade (Molecular Probes).
Confocal microscopy
MDCK cysts were imaged with a Leica DMi8 spinning-disk confocal microscope using a 63× (1.4 NA) oil objective at RT. Images were captured with a Hamamatsu C9100-13 EM-CCD camera and Volocity software (PerkinElmer). Image contrast and brightness were adjusted using ImageJ. For Rab11 colocalization, cysts were imaged with a Leica SP8 laser scanning confocal 63× plan apo (1.4 NA) oil objective lens at RT and images captured with PMT detectors and LAS X software (Leica). Z-stacks were taken in 1-µm increments and colocalization between Tag-RFPT-SH3BP5/SH3BP5L and Rab11a was calculated using Imaris (Oxford Instruments). The percent colocalized 3D volume (± SD) of Tag-RFP-T-SH3BP5/SH3BP5L with Rab11a above threshold intensity was calculated and modeled using the Coloc function. The same absolute threshold value for positive staining for each channel was applied for all images within an experiment for the calculation. Image contrast and brightness was adjusted in Imaris.
MDCK cyst quantification
MDCK cysts were quantified as previously described (Martin-Belmonte et al., 2007). A minimum of 100 cysts per condition were counted and classified as having single or multiple lumens. Cysts that were derived from a single cell and displayed correct basolateral polarity (via DAPI and b-catenin staining) were counted using the epifluorescence mode on an Olympus IX81 inverted microscope using a 20× air (0.75) plan APO objective (Nikon). Single lumen percentage was calculated by determining the percentage of cysts with a single lumen versus multiple lumens for each condition. Values for each condition were normalized to the single lumen percentage in control cells. Single lumen percentages are displayed as mean ± SD of three independent experiments. Statistical significance was calculated using a two-tailed paired t test using GraphPad Prism 8. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
Protein expression and purification
6xHis-Rab11a 1–183-6xHis was expressed in BL21 codon plus cells. A 15N-labeled Rab11a 1–183-6xHis was prepared as follows: A 50-ml culture in Luria-Bertani broth was grown at 37°C overnight, pelleted the next day, and resuspended in 2 liters of M9 minimal media (42 mM Na2HPO4, 22 mM KH2PO4, 9 mM NaCl, 1% glucose, 0.3 mM CaCl2, 1 mM MgSO4, 1 mg/ml biotin, 1 mg/ml thiamine, and 1× trace elements). M9 labeling media was supplemented with 19 mM 15N-ammonium chloride. Cells were grown until an OD at 600 nm wavelength of 0.6, cooled to 15°C, and protein expression induced with 0.25 mM IPTG overnight at 15°C. Cells were centrifuged and stored at −80°C. Frozen pellet was resuspended in ice-cold lysis buffer (50 mM Tris [pH 8], 150 mM NaCl, 15 mM imidazole, 2 mM MgCl2, 1 mM PMSF, 0.2 mg/ml lysozyme, and 10 mM β-mercaptoethanol). Cells were lysed by sonication and lysate was cleared by centrifugation at 20,000 rpm for 30 min at 4°C. Cleared lysate was passed through a 0.45-µm filter before incubation with 5ml Ni-NTA resin (Qiagen) for 1 h at 4°C. The resin was washed with 25 column volumes of lysis buffer (without PMSF and lysozyme) and protein was eluted with six column volumes of elution buffer (50 mM Tris [pH 8], 150 mM NaCl, 250 mM imidazole, 2 mM MgCl2, and 10 mM β-mercaptoethanol). The N-terminal 6xHis tag was removed by incubating eluted protein with thrombin (10 μ/ml of protein) overnight by dialysis in buffer containing 50 mM Tris (pH 8), 150 mM NaCl, 5 mM MgCl2, and 10 mM β-mercaptoethanol. Protein was purified by size exclusion on a Superdex S75 26/60 column in dialysis buffer and elution peak fractions were pooled and concentrated to 6.5 mg/ml and flash frozen in liquid nitrogen and stored at −80°C.
