


Epigenetic histone trimethylation on lysine 9 (H3K9me3) represents a major molecular signal for genome stability and gene silencing conserved from worms to man. However, the functional role of the H3K9 trimethylases SUV39H1/2 in mammalian tissue homeostasis remains largely unknown. Here, we use a spontaneous dog model with monogenic inheritance of a recessive SUV39H2 loss-of-function variant and impaired differentiation in the epidermis, a self-renewing tissue fueled by stem and progenitor cell proliferation and differentiation. Our results demonstrate that SUV39H2 maintains the stem and progenitor cell pool by restricting fate conversion through H3K9me3 repressive marks on gene promoters encoding components of the Wnt/p63/adhesion axis. When SUV39H2 function is lost, repression is relieved, and enhanced Wnt activity causes progenitor cells to prematurely exit the cell cycle, a process mimicked by pharmacological Wnt activation in primary canine, human, and mouse keratinocytes. As a consequence, the stem cell growth potential of cultured SUV39H2-deficient canine keratinocytes is exhausted while epidermal differentiation and genome stability are compromised. Collectively, our data identify SUV39H2 and potentially also SUV39H1 as major gatekeepers in the delicate balance of progenitor fate conversion through H3K9me3 rate-limiting road blocks in basal layer keratinocytes.
Introduction
Disruption of chromatin regulation in inherited and acquired diseases severely affects homeostatic processes. Nevertheless, safe and efficacious therapeutic interventions remain challenging but possible when molecular mechanisms controlling alterations in the epigenetic landscape are better understood (Kim et al., 2017; Valencia and Kadoch, 2019; Zoghbi and Beaudet, 2016).
Spontaneously occurring monogenetic disorders allow to backtrack clinical signs to default tissue homeostasis, associate them with a genetic variant, and detect thus far unknown underlying mechanisms. In particular, they offer the possibility of new in-depth knowledge through study of site-specific requirements such as epidermal skin adaptations (Balmer et al., 2019; Maruthappu et al., 2018).
The epidermis, the outermost layer of the skin, is an attractive model system for studying homeostatic processes, as it self-renews in a reproducibly short, roughly tri-weekly fashion. As a paradigm, a fine-tuned homeostatic program is initiated by stem or progenitor cell proliferation in the basal (innermost) layer, followed by cell cycle exit in early G1 and delamination of keratinocytes from the basement membrane (Miroshnikova et al., 2018; Müller et al., 2008; Watt, 2016). Concomitant with migration to the suprabasal layer, a stepwise, highly coordinated differentiation program is initiated, allowing the upward migration and functional integration of keratinocytes into the different epidermal layers. Finally, terminal differentiation accompanied by a rise in transepidermal calcium concentrations involves transcription of the epidermal differentiation complex, producing the tight sealing of the outer body shell (Lee and Lee, 2018).
Epidermal homeostatic processes can be modeled in cultured keratinocytes that are known to follow the predefined epidermal differentiation program (Kolly et al., 2005; Watt, 2016). An increase in calcium was shown to coordinate the fate conversion of cultured keratinocytes, which follows a stereotypic homeostatic time line under standardized culture conditions (Kolly et al., 2005). The conversion between proliferating and differentiating keratinocytes was suggested to be triggered by crowding (cell density) and cell–cell adhesion (Kolly et al., 2005; Miroshnikova et al., 2018), with predominant roles of E-cadherin and desmosomal cadherin Dsg3 receptor signaling (Miroshnikova et al., 2018; Müller et al., 2008; Waschke, 2019). Other key signaling pathways in this process are stem-, progenitor-, or differentiation-promoting Wnt and p63 as well as Notch (Choi et al., 2013; Klein and Andersen, 2015; Koster et al., 2004; Lim and Nusse, 2013; Negri et al., 2019; Nowell and Radtke, 2013; Truong et al., 2006; Williamson et al., 2006).
