


Histone posttranslational modifications (PTMs) are dynamic, context-dependent signals that modulate chromatin structure and function. Ubiquitin (Ub) conjugation to different lysines of histones H2A and H2B is used to regulate diverse processes such as gene silencing, transcriptional elongation, and DNA repair. Despite considerable progress made to elucidate the players and mechanisms involved in histone ubiquitination, there remains a lack of tools to monitor these PTMs, especially in live cells. To address this, we combined an avidity-based strategy with in silico approaches to design sensors for specifically ubiquitinated nucleosomes. By linking Ub-binding domains to nucleosome-binding peptides, we engineered proteins that target H2AK13/15Ub and H2BK120Ub with Kd values from 10−8 to 10−6 M; when fused to fluorescent proteins, they work as PTM sensors in cells. The H2AK13/15Ub-specific sensor, employed to monitor signaling from endogenous DNA damage through the cell cycle, identified and differentiated roles for 53BP1 and BARD1 as mediators of this histone PTM.
Introduction
Much of biomedical research depends on reagents able to bind proteins with high affinity, specificity, and reproducibility. Antibodies and their derivatives, by far, have filled most of these needs, but deficiencies regarding antibody availability, performance, or suitability have created a critical need for alternatives (Bradbury and Plückthun, 2015). In particular, for use as live-cell probes, misfolding and instability of conventional antibodies are major problems. As one alternative, fluorescently labeled antibody fragments made in vitro can be introduced into mammalian cells via transient membrane disruption, but these fluorescently labeled antibody fragments typically have low affinity for their targets, and suitability for use in live cells must be determined empirically (Hayashi-Takanaka et al., 2011; Stasevich et al., 2014). Other antibody derivatives and mimetics, such as single-chain variable fragment proteins, aptamers, Designed Ankyrin Repeat Proteins (DARPins), and Affimers, rely on selection from large libraries followed by iterations of mutagenesis and screening to identify desirable variants (Azhar et al., 2017; Crivianu-Gaita and Thompson, 2016; Ruigrok et al., 2011; Simeon and Chen, 2018). As an alternative route to develop binding proteins for use as live-cell probes, in this study we have exploited avidity-based recognition to design genetically encoded sensors specific for ubiquitinated (Ub) nucleosomes. The longstanding difficulties to generate antibodies to detect site-specific protein ubiquitination (van Kruijsbergen et al., 2020) make Ub–nucleosomes especially attractive molecular targets for this proof-of-concept evaluation. To our knowledge, no sensors able to monitor protein ubiquitination in live cells have been reported previously.
Ub functions as a protein posttranslational modification (PTM) that signals a large variety of downstream events (Komander and Rape, 2012; Swatek and Komander, 2016). Histones in nucleosomes are among the most abundant Ub substrates, with the consequences depending on both the type of histone and the lysine residue that is modified. For example, Ub attachment to histone H2A at Lys119 (H2AK119Ub) or H2B at Lys120 (H2BK120Ub) installs Ub at opposite ends of a nucleosome where it promotes transcriptional silencing or activation, respectively (Cao and Yan, 2012; de Napoles et al., 2004; Fang et al., 2004; Minsky et al., 2008). In contrast, N-terminal ubiquitination of H2A at K13 or K15 (H2AK13/15Ub) is a key signal in DNA double-strand break (DSB) repair (Mattiroli et al., 2012). In addition, BRCA1-BARD1 acts as an E3 ligase to form H2AK125/127/129Ub (Kalb et al., 2014), which has been suggested to play a role in DSB repair pathway choice (Densham et al., 2016; Uckelmann et al., 2018). Defects in enzymes that either assemble or disassemble these histone ubiquitination signals (e.g., RNF168, polycomb, BRCA1, USP22, and BAP1) are strongly implicated in cancer development (Jeusset and McManus, 2019).
Like other PTMs, ubiquitination is reversible. Although many proteins have been identified that regulate histone ubiquitination, including, in the parlance of the chromatin field, writers, erasers, and readers, it has been difficult to establish the order and kinetics of their actions because tools to monitor PTMs directly have been lacking. We show here how well-characterized binding modules can be developed into avidity-based sensors for Ub–nucleosomes, and that the sensors can be used to study site-specific histone ubiquitination associated with transcription and DNA repair pathways. With one of the sensors, we show that the H2BK120Ub mark is lost upon inhibition of transcription by RNA polymerase II; with another, we confirm that H2AK13/15Ub is made at sites of DNA damage and show that sensor recruitment is coincident with recruitment of 53BP1. Unexpectedly, we also found that, even without exogenous DNA-damaging agents, H2AK13/15Ub foci are routinely observed. We show that 53BP1 and BARD1 are likely the major effectors of H2AK13/15Ub in vivo, with 53BP1 dominating in G1 phase and BARD1 in G2 phase of the cell cycle. These findings support recent revelations that H2AK13/15Ub is an important signal during DNA replication (Schmid et al., 2018), and BARD1 is potentially a regulator of that signal (Becker et al., 2020).
Results
An avidity-based approach to design sensors targeting Ub–nucleosomes
An avidity-based strategy was developed to generate sensors specific for Ub–nucleosomes. Ub-binding domains (UBDs) were connected by peptide linkers of varying lengths and rigidity to anchor domains that recognize the nucleosome acidic patch (Fig. 1, A and B). Anchors and UBDs were selected based on published structures and on the binding orientations and affinities (dissociation constant [Kd] = 10−6 to 10−3 M) with their respective targets. Linkers were designed to promote avidity by reducing the entropic cost of binding. Two viral peptides interacting with the nucleosome acidic patch, LANA (from the latency-associated nuclear antigen; Barbera et al., 2006) and IE1 (from the cytomegalovirus immediate-early protein 1; Fang et al., 2016), were chosen as anchors for the first generation of Ub–nucleosome sensors. Both peptides contain a conserved arginine residue (i.e., LANA R9 and IE1 R486) that contacts a surface formed by H2A residues E61, D90, and E92. When bound to the acidic patch, LANA assumes a hairpin-like turn with its N- and C-termini pointing to the H2A N-terminal and H2B C-terminal tails, whereas IE1 N- and C-termini face the opposite direction (Fig. 1, A and B). For the UBDs, we used either the engineered fusion protein tSR (Choi et al., 2019b) or the ubiquitin-associated (UBA) domain from ubiquilin-1 (UBQ1UBA; Fig. 1 B; Zhang et al., 2008). Because most naturally occurring UBDs bind Ub with low affinity, we previously developed tSR, which combines S5aUIM and Rabex5Ruz domains, to target both the Ub hydrophobic patch surrounding residue I44 and the surface surrounding residue D58; tSR has a Kd of 4 µM for monoubiquitin (Choi et al., 2019b). UBQ1UBA, which binds to the Ub hydrophobic patch, has a Kd of 22 µM for monoubiquitin (Zhang et al., 2008).

Avidity-based design of sensors targeting H2AK15 and H2BK120 ubiquitination in nucleosomes. (A) Cartoon depicting the main sites of H2A and H2B ubiquitination in the nucleosome. The histone octamer is shown as an electrostatic surface (positive, blue; neutral, white; negative, red), and DNA as a gray surface. Backbone C atoms of Ub residues are shown as spheres for H2AK15 and H2AK119 (orange) and H2BK120 (green). A close-up view of the nucleosome acidic patch shows residues contributed by histones H2A and H2B. (B) The sensors targeting histone ubiquitination have an acidic patch-binding peptide (anchor) and a Ub-binding domain (UBD) connected by a peptide linker (linker). (C) The prototype sensors Reader1.0/1.1 are comprised of a LANA anchor, an antiparallel coiled-coil linker, and tSR UBD. (D) Schematic of the in vitro binding assays used to determine Reader1.0/1.1 interactions with Ub–nucleosomes. (E and F) Binding of Alexa Fluor 488–labeled Reader1.0/1.1 was monitored by fluorescence in titrations with the indicated Ub–nucleosomes from which the dissociation constants (Kd, nM) were determined. R1.0, Reader1.0; R1.1, Reader1.1.
Avidity-based design of sensors targeting H2AK15 and H2BK120 ubiquitination in nucleosomes. (A) Cartoon depicting the main sites of H2A and H2B ubiquitination in the nucleosome. The histone octamer is shown as an electrostatic surface (positive, blue; neutral, white; negative, red), and DNA as a gray surface. Backbone C atoms of Ub residues are shown as spheres for H2AK15 and H2AK119 (orange) and H2BK120 (green). A close-up view of the nucleosome acidic patch shows residues contributed by histones H2A and H2B. (B) The sensors targeting histone ubiquitination have an acidic patch-binding peptide (anchor) and a Ub-binding domain (UBD) connected by a peptide linker (linker). (C) The prototype sensors Reader1.0/1.1 are comprised of a LANA anchor, an antiparallel coiled-coil linker, and tSR UBD. (D) Schematic of the in vitro binding assays used to determine Reader1.0/1.1 interactions with Ub–nucleosomes. (E and F) Binding of Alexa Fluor 488–labeled Reader1.0/1.1 was monitored by fluorescence in titrations with the indicated Ub–nucleosomes from which the dissociation constants (Kd, nM) were determined. R1.0, Reader1.0; R1.1, Reader1.1.
As proof of concept, we applied a structure-guided approach to design sensors targeting H2AK13/15Ub- or H2BK120Ub–nucleosomes. The prototypes, hereafter called Reader1.0 and Reader1.1, contained LANA and tSR linked by an antiparallel α-helical coiled-coil (Hadley et al., 2008). Here, the coiled-coil linker was chosen to limit the conformational mobility of the Ub–nucleosome sensors in an attempt to promote preferential binding to either H2AK13/15Ub or H2K120Ub. Reader1.0 has three additional glycines inserted between LANA and two segments of the linker to increase flexibility (Fig. 1 C and Fig. S1). To measure the affinities of the sensors for different Ub–nucleosomes, Reader1.0/1.1 were labeled with Alexa Fluor 488 (Fig. S2 E) and titrated with reconstituted nucleosomes containing nonhydrolyzable H2AK13Ub, H2AK15Ub, H2AK119Ub, H2AK129Ub, or H2BK120Ub histone mimics (Fig. 1 D; and Fig. S2, A and C; Long et al., 2014). Reader1.0 showed high affinity for H2AK13Ub (Kd = 5 nM), H2AK15Ub (Kd = 12 nM), and H2BK120Ub (Kd = 21 nM), and 7- to 81-fold selectivity over H2AK119Ub (Kd = 405 nM) and H2AK129Ub (Kd = 141 nM). Reader1.1 had a similar selectivity profile but with slightly lower affinities (Fig. 1, E and F). Because native chromatin contains arrays of nucleosomes repeated along the DNA, we also assayed binding with tri-nucleosome arrays (Winkler et al., 2011) containing H2AK15Ub, H2AK119Ub, or H2BK120Ub (Fig. S2, B and D). The designed sensors showed similar affinities and selectivities with the Ub-modified tri-nucleosome arrays as with the mono-nucleosomes (Fig. S2 G), thereby validating our design principle.

Primary sequences of the Ub–nucleosome sensors. His-tagged proteins were expressed in E. coli, and eGFP fusions were expressed in mammalian cells. Residues highlighted in yellow indicate the NLS used for mammalian expression. Anchor residues are highlighted in magenta, UBD residues are highlighted in cyan, linker residues are highlighted in gray, and eGFP residues are highlighted in green. A556 in UBQ1UBA-WT (Reader2.0) and A556E in UBQ1UBA-A556E (Reader2.1) are labeled red. The cysteines depicted in gray indicate the Alexa Fluor 488 conjugation sites used for the in vitro binding assays.
Primary sequences of the Ub–nucleosome sensors. His-tagged proteins were expressed in E. coli, and eGFP fusions were expressed in mammalian cells. Residues highlighted in yellow indicate the NLS used for mammalian expression. Anchor residues are highlighted in magenta, UBD residues are highlighted in cyan, linker residues are highlighted in gray, and eGFP residues are highlighted in green. A556 in UBQ1UBA-WT (Reader2.0) and A556E in UBQ1UBA-A556E (Reader2.1) are labeled red. The cysteines depicted in gray indicate the Alexa Fluor 488 conjugation sites used for the in vitro binding assays.

In vitro characterization of sensors binding to Ub–nucleosomes. (A–H) Histone octamers containing nonhydrolyzable Ub-histone mimics were combined with 147-mer Widom 601 DNA to reconstitute mononucleosomes, whose quality were monitored by (A) native PAGE and (C) SDS-PAGE. Similarly, tri-nucleosome arrays containing H2AK15Ub, H2AK119Ub, and H2BK120Ub were reconstituted with non-linker-ended tri-nucleosomes (NLE-tri) DNA (Carruthers et al., 1999; Winkler et al., 2011), digested with EcoRI, and analyzed by (B) native PAGE and (D) SDS-PAGE. (E) Alexa Fluor 488–labeled Reader1.0 (molecular weight = 14.3 kD) and IE1-tSR (pan-Ub–nucleosome sensor; molecular weight = 18.1 kD) were analyzed by SDS-PAGE and detected by fluorescence or Coomassie staining as indicated. (F) IE1-tSR affinities for mono- or tri-nucleosomes containing H2AK119Ub and mononucleosomes containing H2AK129Ub were determined by measuring fluorescence changes in Alexa Fluor 488–labeled IE1-tSR upon titration with increasing concentrations of the indicated Ub–nucleosomes. Affinities of (G) Reader1.0 and (H) Reader2.1 for mono- and tri-nucleosome arrays containing H2AK15Ub, H2AK119Ub, and H2BK120Ub. Tables in G and H show the results from fitting the data with a single-site binding model. Ki values were determined from half-maximal inhibitory concentrations (IC50) using the Cheng and Prusoff (1973) equation.