pGEX-4T1 SH3BP5 (1–280Δ100-207) and SH3BP5L(1–299Δ115-221) helix 1 and 4 constructs were expressed in BL21 codon plus cells. Protein was expressed as described above. Cell pellets were resuspended with ice-cold lysis buffer (50 mM Tris [pH 8], 150 mM NaCl, 10% glycerol, 0.2 mg/ml lysozyme, 1 mM PMSF, and 1 mM DTT). Cells were lysed by sonication and lysates were cleared by centrifugation. Filtered lysates were incubated with 5 ml glutathione agarose (Thermo Fisher Scientific) overnight. Resin was washed with 25 column volumes of lysis buffer, and the GST tag was cleaved off the resin with thrombin overnight. Cleaved protein was recovered, purified by gel filtration on a Superdex S75 26/60 column, concentrated, and flash frozen with liquid nitrogen and stored at −80°C. For full-length 3BP5 and 3BP5L, the GST tag was left on and proteins were eluted with five column volumes of elution buffer (50 mM Tris [pH 8], 100 mM NaCl, and 10 mM reduced glutathione). Eluted protein was dialyzed overnight in buffer (50 mM Tris [pH 8], 100 mM NaCl, and 1 mM DTT), concentrated, and frozen the next day.
Rab11-GTP pulldown assay
Rab11-GTP levels were detected through immunoprecipitation of cell lysates with the Rab11-binding domain of Rab11FIP3 (FIP3RBD). Amino acids 695–756 of Rab11FIP3 were cloned into pGEX4T1 using EcoRI and XhoI restriction sites (Eathiraj et al., 2006). GST-FIP3RBD was expressed in Escherichia coli BL21 codon plus cells. Protein was expressed as described above. Bacterial cells were resuspended in lysis buffer (50 mM Tris [pH 7.5], 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1 mM PMSF, 0.2 mg/ml, and lysozyme) and lysed using sonication, and lysate was cleared by centrifugation. Cleared lysate was incubated with glutathione agarose for 1 h at 4°C and the resin was subsequently washed with 25 column volumes of lysis buffer (without PMSF and lysozyme). Protein was eluted using three column volumes of buffer containing 50 mM Tris (pH 8), 100 mM NaCl, and 10 mM reduced glutathione. The eluted protein was dialyzed overnight to remove the reduced glutathione using buffer with 50 mM Tris (pH 7.5), 100 mM NaCl, and 5 mM MgCl2.
For Rab11-GTP pulldown experiments, MDCKII cells were lysed in 50 mM Tris (pH 8), 100 mM NaCl, 1% Triton X-100, 5 mM MgCl2, 0.5 mM EDTA, Halt protease, and phosphatase inhibitor cocktails for 25 min on ice. Cleared lysates were quantified using the Pierce 660-nm protein assay reagent, and 500 µg of cell lysate was incubated with 20 µg of GST-FIP3RBD and 20 µl of glutathione agarose for 1 h. The agarose was washed thrice with lysis buffer and then resuspended in 20 µl of 2× Laemmli buffer, boiled for 5 min at 100°C, and then subjected to immunoblot analysis. Rab11a-GTP levels were quantified with ImageJ using blots from three independent experiments. Rab11a-GTP levels were normalized to total Rab11a protein, and each experiment was normalized to the control.
Rab11a nucleotide exchange assay by NMR
Rab11a nucleotide exchange was monitored using a real-time NMR-based assay that was described previously (Marshall et al., 2009; Gasmi-Seabrook et al., 2010). A series of 1H-15N-HSQC spectra of 15N-labeled Rab11a were collected sequentially during the exchange reaction, and the heights of peaks that are specific to the GDP- or GTP-bound forms of the GTPase protein were used to determine exchange rates. A 40-µl sample was prepared by mixing 300 µM GDP-loaded 15N-Rab11a and 4.5 mM excess of GTPγS (guanosine 5′-[γ-thio] triphosphate tetralithium salt; Sigma-Aldrich) in a 1.7-mm NMR tube. Sequential 1H-15N-HSQC NMR experiments (10 min each) were collected throughout the time course of the exchange reaction using a Bruker 600-MHz Avance III NMR spectrometer equipped with a 1.7-mm cryogenic TCI MicroCryoProbe. Data were analyzed using Bruker TopSpin. The half-life of the exchange reaction was determined as the point at which GDP- and GTP-specific peaks exhibited equal heights, and nucleotide exchange rates were calculated from the half-life based on single-phase exponential decay (rate = ln2/half-life). To determine whether SH3BP5 and SH3BP5L possessed GEF activity and to investigate which parts of the protein were required to mediate nucleotide exchange, the experiment was repeated with the addition of a series of purified recombinant fragments as indicated.