We reported an N324K missense variant of an evolutionary conserved amino acid in the catalytic domain of the H3K9 trimethylase SUV39H2 as likely causal for hereditary nasal parakeratosis (HNPK) in Labrador retrievers (Jagannathan et al., 2013). The N324K variant leads to loss of SUV39H2 function (Schuhmacher et al., 2015). Affected homozygous mutant dogs exhibit crusts and fissures of the nasal planum, which suggests a role of SUV39H2 in keratinocyte differentiation and formation of cornified sealing. Support for a causal role of loss of SUV39H2 function in HNPK was further provided by a comparable HNPK phenotype in greyhounds with an independent SUV39H2 splice-site variant with predicted loss of function (Bauer et al., 2018). These observations suggested that H3K9me3 chromatin modifications introduced by SUV39H2 have a high impact on the homeostatic program of keratinocytes.
Repressive H3K9me3 marks are known to represent a heritable master switch in the functional organization of chromosomal subdomains, conserved from worms to men (Becker et al., 2016; Greer et al., 2014; Rao et al., 2017). The SUV39H1 and SUV39H2 lysine methyltransferases are two of seven mammalian enzymes catalyzing the addition of H3K9me3 marks in euchromatin, pericentric heterochromatin, and at telomeres (García-Cao et al., 2004; Nielsen et al., 2001; Peters et al., 2003). Little is known about how these enzymes coordinate homeostatic processes, particularly in the skin. However, the developmentally lethal phenotype of Suv39h1/2 double-null mutant mice suggested a key role in gene regulation and genomic stability (Peters et al., 2001; Rea et al., 2000). Although the preferred substrates of SUV39H1 and SUV39H2 vary, these two enzymes are thought to exhibit functional redundancy (O’Carroll et al., 2000; Peters et al., 2001; Rea et al., 2000).
In this study, we investigate the functional activity and significance of SUV39H2-mediated H3K9me3 repressive marks in epidermal homeostasis. For the first time, our mechanistic insights highlight that the epidermal homeostatic process follows its course based on a complex pattern of SUV39H1/2–mediated epigenetic imprints on genes of the progenitor-promoting Wnt/p63/adhesion axis as well as the epidermal differentiation complex in basal layer keratinocytes.
While genetic testing to detect SUV39H2 variants has been developed as a preventive measure for dog breeders, this study stipulates that the epigenetic molecular activity of SUV39H2 is of general relevance in mammalian species, including humans, where inhibition of SUV39H2 is currently investigated as a treatment for certain cancer types (Piao et al., 2019; Vougiouklakis et al., 2018).
Results
SUV39H2 is the major enzyme introducing repressive H3K9me3 marks in the epidermis of the nasal planum
To start addressing the mechanistic role of SUV39H2 and consequences of the N324K SUV39H2 loss-of-function variant, we screened biopsies of the nasal planum epidermis for H3K9me3 marks by immunofluorescence microscopy of three control (324N/324N) and three HNPK Labrador retrievers (324K/324K; Fig. 1). The biopsies of control Labrador retrievers invariably revealed a graded pattern of H3K9me3 marks, which were highest in the nuclei of basal epidermal keratinocytes. In contrast, in nasal planum epidermis of all HNPK dogs, H3K9me3 marks were absent, with the exception of a very faint H3K9me3 signal in basal keratinocytes (Fig. S1 A). In contrast, H3K27me3 marks were not affected. The nearly complete loss of H3K9me3 suggests that SUV39H2 is the major H3K9 trimethylating enzyme in nasal planum epidermis and supports previous findings that the SUV39H2324K allele is functionally inactive (Schuhmacher et al., 2015).

Loss of H3K9me3 marks in HNPK nasal epidermis. Immunofluorescence detection of H3K9me3 in control and HNPK nasal epidermis. Note that H3K9me3 marks are mainly present in the basal layer and decrease along the spinous to the corneal layers in control nasal epidermis, while H3K9me3 is only present at a low level in the HNPK basal layer, seen in the zoomed-in inserts. Data are generated from n = 3 independent experiments on three different control (shown are two dogs) and three different HNPK Labrador retriever dogs in triplicate. Inserts represent 2.5× magnification of the selected areas indicated by small squares. Scale bars, 100 µm. Dashed line indicates the basement membrane zone.