In vitro characterization of sensors binding to Ub–nucleosomes. (A–H) Histone octamers containing nonhydrolyzable Ub-histone mimics were combined with 147-mer Widom 601 DNA to reconstitute mononucleosomes, whose quality were monitored by (A) native PAGE and (C) SDS-PAGE. Similarly, tri-nucleosome arrays containing H2AK15Ub, H2AK119Ub, and H2BK120Ub were reconstituted with non-linker-ended tri-nucleosomes (NLE-tri) DNA (Carruthers et al., 1999; Winkler et al., 2011), digested with EcoRI, and analyzed by (B) native PAGE and (D) SDS-PAGE. (E) Alexa Fluor 488–labeled Reader1.0 (molecular weight = 14.3 kD) and IE1-tSR (pan-Ub–nucleosome sensor; molecular weight = 18.1 kD) were analyzed by SDS-PAGE and detected by fluorescence or Coomassie staining as indicated. (F) IE1-tSR affinities for mono- or tri-nucleosomes containing H2AK119Ub and mononucleosomes containing H2AK129Ub were determined by measuring fluorescence changes in Alexa Fluor 488–labeled IE1-tSR upon titration with increasing concentrations of the indicated Ub–nucleosomes. Affinities of (G) Reader1.0 and (H) Reader2.1 for mono- and tri-nucleosome arrays containing H2AK15Ub, H2AK119Ub, and H2BK120Ub. Tables in G and H show the results from fitting the data with a single-site binding model. Ki values were determined from half-maximal inhibitory concentrations (IC50) using the Cheng and Prusoff (1973) equation.
Molecular dynamics (MD) simulations of Ub–nucleosomes guided the design of H2BK120Ub-selective sensors
The Ub C-terminal tail is highly flexible. In combination with all rotamers of the substrate Lys side chain modified by Ub, this tail allows Ub protein conjugates to sample a very large conformational space. Therefore, to guide the design of sensors with better selectivity profiles, we conducted MD simulations to identify conformations adopted by Ub when covalently attached to either H2AK15 or H2BK120 in nucleosomes. Despite the wide use of MD to examine the flexibility of proteins and nucleic acids, application of MD is often limited by the size of the conformational space and the complexity of the free energy landscapes of macromolecules. An approach to overcome sampling limitations is the analysis of an ensemble of multiple independent MD simulations done at high virtual temperatures (Day and Daggett, 2005a; Day and Daggett, 2005b). Here, we used all-atom MD simulations to study minimal systems extracted from 3D nucleosome structures available from the Protein Data Bank (Morgan et al., 2016; Wilson et al., 2016); specifically, one copy of the H2A/H2BK120Ub or H2AK15Ub/H2B dimer with the surrounding DNA double helix was employed (Fig. 2 A); a superposition algorithm then docked selected anchors and UBDs into the aligned MD snapshots. After discarding conformations where anchor or UBD docking led to steric clashes, we observed that, whereas 70–96% of the H2AK15Ub and H2BK120Ub conformation snapshots could bind to either the LANA or IE1 anchors, only 5% of the H2AK15Ub conformers showed simultaneous nucleosome acidic patch accessibility to IE1 and Ub accessibility to UBQ1UBA. On the other hand, 50% of the H2BK120Ub conformations showed accessibility to both domains (Fig. 2, B and C). For the subset of conformations simultaneously accessible to IE1 and UBQ1UBA, we measured the distances separating H2AE61 from UbI44 and found that the allowable H2BK120Ub and H2AK15Ub conformations showed distinct distributions (Fig. 2 D). In particular, for H2AE61 to UbI44 distances of 30–35 Å, accessible H2BK120Ub conformations did not overlap with those of H2AK15Ub. This prediction allowed us to design H2BK120Ub-specific sensors in which IE1 was linked directly to UBQ1UBA (Reader2.0) or UBQ1UBA-A556E (Reader2.1). The A556E mutation was made to increase the UBD•Ub affinity threefold (Kd = 7.4 µM; Fig. 2 E and Fig. S1; Choi et al., 2019b).

MD simulations of Ub–nucleosomes guided design of sensors for H2BK120Ub. (A) Starting structures for all-atom MD simulations of H2AK15Ub– and H2BK120Ub–nucleosomes included one H2A/H2B dimer, the surrounding DNA double helix, and Ub linked through an isopeptide bond to either H2AK15 or H2BK120. DNA is shown as a gray molecular surface; the histone octamer is dark gray, except for the H2A/H2B dimer used for simulations (H2A, light orange; H2B, pale green). Ub is shown as an orange (H2AK15Ub) or green (H2BK120Ub) surface. (B) Percentage of MD conformations accessible with the indicated anchors and UBDs after alignment to a reference nucleosome, docking, and filtering to remove steric clashes. (C) Positions of Ub residue I44 in all allowable conformers of H2AK15Ub (orange) or H2BK120Ub (green) when both the IE1 anchor and UBQ1UBA UBD are docked. (D) For the MD conformers in C, distributions of distances between the nucleosome acidic patch (H2AE61, δ carbon) and the Ub hydrophobic patch (I44, β carbon) revealed a cluster of conformers unique to H2BK120Ub. (E) Reader2.0/2.1 were generated by connecting the IE1 anchor to a UBD (UBQ1UBA-WT or UBQ1UBA-A556E) without a linker. (F) Schematic of the competition assays used to measure affinities between Reader2.0/2.1 and Ub–nucleosomes in vitro. (G) Reader2.0/2.1 binding was measured with the indicated Ub–nucleosomes; the affinities are shown in H as Ki (nM) values. conf., conformations; R2.0, Reader2.0; R2.1, Reader2.1.
MD simulations of Ub–nucleosomes guided design of sensors for H2BK120Ub. (A) Starting structures for all-atom MD simulations of H2AK15Ub– and H2BK120Ub–nucleosomes included one H2A/H2B dimer, the surrounding DNA double helix, and Ub linked through an isopeptide bond to either H2AK15 or H2BK120. DNA is shown as a gray molecular surface; the histone octamer is dark gray, except for the H2A/H2B dimer used for simulations (H2A, light orange; H2B, pale green). Ub is shown as an orange (H2AK15Ub) or green (H2BK120Ub) surface. (B) Percentage of MD conformations accessible with the indicated anchors and UBDs after alignment to a reference nucleosome, docking, and filtering to remove steric clashes. (C) Positions of Ub residue I44 in all allowable conformers of H2AK15Ub (orange) or H2BK120Ub (green) when both the IE1 anchor and UBQ1UBA UBD are docked. (D) For the MD conformers in C, distributions of distances between the nucleosome acidic patch (H2AE61, δ carbon) and the Ub hydrophobic patch (I44, β carbon) revealed a cluster of conformers unique to H2BK120Ub. (E) Reader2.0/2.1 were generated by connecting the IE1 anchor to a UBD (UBQ1UBA-WT or UBQ1UBA-A556E) without a linker. (F) Schematic of the competition assays used to measure affinities between Reader2.0/2.1 and Ub–nucleosomes in vitro. (G) Reader2.0/2.1 binding was measured with the indicated Ub–nucleosomes; the affinities are shown in H as Ki (nM) values. conf., conformations; R2.0, Reader2.0; R2.1, Reader2.1.
In vitro characterization of Reader2.0 and Reader2.1 employed competition assays with either Alexa Fluor 488–labeled Reader1.0 (for H2AK15Ub and H2BK120Ub) or a pan-Ub–nucleosome sensor (for H2AK119Ub and H2AK129Ub; Fig. 2 F; and Fig. S2, E and F). Equipped with a higher-affinity UBD, Reader2.1 binds H2BK120Ub with an inhibition constant (Ki) of 35 nM and 18- to 40-fold selectivity over H2AK15Ub, H2AK119Ub, and H2AK129Ub (Fig. 2, G and H). Similar affinities and selectivities were observed with tri-nucleosome arrays (Fig. S2, B, D, and H). In comparison, Reader2.0 has a Ki of 144 nM for H2BK120Ub and, respectively, 9- and 18-fold selectivity over H2AK15Ub and H2AK119Ub (Fig. 2, G and H).
Reader2.0/2.1 are reporters for H2BK120Ub in human osteosarcoma (U-2 OS) cells
To evaluate if the sensors can report chromatin ubiquitination in vivo, we expressed them as eGFP fusions in mammalian cells (Fig. S1). First, U-2 OS cells were transiently transfected with plasmids encoding NLS-Reader1.0/2.0/2.1-eGFP and immunostained with an anti-H2BK120Ub antibody. Unexpectedly, cells expressing high levels of Reader2.0 or Reader2.1 showed increases in H2BK120Ub (Fig. S3, A and B) with a positive correlation between H2BK120Ub levels and Reader2.0/2.1 signals (Fig. S3, D and E). These effects were most likely caused by Reader2.0/2.1 binding to H2BK120Ub and protecting it from deubiquitinating enzymes (DUBs) that otherwise would remove the PTM (Atanassov et al., 2016; Henry et al., 2003; Samara et al., 2010). Unlike Reader2.0/2.1, overexpression of Reader1.0 did not affect H2BK120Ub levels in U-2 OS cells (Fig. S3, A–C).

U-2 OS cells expressing Reader2.0 and Reader2.1 at high levels showed an increase in H2BK120Ub. (A) U-2 OS cells transfected with Reader1.0/2.0/2.1-eGFP were stained with an antibody to H2BK120Ub. Cell nuclei were counterstained with DAPI. Scale bar, 5 µm. (B) MFI of H2BK120Ub and Reader1.0/2.0/2.1-eGFP signals were measured using the ZEN 2.3 imaging software. Cells were clustered into three groups according to sensors’ expression levels: Reader-eGFP (-) or nontransfected controls (MFI < 1,000); Reader-eGFP low (MFI between 1,000 and 10,000); and Reader-eGFP high (MFI > 10,000). Between 30 and 63 cells were analyzed per condition. Bars show mean ± SD. Statistical analyses are as described in Materials and methods. (C–E) Correlation between H2BK120Ub levels and Reader expression levels in cells expressing the indicated sensor at high levels (MFI > 10,000). (F) MFI of Reader1.0/2.0/2.1-eGFP in cells analyzed by FRAP (Fig. 3). (G) Nuclear mobilities of Reader1.0/2.0/2.1-eGFP and their mutant variants were assessed by FRAP (ROI-1, gray circle). Fluorescence recoveries were monitored at 0.5-s intervals, background-corrected (ROI-2, yellow square), and normalized to pre-bleach fluorescence intensities. ROI-3 (blue square) and ROI-4 (green rectangle) were used for monitoring unintentional bleaching and for image acquisition, respectively. Scale bar, 5 µm. R1.0, Reader1.0; R2.0, Reader2.0; R2.1, Reader2.1; t, time.
U-2 OS cells expressing Reader2.0 and Reader2.1 at high levels showed an increase in H2BK120Ub. (A) U-2 OS cells transfected with Reader1.0/2.0/2.1-eGFP were stained with an antibody to H2BK120Ub. Cell nuclei were counterstained with DAPI. Scale bar, 5 µm. (B) MFI of H2BK120Ub and Reader1.0/2.0/2.1-eGFP signals were measured using the ZEN 2.3 imaging software. Cells were clustered into three groups according to sensors’ expression levels: Reader-eGFP (-) or nontransfected controls (MFI < 1,000); Reader-eGFP low (MFI between 1,000 and 10,000); and Reader-eGFP high (MFI > 10,000). Between 30 and 63 cells were analyzed per condition. Bars show mean ± SD. Statistical analyses are as described in Materials and methods. (C–E) Correlation between H2BK120Ub levels and Reader expression levels in cells expressing the indicated sensor at high levels (MFI > 10,000). (F) MFI of Reader1.0/2.0/2.1-eGFP in cells analyzed by FRAP (Fig. 3). (G) Nuclear mobilities of Reader1.0/2.0/2.1-eGFP and their mutant variants were assessed by FRAP (ROI-1, gray circle). Fluorescence recoveries were monitored at 0.5-s intervals, background-corrected (ROI-2, yellow square), and normalized to pre-bleach fluorescence intensities. ROI-3 (blue square) and ROI-4 (green rectangle) were used for monitoring unintentional bleaching and for image acquisition, respectively. Scale bar, 5 µm. R1.0, Reader1.0; R2.0, Reader2.0; R2.1, Reader2.1; t, time.