BioID assay and mass spectrometry analysis
BioID was performed as previously described (Roux et al., 2012; Coyaud et al., 2015). HEK293 Flp-In T-REx cell lines stably expressing Flag-BirA-RNF146, Flag-BirA-RNF146 R163A, and Flag-BirA-SH3BP5 were generated using the FlpIn system (Invitrogen). HEK293 T-REx cells in 6-well plates were transfected with 100 ng of BirA construct and 900 ng of pOG44, which contains Flp-recombinase. Culture media was changed 24 h after transfection for another 24 h. Cells were placed under selection with 5 µg/ml blasticidin and 200 µg/ml hygromycin B for 2–3 wk until visible colonies emerged and cells were expanded. For the BioID experiment, five 150-cm2 plates of cells were grown to 80% confluency before induction of protein expression with 1 µg/ml tetracycline and 50 µM biotin for 24 h. For WT and R163A lines, an additional five plates were treated with 5 µM MG132 in addition to the induction media for 24 h. The sample was lyophilized and resuspended in buffer A (0.1% formic acid). One fifth of the sample was analyzed per mass spectrometry run.
Mass spectrometry analysis was conducted as previously described using a Thermo Q-Exactive HF quadrupole-orbitrap mass spectrometer (Coyaud et al., 2015). Proteins identified with an iProphet cutoff of 0.89 (SH3BP5) and 0.92 (RNF146; corresponding to ≤1% false discovery rate [FDR]) and at least two unique peptides were analyzed with SAINT Express v.3.6. 10 control runs (from cells expressing the FlagBirA* epitope tag) were collapsed to the two highest spectral counts for each prey and compared with two technical runs of each of the two biological replicates of RNF146 WT and R163A mutant and SH3BP5. High confidence interactors corresponded to a Bayesian FDR (BFDR) of ≤0.01.
Interaction network analysis
The RNF146 and SH3BP5 interaction networks were created using Cytoscape v3.2 and importing known interaction data with the GeneMANIA plugin (Shannon et al., 2003; Warde-Farley et al., 2010). Proteins were grouped according to biological function through gene ontology (http://www.geneontology.org) and by manual literature search curation (Ashburner et al., 2000; Gene Ontology Consortium, 2015).
Online supplemental material
Fig. S1 shows the generation of Flag-BirA SH3BP5 293 Flp-In cell lines. Fig. S2 shows the generation of Flag-BirA RNF146 and RNF146 R163A 293 Flp-In cell lines. Network map of new RNF146 interacting proteins from BioID is shown in Fig. S2 as well. Table S1 and Table S2 list both SH3BP5 and RNF146 BioID experiments, respectively.
Acknowledgments
We thank Dr. Feng Cong (Novartis, Cambridge, MA) and Dr. David Bryant (Beatson Institute, Glasgow, UK) for reagents and cell lines. We thank Dr. Oliver Kent and Dr. Helen Burston for critical reading of the manuscript.
This research was funded by the Canadian Institutes of Health Research Foundation Grant. A.A. Chandrakumar was supported by the Queen Elizabeth II Graduate Scholarship in Science and Technology (Sir Edward Dunlop Medical Research Foundation Scholarship) and MBP Excellence Ontario Student Opportunity Trust Fund. M. Ikura holds a Canada Research Chair in Cancer Structural Biology. NMR spectrometers were funded by the Canada Foundation for Innovation, and the NMR Core Facility is supported by the Princess Margaret Cancer Foundation.
The authors declare no competing financial interests.
Author contributions: A.A. Chandrakumar and R. Rottapel conceptualized the study. A.A. Chandrakumar, É. Coyaud, and C.B. Marshall performed experiments. A.A. Chandrakumar, É. Coyaud, C.B. Marshall, B. Raught, and R. Rottapel analyzed data. M. Ikura and B. Raught provided analytical tools. A.A. Chandrakumar, C.B. Marshall, B. Raught, and R. Rottapel wrote and edited the manuscript.