Loss of H3K9me3 marks in HNPK nasal epidermis. Immunofluorescence detection of H3K9me3 in control and HNPK nasal epidermis. Note that H3K9me3 marks are mainly present in the basal layer and decrease along the spinous to the corneal layers in control nasal epidermis, while H3K9me3 is only present at a low level in the HNPK basal layer, seen in the zoomed-in inserts. Data are generated from n = 3 independent experiments on three different control (shown are two dogs) and three different HNPK Labrador retriever dogs in triplicate. Inserts represent 2.5× magnification of the selected areas indicated by small squares. Scale bars, 100 µm. Dashed line indicates the basement membrane zone.

Histone trimethylation marks and markers for main signaling pathways in HNPK nasal planum and HNPK haired skin. (A) Representative immunofluorescence microscopy pictures on control and HNPK nasal planum probed on paraffin sections (n = 2 independent experiments on three different dogs in duplicate; shown are representative examples from two dogs). While H3K9me3 is severely reduced in HNPK, no difference is observed in the level of H3K27me3 repressive marks, for which limited functional information is currently available for epidermis. With regard to pathway markers, axin2 was selected for Wnt signaling, p63 for early differentiation, and Dsg3 for adhesion. Note that expression of axin2 is significantly enhanced throughout all epidermal layers of the nasal planum in HNPK, while p63 and Dsg3 are increased to a lesser extent. (B) Representative immunofluorescence microscopy pictures of control and HNPK haired skin probed on paraffin sections (n = 2 independent experiments on two different dogs in duplicate). No major differences were found in the H3 trimethylation patterns between genotypes in haired skin, while axin2 and p63 expression was similar and Dsg3 expression increased to some extent in HNPK sections. Dashed line represents the basement membrane. Scale bars, 50 µm.
Histone trimethylation marks and markers for main signaling pathways in HNPK nasal planum and HNPK haired skin. (A) Representative immunofluorescence microscopy pictures on control and HNPK nasal planum probed on paraffin sections (n = 2 independent experiments on three different dogs in duplicate; shown are representative examples from two dogs). While H3K9me3 is severely reduced in HNPK, no difference is observed in the level of H3K27me3 repressive marks, for which limited functional information is currently available for epidermis. With regard to pathway markers, axin2 was selected for Wnt signaling, p63 for early differentiation, and Dsg3 for adhesion. Note that expression of axin2 is significantly enhanced throughout all epidermal layers of the nasal planum in HNPK, while p63 and Dsg3 are increased to a lesser extent. (B) Representative immunofluorescence microscopy pictures of control and HNPK haired skin probed on paraffin sections (n = 2 independent experiments on two different dogs in duplicate). No major differences were found in the H3 trimethylation patterns between genotypes in haired skin, while axin2 and p63 expression was similar and Dsg3 expression increased to some extent in HNPK sections. Dashed line represents the basement membrane. Scale bars, 50 µm.
Expression profiling reveals premature cell cycle exit, deregulated p53, Wnt, and Notch signaling with impaired terminal differentiation in HNPK dogs
Our analyses of RNA sequencing (RNA-seq) data from the nasal epidermis of three HNPK and three control dogs identified a total of 2,154 differentially expressed genes (cutoff twofold change >|2|; P < 0.05; Fig. S2 and Table S1), which were subjected to signal pathway analysis. The Ingenuity Pathway Analysis (IPA) and AMIGO software revealed four major signaling networks with highest numbers of differentially expressed genes, namely (i) activation of canonical and noncanonical Wnt signaling, (ii) impaired cell cycle progression and enhanced cell cycle exit, (iii) activation of p53-dependent pathways, and (iv) inhibition of Notch signaling (Fig. 2 A). Differentially expressed genes attributed to these pathways by IPA were confirmed and manually completed by published literature (Table S1).