In the absence of DNA damage, Ub–nucleosomes predominantly contain H2AK119Ub or H2BK120Ub (Weake and Workman, 2008). We investigated if Reader1.0/2.0/2.1 recognize these nucleosomes in live cells. Guided by the measurements of H2BK120Ub in U-2 OS cells expressing different levels of the sensors (Fig. S3, A and B), we analyzed cells expressing Reader1.0/2.0/2.1-eGFP at relatively low levels (i.e., 500–10,000 mean fluorescence intensity [MFI]) in order to minimize potential interferences with endogenous signaling events (Fig. S3 F). FRAP measurements in the nucleus were used to determine the kinetics of sensor diffusion and binding to chromatin (Fig. S3 G). To evaluate the individual contributions of anchor and UBD, we also introduced mutations to abrogate the function of each domain. For clarity, the mutants were named based on which domain remains functional: mutants lacking anchor-nucleosome interaction are denoted as R1.0/2.1-UBD, mutants lacking UBD-Ub interactions are denoted as R1.0/2.1-anchor, and mutants lacking both the anchor-nucleosome and the UBD-Ub interactions are denoted as nonbinding (R1.0/2.1-NB; Fig. 3 A and Fig. S3 F).

Reader2.0 and Reader2.1 detect H2BK120Ub in the nucleus. (A) Reader1.0/2.0/2.1-eGFP and constructs mutated to eliminate interactions with Ub (Anchor), nucleosome acidic patch (UBD), or both (NB) are listed. Images show nuclear localization of the proteins expressed in U-2 OS cells. Scale bar, 5 µm. (B) FVP or C1 was used to deplete, respectively, H2BK120Ub or all histone Ub conjugation. U-2 OS cells treated with 5 µM FVP or 10 µM C1 for the indicated times were analyzed by Western blotting. (C and D) Live-cell FRAP measurements of Reader1.0/2.0/2.1-eGFP variants expressed in U-2 OS cells with or without 5 µM FVP or 10 µM C1 pretreatment for 1 h. FRAP kinetics were fit best by a single fast recovery rate for R1.0/2.1-NB and R1.0/2.1-UBD(C), or two exponential components for all other constructs (D). Note that Reader2.0/2.1 have the same anchor, which is referred to as R2.1-Anchor. (E) Fractions (%) of the fast and slow components of FRAP recoveries were determined from fits to the data in C and D; calculated recovery t1/2 values are in Table 1. (F) Models depicting the origins of fast and slow components of the FRAP recoveries. R1.0, Reader1.0; R1.1, Reader1.1; R2.0, Reader2.0; R2.1, Reader2.1.
Reader2.0 and Reader2.1 detect H2BK120Ub in the nucleus. (A) Reader1.0/2.0/2.1-eGFP and constructs mutated to eliminate interactions with Ub (Anchor), nucleosome acidic patch (UBD), or both (NB) are listed. Images show nuclear localization of the proteins expressed in U-2 OS cells. Scale bar, 5 µm. (B) FVP or C1 was used to deplete, respectively, H2BK120Ub or all histone Ub conjugation. U-2 OS cells treated with 5 µM FVP or 10 µM C1 for the indicated times were analyzed by Western blotting. (C and D) Live-cell FRAP measurements of Reader1.0/2.0/2.1-eGFP variants expressed in U-2 OS cells with or without 5 µM FVP or 10 µM C1 pretreatment for 1 h. FRAP kinetics were fit best by a single fast recovery rate for R1.0/2.1-NB and R1.0/2.1-UBD(C), or two exponential components for all other constructs (D). Note that Reader2.0/2.1 have the same anchor, which is referred to as R2.1-Anchor. (E) Fractions (%) of the fast and slow components of FRAP recoveries were determined from fits to the data in C and D; calculated recovery t1/2 values are in Table 1. (F) Models depicting the origins of fast and slow components of the FRAP recoveries. R1.0, Reader1.0; R1.1, Reader1.1; R2.0, Reader2.0; R2.1, Reader2.1.
Based on FRAP recovery half-lives, we categorized two types of binding behavior. First, R1.0/2.1-UBD and R1.0/2.1-NB have similarly fast FRAP recoveries (t1/2 < 1.0 s), indicating that the UBD alone does not contribute significantly to the nuclear mobility of the sensors (Fig. 3, C, E, and F; and Table 1). This is not surprising given that tSR and UBQ1UBA bind Ub with modest affinities and that competition by unconjugated Ub, which is abundant and distributed throughout the nucleus under basal conditions (Choi et al., 2019a; Clague et al., 2015), will reduce Ub–nucleosome binding even further. Second, FRAP behavior of the R1.0/2.1-Anchor mutants showed at least two components, with 20–30% of the population bound to chromatin (LANA t1/2 = 10.2 ± 7.8 s; IE1 t1/2 = 10.2 ± 6.6 s) and the rest freely diffused (Fig. 3, D–F; and Table 1). FRAP of the three readers showed different combinations of fast and slow components. Comparison of the half-lives together with the fractions estimated for the fast and slow components indicates that Reader2.0/2.1 interact more tightly with chromatin than the IE1 anchor alone. The slower rates (Reader2.0 t1/2 = 20.2 ± 6.8 s; Reader2.1 t1/2 = 31.6 ± 6.1 s) and the large fractions of the slow components (57% and 74%, respectively) suggest avid binding to Ub–nucleosomes. In contrast, Reader1.0 and Reader1.0-Anchor had similar mobilities (Fig. 3, D–F; and Table 1). Thus, in live cells, neither H2AK119Ub nor H2BK120Ub recruits Reader1.0 to nucleosomes.
Summary of fits for Reader1.0/2.0/2.1-eGFP in the nucleoplasm and Reader1.0-eGFP in the nucleoplasm and at laser-induced DSBs
| FRAP t1/2 (s)f | |||||||
|---|---|---|---|---|---|---|---|
| Kinetic component | NB | UBD | Anchor | Reader | Reader + FVP | Reader + C1 | |
| Reader1.0 Nucleoplasma | Fast | 0.6 ± 0.3 (100%)c | 0.7 ± 0.4 (100%)c | 0.7 ± 0.3 (80.5%)d | 1.1 ± 0.5 (72.6%)d | 0.9 ± 0.5 (68.4%)d | 0.8 ± 0.4 (79.1%)d |
| Slow | 10.2 ± 7.8 (19.5%)d | 11.0 ± 7.2 (27.4%)d | 6.5 ± 4.0 (31.6%)d | 8.3 ± 7.2 (20.9%)d | |||
| Reader2.0 Nucleoplasma | Fast | 1.9 ± 1.2 (43.1%)d | 1.6 ± 0.9 (60.8%)d | 1.5 ± 0.5 (74.9%)d | |||
| Slow | 20.2 ± 6.8 (56.9%)d | 11.0 ± 5.9 (39.2%)d | 14.6 ± 9.5 (25.1%)d | ||||
| Reader2.1 Nucleoplasma | Fast | 0.7 ± 0.4 (100%)c | 0.6 ± 0.3 (100%)c | 1.4 ± 0.7 (69.3%)d | 1.3 ± 0.9 (26.1%)d | 1.5 ± 0.6 (59.2%)d | 1.2 ± 0.5 (74.3%)d |
| Slow | 10.2 ± 6.6 (30.7%)d | 31.6 ± 6.1 (73.9%)d | 15.9 ± 5.5 (40.8%)d | 10.6 ± 6.9 (25.7%)d | |||
| Reader1.0 Nucleoplasmb | Fast | 0.6 ± 1.2 (69.6%)e | |||||
| Slow | 4.5 ± 9.8 (30.4%)e | ||||||
| Reader1.0 DSBsb | Fast | 0.9 ± 1.7 (16.4%)e | |||||
| Slow | 15.1 ± 2.9 (83.6%)e | ||||||
FRAP of Reader1.0/2.0/2.1-eGFP transiently transfected in U-2 OS cells; n ≥ 6 cells per condition (two to five measurements per cell); photobleaching of circular ROIs in the nucleus (diameter = 30 pixels; 1 pixel = 0.09 × 0.09 µm).
FRAP of Reader1.0-eGFP stably expressed in U-2 OS cells; n = 6 cells (two measurements per cell in both the nucleoplasm and at laser-induced DSBs); rectangular ROI (0.75 µm2). Note that the ROI in these experiments is ∼7.5-fold smaller than the ROI used in the FRAP measurements of Reader1.0/2.0/2.1 in the nucleoplasm. The differences observed for the slow FRAP t1/2 of Reader1.0 in the nucleoplasm suggest that sensor’s diffusion rate has an impact in its recovery kinetics.
Fit to one exponential; time = 25 s; constraints: plateau < 1 and normalized fluorescence set to 0 at time zero.
Fit to two exponentials; time = 120 s; constraints: plateau < 1 and normalized fluorescence set to 0 at time zero.
Fit to two exponentials; time = 90 s; constraints: plateau < 1 and normalized fluorescence set to 0 at time zero.
FRAP t1/2 for fast and slow components of the recovery curves. SDs were calculated from the 95% CIs. Percentages corresponding to fast and slow recoveries are shown in parentheses.
To validate the specificities of the Readers with Ub–nucleosomes, we performed FRAP measurements after cells were treated with flavopiridol (FVP) or compound 1 (C1). FVP potently inhibits phosphorylation of the RNA polymerase II C-terminal domain in transcriptionally active chromatin and is required to maintain H2BK120Ub (Pirngruber et al., 2009); C1 inhibits the E1 Ub-activating enzyme (Chen et al., 2011), thereby blocking all protein ubiquitination. Western blot analysis of cell lysates confirmed that, as expected, FVP reduced H2BK120Ub without affecting H2AK119Ub (Fig. 3 B), whereas C1 depleted both H2BK120Ub and H2AK119Ub (Fig. 3 B). In C1-treated cells, Reader1.0/2.0/2.1 showed FRAP behavior similar to the R1.0/2.1-Anchor (Fig. 3, D and E; and Table 1). For Reader2.0/2.1, FVP treatment increased Reader2.0/2.1 nuclear mobilities overall (Fig. 3 D and Table 1) and markedly decreased the slow component (Fig. 3 E and Table 1), suggesting that those sensors bind predominantly to H2BK120Ub in vivo. However, differences observed between the treatments with C1 and FVP suggest that there are minor albeit detectable off-target interactions with chromatin, possibly due to Reader2.0/2.1 binding to H2AK119Ub. Note that although Reader2.1 has higher affinity than Reader2.0 to H2BK120Ub, it is not more selective (Fig. 3 D and Table 1). For Reader1.0, FRAP was slightly affected by C1 treatment and unaffected by FVP, suggesting that, although Reader1.0 might weakly interact with unknown Ub species in chromatin, interactions with H2BK120Ub were negligible (Fig. 3, D–F; and Table 1). Taken together, our results suggest that, in cells, Reader2.0/2.1 are selective for H2BK120Ub, whereas neither H2BK120Ub nor H2AK119Ub is reported by Reader1.0.
Reader1.0 is recruited to DNA DSBs by RNF168-dependent H2AK13/15 ubiquitination
Ubiquitination of H2AK13/15 by RNF168 is a DNA damage-induced histone PTM that occurs downstream of phosphorylation of H2AX by the ataxia-telangiectasia mutated kinase (Mattiroli et al., 2012). We used laser microirradiation to damage DNA in nuclei of U-2 OS cells stably expressing Reader1.0/2.0/2.1-eGFP under the control of doxycycline (Dox)-inducible promoters. Reader1.0 was efficiently recruited to DNA lesions where, on average, sensor fluorescence increased 3.5-fold in 15 min after microirradiation (Fig. 4, A and B). This accumulation of Reader1.0 at the DNA damage tracks was abolished in cells transfected with RNF168 siRNA (Fig. 4 A), indicating that Reader1.0 recognizes the H2AK13/15Ub signal generated by this E3 ligase. FRAP measurements revealed that Reader1.0 has a slower average mobility at the DNA DSBs (slow t1/2 of 15.1 ± 2.9 s accounts for 83.6% of the recovery) than in undamaged regions of the nucleus (slow t1/2 of 4.5 ± 9.8 s accounts for 30.4%; Fig. 4 C and Table 1). These results are evidence that Reader1.0 recruitment to DSBs is due to tight, avid binding to H2AK13/15Ub (Fig. 4 D) and are consistent with high density of this histone PTM at the damage sites. In contrast, Reader2.0/2.1 accumulated far less at laser-induced DSBs (Fig. 4, A and B). As with Reader1.0, the Reader2.0/2.1 recruitment was absent in cells transfected with siRNF168 (Fig. 4 A). Thus, despite selectivity for H2BK120Ub in undamaged chromatin, Reader2.0/2.1 can interact with other types of Ub–nucleosomes if they are at high local concentrations.