Volcano plot of RNA-seq data. The volcano plot shows comparison of differentially expressed genes from three control and three HNPK dogs. The vertical axis (y axis) corresponds to the mean expression value of log10 (P value) with a P value cutoff threshold of 0.05 (horizontal dashed line). The horizontal axis (x axis) displays the log2 fold change with a cutoff value of >|2| (vertical dashed line). Positive x values represent up-regulated and negative x values down-regulated genes. From a total of 24,580 genes, 2,154 genes were considered differentially expressed (red points; log2 fold change of >|2| and P < 0.05). The blue points pass for the P value cutoff but fail for log2 fold change cutoff.
Volcano plot of RNA-seq data. The volcano plot shows comparison of differentially expressed genes from three control and three HNPK dogs. The vertical axis (y axis) corresponds to the mean expression value of log10 (P value) with a P value cutoff threshold of 0.05 (horizontal dashed line). The horizontal axis (x axis) displays the log2 fold change with a cutoff value of >|2| (vertical dashed line). Positive x values represent up-regulated and negative x values down-regulated genes. From a total of 24,580 genes, 2,154 genes were considered differentially expressed (red points; log2 fold change of >|2| and P < 0.05). The blue points pass for the P value cutoff but fail for log2 fold change cutoff.
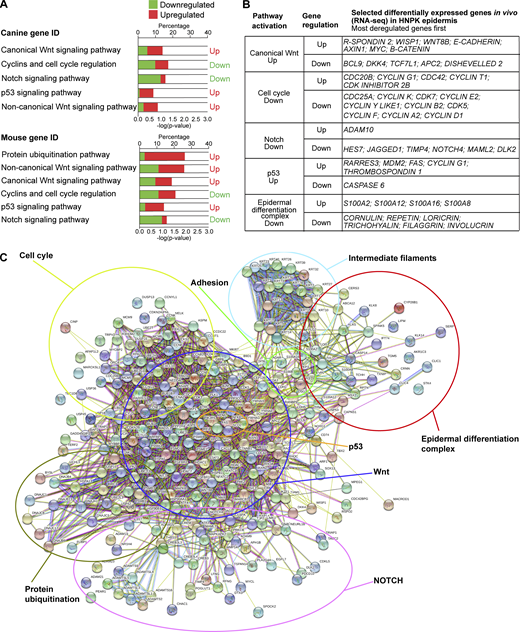
RNA-seq analysis reveals premature cell cycle exit; altered Wnt, Notch, and p53 signaling; and aberrant terminal differentiation in HNPK dogs.(A) Top panel: Hierarchical deregulated pathways generated by IPA software with canine gene identification (ID) input from RNA-seq of significantly deregulated genes (Table S1). Bottom panel: Hierarchical deregulated pathways generated by IPA using converted canine to mouse gene ID. Note that in addition to protein ubiquitination pathway, the same pathways were generated by IPA with different hierarchy. (B) List of representative differentially expressed genes involved in pathways generated by IPA and STRING software. Genes are ranked from most to least deregulated (most deregulated genes first). Data are generated from n = 3 independent experiments on three different control and three different HNPK Labrador retriever dogs. (C) Functional protein connection network generated by STRING software with the 2,154 differentially expressed gene products (Table S1). Same pathways generated by IPA were found with STRING, in addition to adhesion, intermediate filament, and epidermal differentiation complex.
RNA-seq analysis reveals premature cell cycle exit; altered Wnt, Notch, and p53 signaling; and aberrant terminal differentiation in HNPK dogs.(A) Top panel: Hierarchical deregulated pathways generated by IPA software with canine gene identification (ID) input from RNA-seq of significantly deregulated genes (Table S1). Bottom panel: Hierarchical deregulated pathways generated by IPA using converted canine to mouse gene ID. Note that in addition to protein ubiquitination pathway, the same pathways were generated by IPA with different hierarchy. (B) List of representative differentially expressed genes involved in pathways generated by IPA and STRING software. Genes are ranked from most to least deregulated (most deregulated genes first). Data are generated from n = 3 independent experiments on three different control and three different HNPK Labrador retriever dogs. (C) Functional protein connection network generated by STRING software with the 2,154 differentially expressed gene products (Table S1). Same pathways generated by IPA were found with STRING, in addition to adhesion, intermediate filament, and epidermal differentiation complex.
Notable examples supporting Wnt activation were a >100-fold up-regulation of steady-state mRNA levels of R-spondin (a Wnt activator) and an up to 2.7-fold increase in axin1 and c-Myc (Wnt target genes) as well as the Wnt effector β-catenin (Fig. 2, B and C; and Table S1). In contrast and notably, expression of typical growth-promoting and transforming Wnt coactivators such as Bcl9 (Mani et al., 2009), the skin tumor promoter Tcf7L1 (Ku et al., 2017), Wnt antagonists such as APC2, a component of the destruction complex targeting β-catenin, and DKK4 were down-regulated (up to 3.5-fold). In support of Wnt signaling promoting premature cell cycle exit of progenitor cells, pro-proliferative gene products implicated in cell cycle progression were down-regulated (up to 16-fold). These comprised most of the cyclins and Cdk’s as well as CDC25A, a major activator of G1/S transition and mitosis (Rudolph, 2007). On the other hand, gene products involved in cell cycle exit at G0, such as CDC20B and CDK inhibitor 2B, were up-regulated (up to 11-fold; Kapanidou et al., 2017) in parallel with the tumor suppressor p53 network. Exuberantly increased mRNA levels of p53 targets (up to 143-fold) were, for example, cyclin G1, an atypical cyclin involved in G2-M arrest in the cell cycle, and Rarres3, a facilitator of keratinocyte differentiation (Sturniolo et al., 2003). Furthermore, MDM2 and thrombospondin, which can act as positive regulators of p53 during cytotoxic stress (Gajjar et al., 2012) or Ras-induced replicative senescence (Baek et al., 2013), were also increased. Supporting entry into a senescence-like state and premature skin aging, which is likely due to chromosomal instability, mRNA levels of S100A family members, required for calcium ion binding (Halawi et al., 2014), were up-regulated (up to 25-fold). Concomitantly, Notch signaling and terminal differentiation were compromised; gene products encoding major proteins for the formation and proper sealing of the cornfield envelope, such as cornulin, repetin, loricrin, and involucrin, were all severely reduced (up to 81-fold; Negri et al., 2019; Nowell and Radtke, 2013).
When the murine orthologues of the identified differentially expressed canine genes were submitted to IPA, a more extensive gene list of the same pathways was generated, including up-regulation of the protein ubiquitination pathway, which is critically involved in modulating cell cycle progression through degradation of Cdk’s (Nakayama and Nakayama, 2006; Fig. 2 A and Table S1).
We then submitted the 2,154 differentially expressed canine genes (Table S1) to the STRING algorithm (Szklarczyk et al., 2015) to generate a functional protein association network of deregulated effector groups. An interaction prediction for the same groups identified by IPA scored with the highest confidence, positioned Wnt signaling as a core pathway among these events, and further highlighted additional, differentially up-regulated gene products belonging to the adhesion/intermediate filament and epidermal differentiation complex not represented in IPA (Fig. 2, B and C; and Table S1). The former two networks comprised a variety of potential gene targets of the p53 family member p63, which is considered a master regulator of epidermal homeostasis and upstream effector of many structural proteins, including keratins and cell–cell adhesion molecules (Nowell and Radtke, 2013; Soares and Zhou, 2018; Truong et al., 2006). Accordingly, in HNPK, mRNA levels of the major epidermal keratins KRT1, 5, 10, and 14, atypical epidermal keratins such as KRT17 (confined to activated keratinocytes, e.g., during wound healing), and KRT26 (expressed in hair follicles) were increased (up to 20-fold; Moll et al., 2008; Table S1). Noticeably, however, KRT6A, a marker for hyperproliferative skin, was unchanged. Up-regulated cell–cell adhesion molecules comprised components of desmosomes and adherens junctions. Interestingly, p63 (TP63) itself was not identified as a significantly deregulated gene product in the RNA-seq study. Various alternatively expressed ΔNp63 and TAp63 isoforms are involved in epidermal homeostasis, and altered expression of p63 may thus not be resolved in the RNA-seq analysis.