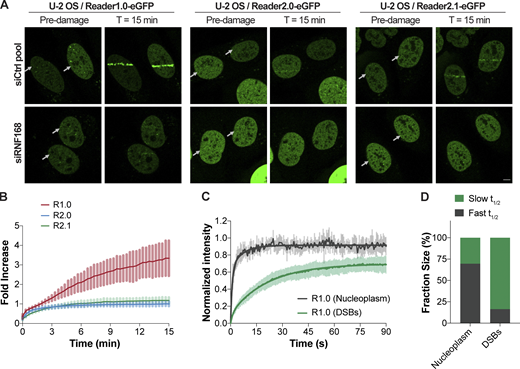
Reader1.0 is recruited to DNA damage sites by RNF168-dependent H2A ubiquitination.(A) U-2 OS Reader1.0/2.0/2.1-eGFP cells were transfected twice with 10 nM siCtrl pool or siRNF168 (Table S1) at 0 h and 24 h, and analyzed at 72 h. Reader1.0/2.0/2.1 expression was induced (100 ng/ml Dox, 4–24 h), and cells were subjected to 405-nm laser microirradiation and monitored for sensor recruitment to the laser tracks. Arrows indicate positions of laser damage (horizontal tracks). Scale bar, 5 µm. (B) eGFP fluorescence within the laser tracks was monitored over time after microirradiation. Mean ± SD are plotted for n ≥ 6 cells per condition. (C) FRAP of Reader1.0-eGFP showed slower recovery at the laser-induced DNA lesions than in areas of undamaged nucleoplasm. Mean ± SD determined from two measurements at the damage site and two measurements in the nucleoplasm per cell; n = 6 cells. (D) Fractions of the fast and slow components accounting for the FRAP kinetics at DNA damage sites and in the nucleoplasm. Fractions correspond to curves shown in C; FRAP t1/2s are in Table 1. R1.0, Reader1.0; R2.0, Reader2.0; R2.1, Reader2.1.
Reader1.0 is recruited to DNA damage sites by RNF168-dependent H2A ubiquitination.(A) U-2 OS Reader1.0/2.0/2.1-eGFP cells were transfected twice with 10 nM siCtrl pool or siRNF168 (Table S1) at 0 h and 24 h, and analyzed at 72 h. Reader1.0/2.0/2.1 expression was induced (100 ng/ml Dox, 4–24 h), and cells were subjected to 405-nm laser microirradiation and monitored for sensor recruitment to the laser tracks. Arrows indicate positions of laser damage (horizontal tracks). Scale bar, 5 µm. (B) eGFP fluorescence within the laser tracks was monitored over time after microirradiation. Mean ± SD are plotted for n ≥ 6 cells per condition. (C) FRAP of Reader1.0-eGFP showed slower recovery at the laser-induced DNA lesions than in areas of undamaged nucleoplasm. Mean ± SD determined from two measurements at the damage site and two measurements in the nucleoplasm per cell; n = 6 cells. (D) Fractions of the fast and slow components accounting for the FRAP kinetics at DNA damage sites and in the nucleoplasm. Fractions correspond to curves shown in C; FRAP t1/2s are in Table 1. R1.0, Reader1.0; R2.0, Reader2.0; R2.1, Reader2.1.
Reader1.0 competes with 53BP1 for binding to H2AK13/15Ub at DSBs
The H2AK13/15Ub epigenetic mark formed by RNF168 is needed to recruit downstream repair factors of which 53BP1 is best understood; 53BP1 has been used as a surrogate for detection of the H2AK15Ub signal (Fradet-Turcotte et al., 2013). To investigate the predicted competition between Reader1.0 and 53BP1 for binding to sites of RNF168-mediated chromatin ubiquitination, we coexpressed Reader1.0-eGFP and the minimal focus-forming region (FFR) of 53BP1 (53BP1-FFR) fused to mCherry (Fradet-Turcotte et al., 2013) in U-2 OS cells (Fig. 5 A). Reader1.0 and 53BP1-FFR colocalized to laser-induced DNA lesions with similar kinetics (Fig. 5, B and D); the inverse correlation between the fluorescence intensities of 53BP1-FFR at the DNA lesions and Reader1.0 expression levels suggests that the sensor competes with 53BP1 for H2AK13/15Ub (Fig. 5, B and C). Increases of 1.5, 1.7, and 4.5-fold in 53BP1-FFR FRAP recovery rates were observed for cells in which Reader1.0 was expressed at low, medium, and high levels, respectively (Fig. 5 E). With high Reader1.0 expression, recruitment of 53BP1-FFR to laser-induced DSBs was greatly reduced, and 53BP1-FFR FRAP mobilities in damaged and undamaged regions of the nuclei were nearly identical (Fig. 5 E).

Reader1.0 competes with 53BP1 for binding to H2AK15Ub at DNA DSBs. (A) Experimental scheme to evaluate Reader1.0-eGFP competition with mCherry-53BP1-FFR at DNA DSBs. (B) U-2 OS cells were microirradiated (horizontal tracks at the arrows), and the recruitment of mCherry-53BP1-FFR and Reader1.0-eGFP to the damaged regions was monitored. Cells were grouped according to Reader1.0-eGFP expression levels evaluated as MFI (i.e., low: MFI < 4,000; medium: MFI = 4,000–10,000; high: MFI > 10,000). Scale bar, 5 µm. (C) Fold-increase in mCherry-53BP1-FFR fluorescence 15 min after 405-nm laser-induced DNA damage in cells with different levels of Reader1.0. (D) Reader1.0-eGFP and mCherry-53BP1-FFR showed similar rates of recruitment to DNA lesions in cells (n = 12) expressing low and medium levels of Reader1.0. (E) FRAP measurements of mCherry-53BP1-FFR in cells expressing different levels of Reader1.0. R1.0 (-) indicates parental U-2 OS cells only expressing mCherry-53BP1-FFR. Mean ± SD of n ≥ 3 cells per condition with two to five measurements per cell. (F) U-2 OS cells stably expressing Reader1.0-eGFP were treated with 0 or 100 ng/ml Dox for 24 h and then exposed to 1.5 Gy γ-irradiation. Cells were fixed at 2, 4, 8, 12, and 24 h after irradiation and immunostained for γH2AX. Scale bar, 5 µm. (G) Quantification of cells with >5 γH2AX foci. Mean ± SD of two or three independent replicates are plotted; n = 40–140 cells per replicate per condition. R1.0, Reader1.0.
Reader1.0 competes with 53BP1 for binding to H2AK15Ub at DNA DSBs. (A) Experimental scheme to evaluate Reader1.0-eGFP competition with mCherry-53BP1-FFR at DNA DSBs. (B) U-2 OS cells were microirradiated (horizontal tracks at the arrows), and the recruitment of mCherry-53BP1-FFR and Reader1.0-eGFP to the damaged regions was monitored. Cells were grouped according to Reader1.0-eGFP expression levels evaluated as MFI (i.e., low: MFI < 4,000; medium: MFI = 4,000–10,000; high: MFI > 10,000). Scale bar, 5 µm. (C) Fold-increase in mCherry-53BP1-FFR fluorescence 15 min after 405-nm laser-induced DNA damage in cells with different levels of Reader1.0. (D) Reader1.0-eGFP and mCherry-53BP1-FFR showed similar rates of recruitment to DNA lesions in cells (n = 12) expressing low and medium levels of Reader1.0. (E) FRAP measurements of mCherry-53BP1-FFR in cells expressing different levels of Reader1.0. R1.0 (-) indicates parental U-2 OS cells only expressing mCherry-53BP1-FFR. Mean ± SD of n ≥ 3 cells per condition with two to five measurements per cell. (F) U-2 OS cells stably expressing Reader1.0-eGFP were treated with 0 or 100 ng/ml Dox for 24 h and then exposed to 1.5 Gy γ-irradiation. Cells were fixed at 2, 4, 8, 12, and 24 h after irradiation and immunostained for γH2AX. Scale bar, 5 µm. (G) Quantification of cells with >5 γH2AX foci. Mean ± SD of two or three independent replicates are plotted; n = 40–140 cells per replicate per condition. R1.0, Reader1.0.
These results highlight the potential for competition between endogenous DNA repair machinery and Reader1.0 and the importance of controlling sensor expression levels. Therefore, we sought to determine if low Reader1.0 expression affects the repair process in cells exposed to ionizing radiation (IR). For this purpose, we assessed the kinetics of phosphorylated histone H2AX (γH2AX) focus dissolution in U-2 OS cells stably expressing Dox-inducible Reader1.0-eGFP. Control cells with sensor expression repressed (i.e., no Dox) and Reader1.0-expressing cells showed similar repair kinetics characterized by maximal γH2AX focal accumulation 2 h after IR and recovery to preirradiation levels after 24 h (Fig. 5, F and G). Thus, despite detectable competition with effectors of DNA repair, Reader1.0 can be used to monitor DNA damage–associated signaling without significant interference with the repair process.
Reader1.0 reports cell cycle–dependent changes in H2AK13/15Ub in unperturbed cells
When expressed in U-2 OS or RPE-1 cells, Reader1.0-eGFP accumulated in nuclear foci even without treatments to damage DNA (Fig. S4 A). RNF168 depletion largely abrogated these Reader1.0 foci (Fig. S4, B and C); thus, the sensor could be detecting H2AK13/15Ub formed at transiently stalled DNA replication forks (Schmid et al., 2018). To investigate how these H2AK13/15Ub signals may be regulated across the cell cycle, we generated a stable U-2 OS cell line in which Dox-inducible Reader1.0-eGFP was coexpressed with the cell cycle reporter PCNA-interacting protein (PIP)–fluorescent ubiquitination-based cell cycle indicator (FUCCI; Grant et al., 2018). In these live-cell experiments, Reader1.0 was used to detect nuclear foci enriched in H2AK13/15Ub while the cell cycle phases were identified based on the levels of fluorescently tagged PIP degron (Cdt11-17-iRFP682) and Geminin (mCherry-Geminin1-110; Fig. 6, A and B). To elucidate which DNA repair factors are involved in basal signaling (i.e., without exogenous DNA damage agents) by H2AK13/15Ub, we quantified numbers, intensities, and sizes of Reader1.0-eGFP foci in G1-, S-, and G2-phase in cells depleted of putative Ub–nucleosome writers (RNF8 and RNF168), readers (53BP1, BARD1, RNF169, and RAD18), and erasers (USP3, USP11, USP16, USP48, and USP51; Fig. 6 C, Fig. 7, and Data S1).

Reader1.0-eGFP accumulates in nuclear foci without treatments to induce DNA damage. (A) Reader1.0-eGFP accumulates as nuclear foci in unperturbed U-2 OS and RPE-1 cells. Scale bar, 5 µm. (B) Reader1.0-eGFP foci were markedly decreased in U-2 OS cells transfected with siRNF168. Scale bars, 5 µm. (C) Reader1.0-eGFP foci were stable after 4 h treatment with 5 µM FVP and decreased after treatment with 10 µM C1 (C1). Scale bars, 20 µm. (D) Levels of Cdt11-17-iRFP682 and mCherry-Geminin1-110 in most of the cells transfected with siRNF8 were below the established thresholds, thus precluding cell cycle classification. Scale bars, 20 µm. (E) Knockdown efficiency by siRNA in U-2 OS/PIP-FUCCI/Reader1.0 cells was assessed by RT-qPCR; plots show mean ± SEM and indicate mRNA expression levels relative to siCtrl #1 (genes: BARD1, USP3, USP16, USP48, and USP51) or siCtrl #2 (genes: RNF169 and RAD18); n = 2 technical replicates (siCtrl #1, siBARD1, siUSP3, siUSP16, siUSP48, and siUSP51) or n = 2 biological replicates (siCtrl #2, siRNF169, and siRAD18); GAPDH was used as a reference gene for normalization. (F) U-2 OS/Reader1.0 cells transfected with the indicated siRNAs and treated with 10 ng/ml Dox for 24 h before fixation were stained with an antibody to 53BP1. Scale bar, 10 µm. 53BP1 foci number (G) and nuclear 53BP1 fluorescence intensity (H) were quantified from at least 100 cells. Bars show mean ± SD. Statistical analyses are as described in Materials and methods. ***, P < 0.0001. Note that Reader 1.0-eGFP foci are difficult to observe in formaldehyde-fixed cells.
Reader1.0-eGFP accumulates in nuclear foci without treatments to induce DNA damage. (A) Reader1.0-eGFP accumulates as nuclear foci in unperturbed U-2 OS and RPE-1 cells. Scale bar, 5 µm. (B) Reader1.0-eGFP foci were markedly decreased in U-2 OS cells transfected with siRNF168. Scale bars, 5 µm. (C) Reader1.0-eGFP foci were stable after 4 h treatment with 5 µM FVP and decreased after treatment with 10 µM C1 (C1). Scale bars, 20 µm. (D) Levels of Cdt11-17-iRFP682 and mCherry-Geminin1-110 in most of the cells transfected with siRNF8 were below the established thresholds, thus precluding cell cycle classification. Scale bars, 20 µm. (E) Knockdown efficiency by siRNA in U-2 OS/PIP-FUCCI/Reader1.0 cells was assessed by RT-qPCR; plots show mean ± SEM and indicate mRNA expression levels relative to siCtrl #1 (genes: BARD1, USP3, USP16, USP48, and USP51) or siCtrl #2 (genes: RNF169 and RAD18); n = 2 technical replicates (siCtrl #1, siBARD1, siUSP3, siUSP16, siUSP48, and siUSP51) or n = 2 biological replicates (siCtrl #2, siRNF169, and siRAD18); GAPDH was used as a reference gene for normalization. (F) U-2 OS/Reader1.0 cells transfected with the indicated siRNAs and treated with 10 ng/ml Dox for 24 h before fixation were stained with an antibody to 53BP1. Scale bar, 10 µm. 53BP1 foci number (G) and nuclear 53BP1 fluorescence intensity (H) were quantified from at least 100 cells. Bars show mean ± SD. Statistical analyses are as described in Materials and methods. ***, P < 0.0001. Note that Reader 1.0-eGFP foci are difficult to observe in formaldehyde-fixed cells.

A system to monitor H2AK13/K15Ub across the cell cycle in unperturbed cells. (A) Live-cell imaging of U-2 OS cells expressing PIP-FUCCI and Reader1.0. Cell cycle phases were assigned based on the levels of Cdt11-17-iRFP682 and mCherry-Geminin1-110 (Grant et al., 2018). (B) Reader1.0-eGFP foci numbers (mean ± SD), normalized fluorescence intensities (mean ± SD), and sizes (median with interquartile range) are shown for cells transfected with siCtrl RNAs (n = 3–8 biological replicates). (C) Number of Reader1.0 foci (mean ± SD), normalized fluorescence intensities of Reader1.0 at foci (mean ± SD), and foci sizes (median with interquartile range) were measured for asynchronous cells in G1, S, and G2 phases transfected with the indicated siRNAs (n = 2–8 biological replicates). Multiple (B) or pairwise (C) comparisons are described in Materials and methods: *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant. Gem, Geminin; R1.0, Reader1.0.
A system to monitor H2AK13/K15Ub across the cell cycle in unperturbed cells. (A) Live-cell imaging of U-2 OS cells expressing PIP-FUCCI and Reader1.0. Cell cycle phases were assigned based on the levels of Cdt11-17-iRFP682 and mCherry-Geminin1-110 (Grant et al., 2018). (B) Reader1.0-eGFP foci numbers (mean ± SD), normalized fluorescence intensities (mean ± SD), and sizes (median with interquartile range) are shown for cells transfected with siCtrl RNAs (n = 3–8 biological replicates). (C) Number of Reader1.0 foci (mean ± SD), normalized fluorescence intensities of Reader1.0 at foci (mean ± SD), and foci sizes (median with interquartile range) were measured for asynchronous cells in G1, S, and G2 phases transfected with the indicated siRNAs (n = 2–8 biological replicates). Multiple (B) or pairwise (C) comparisons are described in Materials and methods: *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant. Gem, Geminin; R1.0, Reader1.0.

Use of Reader1.0 to identify regulators of H2AK13/15Ub. (A) Cell cycle distribution of U-2 OS cells expressing PIP-FUCCI and Reader1.0 and transfected with the indicated siRNAs. Stacked bar chart shows mean ± SD (n = 2–8 biological replicates) for each siRNA; for each cell cycle phase, P values from pairwise comparisons with siCtrl are shown, with significant (P < 0.01) increases highlighted in red and significant decreases in green. (B–D) Representative images of (B) G1 phase, (C) S phase, and (D) G2 phase cells transfected with the indicated siRNAs. (E) Reader1.0-eGFP foci numbers (mean ± SD), normalized fluorescence intensities (mean ± SD), and sizes (median with interquartile range) are shown grouped based on cell cycle phase. P values from pairwise comparisons with siCtrl: *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant; n.d., not determined. Data for siCtrl #1 and siCtrl #2 are shared with Fig. 6 B. Gem, Geminin; R1.0, Reader1.0.
Use of Reader1.0 to identify regulators of H2AK13/15Ub. (A) Cell cycle distribution of U-2 OS cells expressing PIP-FUCCI and Reader1.0 and transfected with the indicated siRNAs. Stacked bar chart shows mean ± SD (n = 2–8 biological replicates) for each siRNA; for each cell cycle phase, P values from pairwise comparisons with siCtrl are shown, with significant (P < 0.01) increases highlighted in red and significant decreases in green. (B–D) Representative images of (B) G1 phase, (C) S phase, and (D) G2 phase cells transfected with the indicated siRNAs. (E) Reader1.0-eGFP foci numbers (mean ± SD), normalized fluorescence intensities (mean ± SD), and sizes (median with interquartile range) are shown grouped based on cell cycle phase. P values from pairwise comparisons with siCtrl: *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant; n.d., not determined. Data for siCtrl #1 and siCtrl #2 are shared with Fig. 6 B. Gem, Geminin; R1.0, Reader1.0.
It has been reported that foci sizes of different DNA repair factors correlate with repair pathway choice, with factors involved in nonhomologous end joining (NHEJ) forming larger foci than those in homologous recombination (HR; Luijsterburg et al., 2017). Indeed, we observed that average Reader1.0 foci size follows the trend G1 > S > G2, consistent with preferred repair pathway choice associated with each cell cycle phase (Fig. 6 B and Data S1).
In asynchronous cell populations without cell cycle classification, depletion of RNF8 or RNF168 resulted in decreased numbers and intensities of Reader1.0 foci (Fig. 6 C and Data S1). Whereas RNF168 knockdown was characterized by an increased fraction of cells in G1 as previously reported (Schmid et al., 2018; Fig. 7 A and Data S1), cells depleted of RNF8 showed a global reduction of both Cdt11-17-iRFP682 and mCherry-Geminin1-110, thereby precluding their cell cycle classification (Fig. S4 D). Shifts in cell cycle distribution marked by an increased G1 fraction with concomitant decrease in S-phase cells were also observed with 53BP1 and BARD1 depletion. USP3 knockdown, by contrast, shifted the distribution from S to G2 phase, in agreement with a previous report (Nicassio et al., 2007). Finally, RNF169 knockdown moderately increased the fraction of cells in G1, and RAD18 knockdown led to a small decrease in the fraction of cells in G2 (Fig. 7 A and Data S1).
In addition to RNF8 and RNF168 knockdowns, prominent effects on Reader1.0 focal accumulation were observed in cells depleted of 53BP1, BARD1, RNF169, or USP51 (Fig. 6 C, Fig. 7 E, and Data S1). 53BP1 depletion significantly increased the number of Reader1.0 foci in G1 and the foci intensities in both G1 and S phases. Statistically significant increases in foci size were also observed, although the magnitudes of the changes were small (Fig. 7, B–E). These findings are consistent with two possibilities: (1) loss of 53BP1 may promote an increase in H2AK13/15Ub in G1 and S phases, or (2) reduced competition by endogenous 53BP1 allows more Reader1.0 to bind to H2AK13/15Ub-containing nucleosomes. These possibilities are not mutually exclusive. Our results strongly support that 53BP1 is the major reader for the H2AK13/15Ub signal in G1 and possibly early S phase.
A recent manuscript suggests that BARD1 may also be a reader for H2AK15Ub (Becker et al., 2020). We found that BARD1 knockdown led to increases in both the number and intensities of Reader1.0 foci throughout the cell cycle. Unlike 53BP1 depletion, which had its biggest impact in G1 phase, the effect of BARD1 knockdown was greatest in G2 (Fig. 7, D and E; and Data S1), consistent with the roles of BRCA1/BARD1 in HR and replication fork protection (Tarsounas and Sung, 2020). Although our data are consistent with the possibility that BARD1 is a direct reader of H2AK13/15Ub (Becker et al., 2020), BARD1 depletion may also impede resolution of DSBs, thereby increasing H2AK13/15Ub. To test this, we assessed Reader1.0 foci in cells transfected with siRNA targeting another HR repair factor, the CtBP-interacting protein CtIP, which participates in both DNA end resection and protection of stalled replication forks (Przetocka et al., 2018; Sartori et al., 2007). Similar to BARD1 knockdown, CtIP knockdown increased the number of Reader1.0 foci as well as foci intensities and sizes, particularly in the S and G2 phases (Fig. S5 and Data S1).

Depletion of the HR repair factor CtIP results in increase of H2AK15 ubiquitination in interphase cells. (A) Representative images of G1-phase, S-phase, and G2-phase cells transfected with the indicated siRNAs. Scale bar, 5 µm. (B) Cell cycle distribution of U-2 OS cells expressing PIP-FUCCI and Reader1.0 and transfected with the indicated siRNAs. Stacked bar chart shows mean ± SD from n = 2–4 biological replicates for each siRNA; for each cell cycle phase, P values for pairwise comparisons with the siCtrl are shown, with significant (P < 0.01) increases highlighted in red and significant decreases in green. (C) Reader1.0-eGFP foci numbers (mean ± SD), normalized foci intensities (mean ± SD), and sizes (median with interquartile range) measured for cells transfected once with 10 nM siCtrl #1, siBARD1, or siCtIP for 72 h. Statistical analyses are as described in Materials and methods. *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant. For the comparisons of foci numbers and normalized foci intensity, significant increases or decreases with R2 > 0.05 are highlighted in red and green, respectively; for the comparisons of foci size, significant increases or decreases ≥0.1 µm2 are highlighted in red and green, respectively.
Depletion of the HR repair factor CtIP results in increase of H2AK15 ubiquitination in interphase cells. (A) Representative images of G1-phase, S-phase, and G2-phase cells transfected with the indicated siRNAs. Scale bar, 5 µm. (B) Cell cycle distribution of U-2 OS cells expressing PIP-FUCCI and Reader1.0 and transfected with the indicated siRNAs. Stacked bar chart shows mean ± SD from n = 2–4 biological replicates for each siRNA; for each cell cycle phase, P values for pairwise comparisons with the siCtrl are shown, with significant (P < 0.01) increases highlighted in red and significant decreases in green. (C) Reader1.0-eGFP foci numbers (mean ± SD), normalized foci intensities (mean ± SD), and sizes (median with interquartile range) measured for cells transfected once with 10 nM siCtrl #1, siBARD1, or siCtIP for 72 h. Statistical analyses are as described in Materials and methods. *, P < 0.01; **, P < 0.001; ***, P < 0.0001; n.s., not significant. For the comparisons of foci numbers and normalized foci intensity, significant increases or decreases with R2 > 0.05 are highlighted in red and green, respectively; for the comparisons of foci size, significant increases or decreases ≥0.1 µm2 are highlighted in red and green, respectively.
Relative to 53BP1 and BARD1, depletion of the H2AK13/15Ub readers RNF169 and RAD18 had smaller effects. RNF169 knockdown increased Reader1.0 foci number in G1, S, and G2 phases with little impact on foci intensity or size (Fig. 7, B–E; and Data S1). Also, despite the twofold increase in Reader1.0 foci number, RNF169 knockdown cells in G2 showed modestly reduced foci intensity; possibly, rather than competing with 53BP1 or BARD1, RNF169 restricts RNF168 activity (Panier et al., 2012). Finally, RAD18 knockdown had little effect and showed only a small increase in Reader1.0 foci number in G2 phase that may arise from its role in post-replicative DNA repair (Huang et al., 2009).
Among the many DUBs that participate in DNA damage repair pathways, we examined several reported to target Ub–nucleosomes. USP3, USP11, USP16, and USP51 were proposed to be involved in the RNF8–RNF168 signaling axis (Uckelmann and Sixma, 2017), whereas USP48 was reported to target H2AK125/127/129Ub (Uckelmann et al., 2018). Of these, USP51 knockdown showed the most pronounced effects on Reader1.0 foci, causing small increases of foci number in G1- and G2- and marked increases of foci intensity in G1-phase cells (Fig. 7, B–E; and Data S1). These findings point to a potential role of USP51 in NHEJ-mediated repair. In contrast, depletion of USP3, USP11, USP16, and USP48 yielded only minor changes in the profiles of Reader1.0 focal accumulation across the cell cycle (Fig. 7, B–E; and Data S1).
Discussion
Design of an avidity-based sensor
Avidity is commonly used in nature to augment binding sensitivity and selectivity. Adopting this strategy, a previous work combined multiple reader domains into probes for combinatorial histone tail PTMs (Su and Denu, 2015). Recent structural characterizations of several protein–nucleosome complexes have revealed a new mechanism of recruitment characterized by simultaneous interaction with the nucleosome acidic patch and a PTM covalently attached to a histone tail (Anderson et al., 2019; Fradet-Turcotte et al., 2013; Hu et al., 2017; Kitevski-LeBlanc et al., 2017; Valencia-Sánchez et al., 2019; Wilson et al., 2016; Worden et al., 2019; Worden et al., 2020). Inspired by this mode of interaction, we extended an avidity-based strategy used previously for (poly)Ub (Choi et al., 2019a; Sims et al., 2012) to design protein sensors that target Ub–nucleosomes in live cells. The sensors we generated, Reader1.0/1.1 and Reader2.0/2.1, were assembled from independent binding domains, a UBD that binds to Ub and an anchor that binds to the nucleosome acidic patch, connected by a linker chosen to promote avidity (Fig. 1, B, C, and E; and Fig. S1). By choosing anchors and UBDs with Kds in the range of 10−5 to 10−6 M, we are able to generate Ub–nucleosome sensors with Kds of ∼10−8 M. Such affinities are on par with those of published live-cell PTM sensors, such as a single-chain variable fragment derivative that recognizes H4K20me (Zhao et al., 2019). For Reader1.0, binding to H2AK13/15Ub at DNA DSBs was comparable to that of the cognate reader domain from 53BP1 (Fig. 5, D and E; and Table 1), and FRAP analyses showed similar binding by Reader2.0/2.1 for endogenous H2BK120Ub (Fig. 3, D and E; and Table 1). Importantly, when expressed at low levels, Reader1.0 did not detectably affect DNA repair efficiency (Fig. 5, F and G). Thus, it is possible to generate Ub–nucleosome sensors that allow live-cell detection of the signal without significantly perturbing physiology.