In conclusion, differential gene expression in the nasal planum epidermis of HNPK dogs suggests that the loss of H3K9 trimethylation has a profound impact on core homeostatic signaling pathways implicated in fate conversion and differentiation of proliferating progenitor cells. Major changes comprise premature cell cycle exit and increased Wnt and potentially p63 signaling. Furthermore, differentiation is compromised, indicated by decreased Notch signaling, impaired terminal differentiation, and cornification. Up-regulation of stress-induced p53 targets further highlights that the premature exit from the cell cycle occurs on a background of progressive genomic instability and a senescence-like state. This collective phenotype agrees with the severe histological alterations in HNPK nasal epidermis (Bannoehr et al., 2020; Bauer et al., 2018; Jagannathan et al., 2013; Pagé et al., 2003).
Nasal keratinocytes isolated from HNPK dogs exhibit premature progenitor fate conversion paired with a senescence-like state
To gain mechanistic insight into the role of SUV39H2 in epithelial homeostasis, keratinocytes were isolated (Kolly et al., 2005) from the nasal epidermis of two HNPK Labrador retrievers (HNPK I, Fig. 1, and one dog from the RNA-seq study), one healthy Labrador control (Control II, Fig. 1), and two other healthy control dogs of other breeds. In standard medium containing 1.8 mM calcium chloride (Kolly et al., 2005), keratinocytes from HNPK dogs could not be expanded. They stopped proliferating and formed compact islands with cells of flattened, differentiated morphology. However, a comparable growth rate for proliferating HNPK and control keratinocytes could be achieved with a two-component medium (CnT-09; CnT-07) containing only 0.6 mM calcium chloride (Fig. 3 A). Nonetheless, while normal canine keratinocytes could be expanded over >50 passages without significant reduction in population doubling (Kolly et al., 2005), HNPK keratinocyte growth consistently slowed down after passage 12. Cells became flatter with irregular morphology but without increased apoptosis (Fig. 3, B and C). As assessed at passage 15, HNPK cells exhibited 8–10% nuclei with aberrant shape and nuclear disintegration or became multinucleated (Fig. 3, C–F). This indicated that keratinocytes from HNPK dogs exhibited progressive chromosomal instability paired with a senescence-like state, which is compatible with the requirement of SUV39H2 for genome stability (Peters et al., 2001).

HNPK nasal keratinocytes exhibit premature exit from the cell cycle and recapitulate in vivo RNA-seq data. (A) Cumulative population doubling of proliferative nasal keratinocytes during expansion over several passages (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate); note that population doublings from the two HNPK dogs significantly decrease after passage 14. (B) Cell viability of proliferating keratinocytes along passages (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Graph depicts no differences in apoptotic rate. (C) Nasal keratinocyte morphology during expansion. White arrows indicate example of multinucleated cells (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Scale bars, 10 µm. (D) Hoechst staining reveals aberrant nuclear shape and nuclear disintegration in HNPK nasal keratinocytes at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Scale bars, 5 µm. (E) Quantification of cells with aberrant nuclear shape and/or disintegration (590–610 nuclei/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs). (F) Quantification of multinucleated cells in keratinocytes at passage 15 at onset of calcium-induced differentiation (900–1,000 cells/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (G) Quantification of attached cells during calcium-induced differentiation per micrographs, indicating that HNPK nasal keratinocytes detach earlier than control cells (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (H) Quantification of cells in supernatant during calcium-induced differentiation at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (I) Reporter gene assays for Wnt signaling pathway activity before calcium-induced onset of differentiation at passages 6 and 15 (mean of n = 2 independent experiments on nasal keratinocytes from two (passage 6) and five (passage 15) different dogs in triplicate). The six values per cell type and treatment obtained from the two independent experiments done in triplicates were submitted for statistical analysis. (J) RT-qPCR on RNA from cultured cells of passage 15 during calcium-induced epidermal differentiation, assessing SUV39H2, SUV39H1, and selected deregulated gene products indicated by RNA-seq and signaling pathway analyses (n = 3 independent experiments on nasal keratinocytes from two or three different dogs). Note that the expression pattern of SUV39H1 and SUV39H2 are different. Data were calculated relative to control keratinocytes at day 0 set to 1. In the graph comparing SUV39H1 and SUV39H2 expression, data were calculated relative to the level of SUV39H2 in control keratinocytes at day 0 set to 1. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated with Kruskal-Wallis test with Dunn post hoc test for experiments with nasal keratinocytes from three dogs and with Wilcoxon signed-rank test with nasal keratinocytes from two dogs. Ctrl, control; P, cell passage.
HNPK nasal keratinocytes exhibit premature exit from the cell cycle and recapitulate in vivo RNA-seq data. (A) Cumulative population doubling of proliferative nasal keratinocytes during expansion over several passages (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate); note that population doublings from the two HNPK dogs significantly decrease after passage 14. (B) Cell viability of proliferating keratinocytes along passages (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Graph depicts no differences in apoptotic rate. (C) Nasal keratinocyte morphology during expansion. White arrows indicate example of multinucleated cells (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Scale bars, 10 µm. (D) Hoechst staining reveals aberrant nuclear shape and nuclear disintegration in HNPK nasal keratinocytes at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Scale bars, 5 µm. (E) Quantification of cells with aberrant nuclear shape and/or disintegration (590–610 nuclei/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs). (F) Quantification of multinucleated cells in keratinocytes at passage 15 at onset of calcium-induced differentiation (900–1,000 cells/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (G) Quantification of attached cells during calcium-induced differentiation per micrographs, indicating that HNPK nasal keratinocytes detach earlier than control cells (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (H) Quantification of cells in supernatant during calcium-induced differentiation at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (I) Reporter gene assays for Wnt signaling pathway activity before calcium-induced onset of differentiation at passages 6 and 15 (mean of n = 2 independent experiments on nasal keratinocytes from two (passage 6) and five (passage 15) different dogs in triplicate). The six values per cell type and treatment obtained from the two independent experiments done in triplicates were submitted for statistical analysis. (J) RT-qPCR on RNA from cultured cells of passage 15 during calcium-induced epidermal differentiation, assessing SUV39H2, SUV39H1, and selected deregulated gene products indicated by RNA-seq and signaling pathway analyses (n = 3 independent experiments on nasal keratinocytes from two or three different dogs). Note that the expression pattern of SUV39H1 and SUV39H2 are different. Data were calculated relative to control keratinocytes at day 0 set to 1. In the graph comparing SUV39H1 and SUV39H2 expression, data were calculated relative to the level of SUV39H2 in control keratinocytes at day 0 set to 1. Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated with Kruskal-Wallis test with Dunn post hoc test for experiments with nasal keratinocytes from three dogs and with Wilcoxon signed-rank test with nasal keratinocytes from two dogs. Ctrl, control; P, cell passage.
Given that the HNPK phenotype develops in young dogs (Bannoehr et al., 2020; Pagé et al., 2003), it can be assumed that keratinocytes, which have undergone a number of cell divisions, start to enter a metastable state. Cells of passage 15 were therefore chosen for subsequent analyses that are healthy and nonapoptotic but might be prone to phenotypic alterations. To define whether the cultured nasal keratinocytes reflect the HNPK phenotype of the dogs in vivo, including RNA-seq profiling, we assessed several parameters in a differentiation assay (Kolly et al., 2005) over a time course of 12 d. Cells of all groups were seeded at the same density, and a switch to physiological calcium (1.8 mM, day 0) was introduced at 100% confluency (defined by cell counting on photomicrographs). As expected, after around 12–20 d of advanced differentiation, control keratinocytes started to delaminate from the culture dish (Fig. 3 G; Kolly et al., 2005). Strikingly, HNPK nasal keratinocytes lifted up prematurely, already 3 d after the calcium switch, as seen by less attached and more floating cells (Fig. 3, G and H). The delamination behavior of HNPK nasal keratinocytes at day 3, which was comparable to control nose keratinocytes at 12 d, supports premature cell cycle exit and aberrant differentiation in HNPK nasal keratinocytes with loss of SUV39H2 function.