Signaling by a PTM frequently depends on the identity of the modification site. Because this is a particularly prominent feature of chromatin PTMs, site selectivity is a key challenge to development of high-affinity probes for modified nucleosomes. In many naturally occurring readers, docking onto the nucleosome acidic patch serves to limit the search space of a PTM-recognition domain, thereby facilitating interactions between the reader molecule and the modified target. With respect to histone ubiquitination, in vitro selectivity for the prototype sensors Reader1.0/1.1 was partially achieved by taking advantage of the orientation of the LANA anchor peptide bound to a nucleosome (Barbera et al., 2006) and by the use of a linker with high α-helical propensity (i.e., antiparallel coiled-coil) to limit mobility of the tethered UBD (Fig. 1, B, C, and F). Additionally, MD simulations identified unique features of H2BK120Ub–nucleosomes that helped to guide the designs of Reader2.0/2.1 (Fig. 2). Strikingly, the simulations suggested that nucleosomes with H2BK120Ub can sample a greater range of conformations than those with H2AK15Ub (Fig. 2 C). Consistent with our in silico predictions, recent structures of H2BK120Ub–nucleosome complexes illustrate this variability and suggest that H2BK120Ub readers and erasers can engage their target in a variety of orientations (Worden et al., 2020). Of particular importance for sensor design, the MD simulations identified a cluster of conformations in which selected combinations of anchors and UBDs could bind to H2BK120Ub but not to H2AK15Ub (Fig. 2, B–D). Reader2.0/2.1 were constructed accordingly, and determination of their affinities for nucleosomes in vitro (Fig. 2 H) and binding kinetics in vivo (Fig. 3, D and E; and Table 1) confirmed the predictions.
Sensor behavior within cells
Expression of Reader1.0/2.0/2.1 in mammalian cells revealed aspects of affinity and selectivity that were unanticipated from in vitro assays with purified nucleosomes containing nonhydrolyzable Ub–histone mimics (Long et al., 2014). The FRAP analyses suggest that the anchor domains are the primary determinants of Reader1.0/2.0/2.1 binding to Ub–nucleosomes; docking of LANA or IE1 onto the nucleosome acidic patch facilitates UBD interactions, thereby boosting overall affinity via the avidity effect (Fig. 3, C–E; Fig. 4, C and D; and Table 1). Whereas Reader2.0/2.1 bind H2BK120Ub selectively both in vitro and in vivo (Fig. 2 H; Fig. 3, D and E; and Table 1), Reader 1.0 does not distinguish H2AK13/15Ub from H2BK120Ub in vitro (Fig. 1 F) but is selective for H2AK13/15Ub in cells (Fig. 3, D and E; Fig. 4, C and D; and Table 1). Recently it was reported that LANA binding to nucleosomes in vitro is antagonized by Ub conjugation at H2BK120 (Anderson et al., 2019); nonetheless, Reader1.0 binds to purified H2BK120Ub–nucleosomes with high affinity, suggesting that UBD•Ub interactions may compensate for weakened LANA binding. In cells, as suggested by our FRAP results, anchor binding to the nucleosome acidic patch is a major component of the avidity that underlies stable interactions between the sensors and Ub–nucleosomes. Moreover, the UBD contribution will be weakened by competition from the high content of free Ub in the nucleus (5–20 µM; Choi et al., 2019a; Clague et al., 2015). In the case of H2AK13/15Ub, binding in cells may be maintained through the more stable anchor•acidic patch interaction; also, other histone modifications or associated proteins could restrict the ensemble of allowed Ub–nucleosome conformers, thereby reducing the opportunities for binding (i.e., reducing the effective association rate constant, kon) of Reader1.0 to H2BK120Ub. Indeed, modulation of either kon or the dissociation rate koff of the sensor could depend on the nucleosome environment that in vivo might differentiate H2AK13/15Ub from H2BK120Ub with respect to sensor binding. In any event, our results show that estimates of affinity and selectivity based solely on in vitro equilibrium binding can be misleading. First, such approaches do not establish the rates of association and dissociation needed to predict residence times on chromatin. Second, in vitro binding assays are unlikely to capture the complexity of the chromatin environment. For these reasons, characterization of the sensors and their individual components using FRAP in live cells has been most informative.
Reader1.0 is a live-cell sensor for H2AK13/15Ub in unperturbed cells
In both NHEJ and HR pathways for DNA DSB repair, H2AK13/15Ub–nucleosomes are a critical signal. Recently, RNF168 was found to be involved in replication fork progression in the absence of DNA damage inducers (Schmid et al., 2018), suggesting an expanded role for H2AK13/15Ub-dependent signaling. The high affinity and selectivity of Reader1.0 enabled the detection of H2AK13/15Ub-enriched regions in chromatin in cells without the need of exogenous DNA damage agents, thereby allowing us to interrogate how potential effectors of H2AK13/15Ub are regulated throughout the cell cycle. High-content analysis of Reader1.0 foci number, intensity, and size revealed that there may be multiple H2AK13/15Ub readers that are functionally distinct (Fig. 6 and Fig. 7).
Cells depleted of RNF168, 53BP1, or BARD1 showed a G1/S arrest, underscoring the importance of H2AK13/15Ub for S-phase entry and DNA replication (Fig. 7 A). Whereas RNF168 knockdown reduced Reader1.0 foci, depletion of 53BP1, BARD1, or CtIP led to increases in both Reader1.0 foci number and fluorescence intensities, reflecting either increased availability of H2AK13/15Ub for sensor binding or H2AK13/15Ub accumulation due to defective DNA repair (Fig. 7 E and Fig. S5 C). In contrast, knockdown of RNF169 or RAD18 increased Reader1.0 foci number without effects on foci intensity (Fig. 7 E), suggesting a role in restricting RNF168 activities (Hu et al., 2017; Panier et al., 2012). Although we are not certain that the levels of protein depletion were similar among these knockdowns (only quantitative RT-PCR [RT-qPCR] analyses were done; Fig. S4 E), our results suggest that 53BP1 and BARD1 contribute most to H2AK13/15Ub recognition. It is well established that 53BP1 requires both H2AK15Ub and H4K20me2 for binding to nucleosomes (Fradet-Turcotte et al., 2013; Pellegrino et al., 2017; Wilson et al., 2016), but BARD1 only recently has been proposed to be recruited by the combination of H2AK15Ub and the post-replicative mark H4K20me0 (Becker et al., 2020; Nakamura et al., 2019). Thus, 53BP1 appears to play a major role in reading H2AK15Ub in the G1 phase of the cell cycle, whereas BARD1 may have a dominant role in the late S and early G2 phases; this is in line with what we observed (Fig. 7, B–E). However, the BARD1 knockdown phenotype was similar to that observed for cells depleted of CtIP (Fig. S5). This may be similar to the reported increase of 53BP1 foci when HR is defective (Przetocka et al., 2018). Future experiments are needed to test if Reader1.0 directly competes with BARD1 for binding to H2AK15Ub–nucleosomes.
Multiple DUBs have been proposed to remove the Ub–histone signals generated by RNF8 and RNF168 (Mosbech et al., 2013; Nicassio et al., 2007; Sharma et al., 2014; Wang et al., 2016; Yu et al., 2016), but controversies remain as to whether they are H2AK13/15Ub-specific. Also unknown is whether these DUBs function in the absence of induced DNA damage. Depletion of DUBs reported to be involved in DNA DSB repair generally resulted in only minor changes in Reader1.0 accumulation; USP51 knockdown, however, significantly increased Reader1.0 foci intensity in G1 cells (Fig. 7 E), consistent with a previous study (Wang et al., 2016). Interestingly, USP51 knockdown was accompanied by a prominent increase of total nuclear 53BP1 and 53BP1 foci (Fig. S4, F–H). Presumably, competition from the increased 53BP1 reduced Reader1.0 recruitment into foci; notably, this attenuation of the sensor response did not prevent the identification of the USP51 knockdown phenotype, underscoring the excellent dynamic range of Reader1.0 as a reporter.
Limitations
We have found that signals from Reader1.0-eGFP foci generally were much higher in live cells. Upon fixation with paraformaldehyde, Reader1.0 accumulation at endogenous DNA damage sites is barely visible (see Fig. S4 F as an example). Similar observations with other proteins have been attributed to short-lived interactions that, despite being detected in living cells, are not efficiently captured by chemical cross-linking (Schmiedeberg et al., 2009). For Reader1.0, these transient interactions are presumed to reduce interference with the endogenous repair machinery, but a disadvantage is that the sensor is unlikely to perform well as an antibody substitute in standard protocols for chromatin immunoprecipitation, immunoblotting, or immunofluorescence experiments.
Another limitation is that, despite the similar affinities measured in vitro, we do not know if Reader1.0 binds H2AK13Ub and H2AK15Ub equally in cells. Moreover, we anticipate that Reader1.0 is unlikely to distinguish H2A from other histone H2A variants (e.g., H2A.X, H2A.Z, and macroH2A) that also can be ubiquitinated by RNF168 (Kelliher et al., 2020). Finally, as we observed for the H2BK120Ub sensors Reader2.0/2.1, apparent binding selectivity can be affected by the local concentrations of Ub–nucleosomes. Therefore, the possibility of off-target binding must be considered in the experimental design.
That Reader1.0 expressed at increasing levels proportionally inhibited 53BP1-FFR accumulation at DNA DSBs indicates that it will, as expected, compete with the DNA repair machinery recruited by H2AK13/15Ub (Fig. 5). This finding highlights that competition with endogenous proteins is an inescapable feature not only of Reader1.0 but of virtually any live-cell sensor that binds a physiological target. Consequently, tight control of sensor expression levels is critical to minimize interference with endogenous pathways due to competition. In our study, a system using Dox-inducible stable expression afforded Reader1.0 levels that could efficiently report H2AK13/15Ub without causing detectable perturbation to DNA repair (Fig. 5, F and G).
In summary, our design strategy yielded genetically encoded sensors that can directly detect site-specific histone ubiquitination in chromatin. Reader1.0 is a highly sensitive and selective reporter of DNA damage–associated H2AK13/15Ub, whereas Reader2.0/2.1 reported transcription-associated H2BK120Ub in chromatin. When expressed at low levels, the sensors could be used to track H2AK13/15Ub or H2BK120Ub, respectively. We anticipate that this ability to detect changes in histone ubiquitination status can be exploited in assays to screen or validate epigenetic modulators. These sensors are particularly suited to real-time observations of live cells and make possible monitoring of processes in single cells over extended times. With appropriate fluorescent reporters, the sensors also could be suitable for observations in transgenic animal models. Finally, by increasing the repertoire of binding modules, we anticipate that the avidity-based strategy described here can be extended to develop live-cell sensors for other nucleosome PTMs.
Materials and methods
Preparation of Ub–nucleosome sensors
His6-Reader1.0, His6-GB1-Reader2.0, His6-GB1-Reader2.1, and His6-GB1-IE1-tSR (pan-Ub–nucleosome sensor) were cloned into pET28a and transformed into BL21-CodonPlus (DE3) Escherichia coli cells for protein expression. Expression was induced by the addition of 0.4 mM IPTG to cells grown at 37°C to an optical density (OD660 nm) of 0.6–0.8, and then growth was continued at 25°C for 12 h. The cells were harvested by centrifugation at 3,200 g, resuspended in ice-cold buffer A (20 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10 mM imidazole, and 10 mM β-mercaptoethanol), and lysed by sonication. The lysates were clarified by centrifugation for 30 min at 4°C at 20,199 g. A HisTrap HP column (GE Healthcare) was used to isolate the His6-tagged proteins from the lysates. Samples were loaded into the column preequilibrated with buffer A, and, after washing with 20 column volumes, bound proteins were eluted with a linear gradient to 250 mM imidazole in buffer A. Proteins were further purified by gel filtration using a Superdex 75 10/300 GL column (GE Healthcare) eluted with PBS, pH 7.4, and 1 mM tris(2-carboxyethyl)phosphine. Purity was assessed by SDS-PAGE. His6-Reader1.0 and His6-GB1-IE1-tSR were labeled with Alexa Fluor 488 as described elsewhere (Choi et al., 2019a).
Preparation of Ub–histones and reconstitution of nucleosome core particles
Recombinant human histones H2A, H2B, H3.3, and H4 as well as mutant histones H2AK15C, H2AK119C, H2AK129C, and H2BK120C were obtained from The Histone Source – Protein Expression and Purification Facility at Colorado State University. H2AK13C was generated by site-specific mutation of histone H2A cloned into pET21a followed by expression and purification from BL21-CodonPlus (DE3) E. coli cells (Dyer et al., 2004). His6-UbG76C (Long et al., 2014) as well as the 147-mer (Dyer et al., 2004) or the non-linker-ended tri-nucleosomes (Carruthers et al., 1999; Winkler et al., 2011) DNA fragments were expressed and purified as described. Ub–histones were prepared by cross-linking with 1,3-dichloroacetone (DCA; Long et al., 2014). Briefly, His6-UbG76C and H2AK13C/H2AK15C/H2AK119C/H2AK129C/H2BK120C (1:1, Ub:histone molar ratio) were mixed in cross-linking buffer (50 mM sodium tetraborate, pH 8.5, 6 M urea, and 5 mM tris(2-carboxyethyl)phosphine and incubated at room temperature for 30 min to reduce cysteines. The mixture was cooled to 4°C, and DCA (1:1:1, Ub:histone:DCA molar ratio) was added; after 60 min, reactions were stopped with 5 mM β-mercaptoethanol. Next, the reaction mixtures were diluted 1:10 in denaturing binding buffer (50 mM sodium phosphate, pH 7.4, 300 mM NaCl, 6 M urea, 10 mM imidazole, and 5 mM β-mercaptoethanol) and loaded into a HisTrap HP column for purification of the His6-tagged proteins. After washing with 10 column volumes, bound proteins were eluted with a linear gradient to 125 mM imidazole in denaturing binding buffer. Eluates contained cross-linked Ub–histones, unreacted His6-UbG76C, and cross-linked His6-diubiquitin. Because unreacted His6-UbG76C and His6-diubiquitin do not interfere with the refolding and assembly of histones into octamers, the mixtures obtained from the HisTrap column were used as the source of Ub–histones in the subsequent refolding steps.
Reconstitution of histone octamers was performed as described (Dyer et al., 2004; Long et al., 2014). Purified octamers were concentrated with Amicon Ultra Centrifugal filters (Millipore) to ∼40 µM and stored at –80°C in refolding buffer supplemented with 20% glycerol (vol/vol). Mononucleosomes and tri-nucleosomes containing H2AK13Ub, H2AK15Ub, H2AK119Ub, H2AK129Ub, or H2BK120Ub were assembled by mixing the respective histone octamers with the 147-mer or non-linker-ended tri-nucleosomes DNA fragments in high salt (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml BSA, and 2 M NaCl) at 30°C, followed by stepwise dilution to reach a final salt concentration corresponding to 100 mM NaCl (Carruthers et al., 1999; Dyer et al., 2004; Winkler et al., 2011). Mononucleosomes and tri-nucleosomes were analyzed by native PAGE and SDS-PAGE and used for binding assays. Tri-nucleosomes analyzed by native PAGE were first digested with EcoRI to generate mononucleosomes containing 223-, 207-, or 191-bp DNA fragments, as previously described (Winkler et al., 2011).
Binding assays
All binding assays were done at room temperature in PBS buffer, pH 7.4, supplemented with 0.05% Brij35, 0.2 mg/ml ovalbumin, and 1 mM DTT. A FluoroMax-4 spectrofluorometer (HORIBA Scientific) was used to measure fluorescence of the Alexa Fluor 488–labeled ligands. Reader1.0/1.1 affinities for mononucleosomes and tri-nucleosomes containing H2AK13Ub, H2AK15Ub, H2AK119Ub, H2AK129Ub, and H2BK120Ub were determined by measuring fluorescence changes in Alexa Fluor 488–labeled Reader1.0 (5–20 nM) upon titration with increasing concentrations of the respective nucleosomes. Reader2.0/2.1 affinities for mononucleosomes and tri-nucleosomes containing H2AK15Ub and H2BK120Ub were measured by competition with Alexa Fluor 488–labeled Reader1.0 (5–20 nM) in the presence of 20–30 nM H2AK15Ub/H2BK120Ub and titration with Reader2.0/2.1. Reader2.0/2.1 affinities for mononucleosomes and tri-nucleosomes containing H2AK119Ub and H2AK129Ub were measured by competition with Alexa Fluor 488–labeled IE1-tSR (20–50 nM) in the presence of 40–100 nM H2AK119Ub or H2AK129Ub, and titrations with Reader2.0/2.1. Kd and Ki values were calculated by fitting with a single-site binding model using GraphPad Prism v.8 software (Cheng and Prusoff, 1973).
All-atom MD simulations
MD simulations were done using the GROMACS package v4.6.5 (Pronk et al., 2013) with graphics processing unit acceleration and message passing interface parallelization. The Amber ff99SB-ILDN force field for proteins and nucleic acids (Lindorff-Larsen et al., 2010) was modified to simulate the isopeptide bond between either H2AK15 or H2BK120 and UbG76 (Gavrilov et al., 2015). A time step of 2 fs was used for integration of equations of motion. Initial velocities were generated from the Maxwell–Boltzmann distribution with random seeds, and the Verlet cutoff scheme was used for neighbor searching with nstlist set to 20 steps. The van der Waals interactions were treated with a cutoff of 1.4 nm, while long-range electrostatic effects were treated with the particle mesh Ewald method and a cutoff of 1.4 nm (Darden et al., 1993). Bond lengths were constrained by the LINear Constraint Solver (LINCS) algorithm.
Atomic coordinates for H2AK15Ub (RCSB Protein Data Bank [PDB] accession no. 5KGF; Wilson et al., 2016) and H2BK120Ub (PDB accession no. 4ZUX; Morgan et al., 2016) nucleosomes were retrieved from the PDB. Initial models for MD simulations were formed by one copy of the H2A/H2B dimer (H2A: from K9 to L97; H2B: from S32 to K125), the surrounding DNA double helix (29 bases in each DNA strand), and Ub linked through an isopeptide bond to either H2AK15 or H2BK120 (Fig. 2 A). When necessary, side chains were mutated to match amino acid sequences to the human histones (H2A type 1: Uniprot P0C0S8; H2B type 1: Uniprot P62807). Models were placed into rhombic dodecahedral periodic boxes (12 Å distance on each side of the simulated complexes) filled with TIP3P water molecules plus Na+ and Cl− ions, which were added to neutralize DNA phosphate backbones and maintain the salt at 150 mM. Systems were subjected to 5,000 cycles of steepest-descent energy minimization with the maximum step size set to 0.01 nm and the tolerance set to 1,000 kJ • mol−1 • nm−1. After minimization, systems were sequentially equilibrated under position restraints with the constant temperature, constant volume (NVT) ensemble at 393 K for 100 ps using v-rescale temperature coupling, and the isothermal-isobaric (NPT) ensemble for 1 ns using Nosé–Hoover temperature coupling and Parrinello–Rahman pressure coupling. Position restraints were removed, and production MD simulations were performed at 393 K for 20 ns using Nosé–Hoover temperature coupling and Parrinello–Rahman pressure coupling. Each model (i.e., H2AK15Ub and H2BK120Ub) was subjected to a total of 10 independent MD simulations comprising NVT and NPT equilibration plus 20 ns production. Trajectories were post-processed with GROMACS trjconv to fix periodicity effects.
Rigid docking of anchors and UBDs into H2AK15Ub and H2BK120Ub MD ensembles
H2AK15Ub and H2BK120Ub conformations resulting from 10 × 20 ns MD simulations extracted at every 2-ps time interval were superimposed onto the H2A/H2B dimer of a reference nucleosome (PDB accession no. 1AOI; Luger et al., 1997) using pdb-tools and NumPy Python scripts. Next, the LANA (Barbera et al., 2006) and IE1 (Fang et al., 2016) anchors, and the tSR (Choi et al., 2019b; Lee et al., 2006; Wang et al., 2005) and UBQ1UBA (Zhang et al., 2008) UBDs were individually docked into the ensemble of H2AK15Ub and H2BK120Ub conformations by structural superposition. For anchor rigid docking, the dimer H2A/H2B in the LANA–nucleosome (PDB accession no. 1ZLA) or IE1–nucleosome (PDB accession no. 5E5A) complex was superimposed onto the dimer H2A/H2B in the MD ensembles. For UBD rigid docking, Ub in the complexes S5aUIM1-Ub (PDB accession no. 1YX5), Rabex5Ruz-Ub (PDB accession no. 2FIF), and UBQ1UBA-Ub (PDB accession no. 2JY6) was superimposed onto Ub in the MD ensembles; the docked complexes were then filtered for steric clashes using SHARPEN (Loksha et al., 2009). Clash-free conformations, in which the nucleosome acidic patch and Ub were respectively accessible to anchors and UBDs, were defined as those with <20 atom pairs within <2 Å of each other. This approach allowed us to determine the fractions of H2AK15Ub and H2BK120Ub conformations that were accessible to different combinations of anchors and UBDs and to map Ub dynamics when it was linked to either H2AK15 or H2BK120. Finally, a Biopython script was used to calculate the distances separating the nucleosome acidic patch (i.e., H2AE61, δ carbon) from Ub I44 (β carbon) in all nonclashing conformations that were simultaneously accessible to IE1 and UBQ1UBA.
Plasmids for mammalian expression
Reader1.0/2.0/2.1 constructs were made using In-Fusion cloning (Takarabio). For lentiviral expression, Reader1.0/2.0/2.1-eGFP fragments were amplified by PCR and cloned by In-Fusion into XhoI/EcoRI digested pTRIPZ vector. mCherry-BP1-2 pLPC-Puro (mCherry-53BP1-FFR) was from Titia de Lange (Rockefeller University, New York, NY; Addgene plasmid 19835); pLenti-PGK-Neo-PIP-FUCCI was from Jean Cook (University of North Carolina, Chapel Hill, NC; Addgene plasmid 118616). In-Fusion cloning was used to replace mVenus in pLenti-PGK-Neo-PIP-FUCCI by iRFP682. All constructs were verified by sequencing.
Cell culture, transient transfection, and viral transduction
U-2 OS cells (American Type Culture Collection) were cultured in DMEM (Corning) supplemented with 10% FBS (Atlas Biologicals), 1% penicillin plus streptomycin (Corning), and 2 mM L-glutamine (Hyclone). hTERT-RPE-1 cells were cultured in DMEM/F-12 50/50 (Corning) with L-glutamine supplemented with 10% FBS and 1% penicillin plus streptomycin; cells were maintained at 37°C in a humidified incubator with 5% CO2. Transient expression was achieved by transfection with Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To make stable cell lines, HEK293T cells were cotransfected with psPAX2 (Addgene; plasmid 12260), pMD2.G (Addgene; plasmid 12259), and pTRIPZ-Reader1.0/2.0/2.1-eGFP. Viral supernatants were harvested after 48 h, and U-2 OS cells were transduced in the presence of 8 µg/ml polybrene. At 48 h after transduction, infected cells were selected with 2 µg/ml puromycin. Expression of Reader1.0/2.0/2.1-eGFP was induced by 4–24 h treatment with 10–100 ng/ml Dox (Thermo Fisher Scientific). To generate U-2 OS/PIP-FUCCI/Reader1.0 stable cells, lentiviral particles carrying pLenti-PGK-Neo-PIP-FUCCI (with mVenus replaced by iRFP682) were made as described above. At 48 h after transduction, infected cells were selected with 400 µg/ml G-418 (Research Products International; G64000-1.0). Monoclonal cells were isolated by limiting dilution and tested for Cdt11-17-iRFP682 and mCherry-Geminin1-110 expression. One clone was selected for another transduction with lentiviral particles carrying pTRIPZ-Reader1.0-eGFP. Expression of Reader1.0 was induced by 24 h treatment with 10–50 ng/ml Dox.
Western blotting and immunofluorescence
Whole-cell lysates were obtained by lysing cells in 50 mM Tris-HCl, pH 7.6, 10 mM EDTA, 1% SDS, and 1× protease inhibitors (Sigma-Aldrich; P8340), followed by sonication and clarification by centrifugation. Primary antibodies and the dilutions used were as follows: anti-GFP (CST; rabbit monoclonal; 2656, 1:1,000), anti-mCherry (Abcam; mouse monoclonal; 125096; 1:1,000), anti-HA (Sigma-Aldrich; mouse monoclonal; H3663; 1:5,000), anti-H2AK119Ub (CST; rabbit monoclonal; 8240; 1:1,000), anti-H2BK120Ub (CST; rabbit monoclonal; 5546; 1:1,000), and anti-H2B (Abcam; mouse monoclonal; 52484; 1:1,000). Western signals were visualized by fluorescent secondary antibodies and imaged using LI-COR Odyssey.
U-2 OS cells transiently transfected with Reader1.0/2.0/2.1-eGFP for 24 h were fixed with 2.5% PFA in PBS for 15 min at 37°C, and permeablized with 100% methanol for 10 min at −20°C. Cells were then blocked for 1 h with 3% BSA and 0.3% Triton X-100 in PBS. Next, cells were immunostained with a rabbit mAb against H2BK120Ub (CST; mAb; 5546; diluted 1:800 with 1% BSA and 0.1% Triton X-100 in PBS) for 2 h and with an Alexa Fluor 568–conjugated goat anti-rabbit IgG (Thermo Fisher Scientific; diluted 1:500 with 1% BSA and 0.1% Triton X-100 in PBS) for 1 h.
U-2 OS cells stably expressing Reader1.0-eGFP were treated with 0 or 100 ng/ml Dox for 24 h before being exposed to IR (1.5 Gy) using a 137Cs gamma-ray source. Cells were fixed with 3.7% PFA for 20 min at room temperature at times 0, 2, 4, 8, 12, and 24 h after IR, permeabilized with 0.5% Triton X-100 in PBS for 20 min, blocked with 3% BSA in PBS for 1 h, and immunostained with a mouse mAb against γH2AX (Millipore; 05–636; diluted 1:1,750 in blocking buffer) for 2 h and with an Alexa Fluor 568–conjugated goat anti-mouse IgG (Thermo Fisher Scientific; diluted 1:500 with blocking buffer) for 1 h. Staining with anti-53BP1 (Abcam; rabbit monoclonal; 175933, diluted 1:500 in blocking buffer) followed the same procedure. Nuclei were counterstained with DAPI (MP Biomedicals). Coverslips were mounted onto slides using ProLong Diamond Antifade (Thermo Fisher Scientific).
RNAi transfections and RT-qPCR
Cells were transfected twice at 0 h and 24 h with 10 nM silencing RNAs: siCtrl pool, siCtrl #1, siRNF168, siRNF8, si53BP1, siBARD1, siUSP3, siUSP11, siUSP16, siUSP48, and siUSP51, and analyzed at 72 h. For Silencer Select siRNAs (Thermo Fisher Scientific), cells were transfected once with 10 nM siCtrl #2, siRNF169, and siRAD18 and analyzed 72 h later. Single transfection of 10 nM siCtrl #1, siBARD1, and siCtIP were also employed in Fig. S5. siRNA transfections were performed with Lipofectamine RNAiMAX (Thermo Fisher Scientific) following the manufacturer’s instructions. Total RNA was extracted using the Aurum Total RNA Mini Kit (Bio-Rad). First-strand cDNAs were generated from 100 ng total RNA with the iScript Reverse Transcription Supermix (Bio-Rad). Real-time PCR reactions contained 2 µl of diluted cDNA, 250 nM forward and reverse primers, and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). All the reactions were run on a CFX96 quantitative PCR system (Bio-Rad). Relative mRNA levels were normalized against that of GAPDH. The Pffafl formula was applied for efficiency correction (Pfaffl, 2001). Primers used for RT-qPCR are listed in Table S2.
Data for siCtrl #1, siBARD1, siUSP3, sUSP11, siUSP16, siUSP48, and siUSP51 correspond to two technical replicates from a single experiment, while data for siCtrl #2, siRNF169, and siRAD18 correspond to two biological replicates (with two technical replicates for each experiment; Fig. S4 E).
Microscopy
Confocal microscopy of fixed and live cells was performed on a Zeiss LSM 880 confocal microscope with a Plan-Apochromat 63×/1.40 oil-immersion objective. For live-cell imaging, a stage incubator (PECON, PM 2000 RBT) was used to maintain 37°C and a 5% CO2 atmosphere. Live-cell imaging acquisition was performed using cells plated onto 35-mm glass-bottom dishes (MatTek; P35G-1.5-14-C) or four-chamber glass-bottom dishes (Cellvis; D35C4-20-1.5-N) and kept in FluoroBrite DMEM (Thermo Fisher Scientific; A1896701) supplemented with 10% FBS, 1% penicillin plus streptomycin, and 2 mM L-glutamine. The Zen 2.3 (black edition) software (v.14.0.9.201) was used for image capture. The Processing and Analysis modules of the ZEN 2.3 (blue edition) software (v.2.3.69.1000) were used to extract maximum intensity projections of z-stacks, measure fluorescence intensities, and count γH2AX foci.
FRAP in undamaged nuclei
U-2 OS cells were transfected with Reader1.0/2.0/2.1-eGFP or their NB variants for 4–12 h before live-cell imaging. Treatments with 5 µM FVP (Sigma-Aldrich) or 10 µM C1 (provided by Millenium/Takeda) were performed 1 h before starting the measurements. Photobleaching was performed with the 488-nm line from a 35 mW Ar laser operating at 75% power. The pinhole was set to 1.51 Airy Units (1.1 µm z-section). Fluorescence recovery was monitored at intervals of 0.5 s using the 488-nm laser line at 0.5% power and a photomultiplier tube detector. For each construct, data were obtained from two to five independent spots per nucleus (n ≥ 6 cells). Each bleached spot corresponded to a circle with 30-pixel diameter (1 pixel = 0.09 × 0.09 µm). Normalized FRAP curves were generated from raw data after background subtraction (McNally, 2008). After normalization, spatiotemporal FRAP recoveries were fit to one-exponential or two-exponential equations using GraphPad Prism v.8.
Laser microirradiation and real-time recruitment assays
To induce DNA damage, irradiation of a 0.45-µm-high stripe in the nucleus was made with the 405-nm diode laser (30 mW) at 75% power. Time-lapse images were captured at 20-s intervals for ∼15 min. Reader1.0-eGFP fluorescence was detected with the 488-nm laser operating at 1.0% power, while mCherry-53BP1-FFR fluorescence was detected with the 561-nm diode-pumped solid-state laser (20 mW) operating at 0.5% power. Fluorescence redistribution from regions encompassing the irradiated tracks were extracted from background-corrected individual frames as described (Lukas et al., 2004; Bekker-Jensen et al., 2005). Local increases in fluorescence at the DNA lesions were calculated as follows: fold Increase = It/Ipre, where It denotes the fluorescence intensity at the damaged region at time t, and Ipre denotes the fluorescence intensity before microirradiation. For kinetic profiling, fluorescence was normalized using the following formula: normalized intensity = (It – Ipre)/(Ilast – Ipre), where Ilast denotes the fluorescence intensity at the lesion in the last time point.
FRAP mobilities of Reader1.0-eGFP and mCherry-53BP1-FFR were measured in a rectangular region of interest (ROI; 0.75 µm2) at the microirradiated area and in an undamaged area of the nucleus (Ctrl). Photobleaching used either the 561-nm or the 488-nm laser operating at 75% power. Acquisition conditions were the same as used for the real-time recruitment assays; fluorescence recoveries were monitored at 0.5-s intervals. Normalized FRAP curves were generated from raw data after background subtraction, and spatiotemporal recoveries were fit to a two-exponential equation using GraphPad Prism v.8.
Quantification of Reader1.0-eGFP at endogenous DNA lesions
U-2 OS/PIP/FUCCI cells were transfected with siRNAs as described above and treated with 10–50 ng/ml Dox 48 h after the first transfection to induce the expression of Reader1.0-eGFP. Live-cell imaging was performed 24 h after Dox treatment using the 488-nm argon, 561-nm diode-pumped solid-state, and 633-nm HeNe lasers operated at 0.5, 0.5, and 40% power, respectively; pinholes were adjusted to correspond to a 0.9-µm section. Images were captured at 16-bit resolution with z-stacks acquired at 2-µm intervals.
Quantifications of foci in images were done in CellProfiler v.3.1.9 (McQuin et al., 2018). A user-assisted approach was implemented to classify nuclei into two groups based on the intensity of Reader1.0-eGFP. For each group, nuclei were segmented by the Otsu threshold method using an adaptative strategy in which pixels in the middle intensity class were assigned to foreground. Fluorescence intensities of Reader1.0-eGFP, mCherry-Geminin1-110, and Cdt11-17-iRFP682 within nuclear ROIs were measured after subtracting the respective intensities by the lower-quartile intensities of the individual channels. To identify Reader1.0-eGFP foci within the nuclear ROIs, images were first subjected to White Top-Hat transformation (structuring elements were defined as disks with 5-pixel radius) followed by speckles enhancement to intensify the foci signals. Next, foci were defined as circular objects 4–20 pixels in diameter (scaling per pixel: 0.16 × 0.16 µm) and segmented by the Robust Background method using a global strategy. The averaging method was set as mean and the variance set at three SDs.
Cell cycle classification was performed based on the MFIs of mCherry-Geminin1-110, and Cdt11-17-iRFP682 within each individual nucleus. G1 cells were defined as those with iRFP682 MFI > 0.07 and mCherry MFI < 0.02; S cells were defined as those with iRFP682 MFI < 0.07 and mCherry MFI > 0.02; and G2 cells were defined as those with iRFP682 MFI > 0.07 and mCherry MFI > 0.02. Cells with mCherry-Geminin1-110 and Cdt11-17-iRFP682 expressed below these thresholds were not considered for the cell cycle assignments. After cell cycle classification, cells were filtered based on Reader1.0-expression levels; only cells showing Reader1.0-eGFP MFI between 0.08 and 0.24 MFI were taken into account for quantification of foci number and intensity. Normalized fluorescence intensities are defined as the ratio of Reader1.0-eGFP MFI at foci/Reader1.0-eGFP fluorescence in the nucleus. Foci sizes in square micrometers were calculated by multiplying the foci areas provided by CellProfiler (pixels) by 0.0256 µm2, the pixel size in the acquired images. Data analysis scripts are available upon request.
Statistical analysis
Statistical analyses of immunofluorescence and live-cell imaging measurements as well as the fitting to binding, FRAP, and real-time recruitment data were performed using GraphPad Prism v.8 software. Correlation analyses were computed using a two-tailed P value. Multiple comparisons for number of Reader1.0 foci and normalized foci intensity were performed using Brown–Forsythe and Welch ANOVA tests with Dunnet’s T3 test. Foci sizes, which had a minimum cutoff of 0.33 µm2, were compared using the nonparametric Kruskal–Wallis ANOVA test with Dunn’s test for multiple comparisons. Pairwise analyses for number of Reader1.0 foci and normalized foci intensity were performed by a two-tailed unpaired t test for comparisons between siRNAs and siCtrl. Welch’s correction for unequal variances was used when applicable. Data for foci sizes were analyzed using the nonparametric Mann–Whitney test for comparisons between the indicated siRNAs and siCtrl: siRNF168, si53BP1, siBARD1, siUSP3, siUSP11, siUSP16, siUSP48, and siUSP51 were compared with siCtrl #1; siRNF169 and siRAD18 were compared with siCtrl #2 (Table S1). For data analyzed by unpaired t test, R2 values were used to estimate effect size. Significant increases or decreases with R2 > 0.05 were highlighted in red and green, respectively; for the comparisons of foci size, significant increases or decreases ≥0.1 µm2 were highlighted in red and green, respectively. P values <0.01 were considered significant.
PBD entries
Online supplemental material
Fig. S1 shows the amino acid sequences of the Ub–nucleosome Readers. Fig. S2 shows characterization by gel electrophoresis of the Ub–nucleosomes and Alexa Fluor 488–labeled Reader1.0 and IE1-tSR used in the experiments in Fig. 1 and Fig. 2. Fig. S2 also shows the results of in vitro binding assays for IE1-tSR and tri-nucleosome arrays. Fig. S3 shows measurements of H2BK120Ub in U-2 OS cells transiently expressing Reader1.0/2.0/2.1 that are relevant for Fig. 3. Fig. S4 shows examples of Reader1.0 focal accumulation in nontreated cells, in cells treated with FVP or C1, and in cells transfected with siRNAs targeting RNF168 and RNF8. Fig. S4 also shows the evaluation of siRNA transfection efficiency relevant for Fig. 6 and Fig. 7. Fig. S5 shows focal accumulation of Reader1.0 in cells treated with siCtrl #1, siBARD1, or siCtIP. Table S1 shows the sequences of the siRNA oligos used in this study. Table S2 shows the sequences of the primers used for RT-qPCR (Fig. S4 E). Data S1 shows the detailed statistical analyses for the results presented in Fig. 6, Fig. 7, and Fig. S5.
Acknowledgments
We thank members of the Cohen and Yao laboratories for helpful discussions throughout the course of this project. N. Ahuja, E. Beyers, B. Schmitt, and L. Yarfi in our laboratories contributed purified proteins and nucleosomes, E. Beyers performed initial in vitro and live-cell characterizations of the sensors, N. Ahuja helped with in vitro binding experiments, and J. Dortch helped with PDB database searches. The authors thank Prof. C. Wiese for discussions about DNA damage repair, and Prof. A. Hess of the Graybill Statistics and Data Science Laboratory at Colorado State University for statistical consulting.
This research was supported by National Institutes of Health grants R21ES029150 to R.E. Cohen and T. Yao, R01GM115997 to R.E. Cohen, and R01GM098401 to T. Yao.
The authors declare no competing financial interests.
Author contributions: C. dos Santos Passos and Y.-S. Choi designed and prepared the Reader plasmids and proteins and performed binding assays with in vitro–assembled nucleosomes. C. dos Santos Passos constructed the stable cell lines and performed all experiments that used live and fixed cells. Under the guidance of C.D. Snow, C. dos Santos Passos also performed the MD analyses. The project was conceived and supervised by R.E. Cohen and T. Yao. The manuscript was prepared by C. dos Santos Passos, R.E. Cohen, and T. Yao with input from all of the authors.