Quantitative RT-PCR (RT-qPCR) performed over the entire time course of 12 d confirmed, consistent with the RNA-seq data in vivo (Table S1), that the relative SUV39H2 and SUV39H1 mRNA expression levels were not affected in HNPK nasal keratinocytes (Fig. 3 J). Remarkably, however, in control and HNPK keratinocytes, the relative SUV39H2 mRNA levels were, on average, 77 times higher than those of SUV39H1. This reflects our results in vivo (Fig. 1), which show that SUV39H2 is the major H3K9 trimethyltransferase in nasal epidermis. Furthermore, SUV39H2 mRNA expression levels peaked around day 3, suggesting that SUV39H2 has a main window of activity around the exit from the cell cycle and fate conversion of epidermal progenitor cells (Kolly et al., 2005).
We went on to test premature cell cycle exit and fate conversion linked to increased Wnt signaling as well as aberrant differentiation in these cells, as suggested from the RNA-seq data (Fig. 2). A Wnt reporter gene assay set up 1 d before calcium switch for 24 h (up to day 0) revealed sixfold to 10-fold higher Wnt activity around cell cycle exit in HNPK nasal keratinocytes than in control cells (Fig. 3 I). Wnt activity was in a similar range in HNPK cells of lower and higher passages (P6, P15). Consistent with these results, the Wnt target genes axin1, axin2, and WISP3 were up-regulated at the RNA steady-state level from sevenfold to 30-fold at day 0 in HNPK nasal keratinocytes, in parallel with R-spondin, an activator of the Wnt pathway (Fig. 3 J). Noticeably, these transcripts also peaked in control cells around day 3, albeit at a lower level, indicating that Wnt activation occurs also at fate transition in canine nose keratinocytes.
In alignment with the RNA-seq analysis and premature cell cycle exit (Fig. 2 B and Table S1), CDC25A was down-regulated by 2.5-fold at day 0 in cultured HNPK cells (Fig. 3 J). Furthermore, compromised differentiation was substantiated by impaired Notch signaling with reduced expression levels of Notch targets Hes5, Hey1, and Hey2, as well as the ligand Jagged1 and the receptor Notch4. In contrast to in vivo profiling (Fig. 2) and HNPK keratinocytes at day 12 (Fig. 3 J), cycling HNPK cells of day 3 did not appear to exhibit activation but rather inhibition of a p53-mediated stress response, as seen by lower expression levels of the transcriptional activator of p53, Rarres3, and 30 times–increased levels of Park2, a E3 ubiquitin ligase and transcriptional repressor of p53 (Sunico et al., 2013). However, as in skin biopsies of HNPK dogs (Table S1), potential p63 targets (Truong et al., 2006; Soares and Zhou, 2018) such as the cell–cell adhesion molecule desmoglein 3, governing proliferation in basal epithelia (Müller et al., 2008; Williamson et al., 2006), KRT17, and S100A2 were increased around cell cycle exit while KRT6A was unaffected. Furthermore, repetin and cornulin were decreased up to 10-fold (Fig. 3 J), in a similar range as RNA-seq data obtained from epidermal biopsies (Fig. 2 B). In contrast, most other late epidermal differentiation markers, such as involucrin, loricrin, and filaggrin, were prematurely expressed (Fig. 2 B and Table S1).
Collectively, the cultured HNPK nasal keratinocytes largely reproduced the RNA-seq data obtained in vivo with regard to premature cell cycle exit and compromised differentiation. The notable exception was lack of p53 activation at cell cycle exit, pointing toward a stress response due to genome instability confined to more advanced time points. Moreover, increased expression of terminal differentiation markers, compatible with premature cell cycle exit, may also occur in the epidermis but cannot be extracted from the RNA-seq data. In summary, the comparable phenotype of keratinocytes in vivo and in vitro supports the notion that the epigenetic landscape is preserved when cells are taken in culture.
The SUV39H2 variant is the functional cause of HNPK
To define the direct mechanistic consequences of SUV39H2 inactivation, we knocked down SUV39H2 mRNA in control keratinocytes 1 d before calcium switch using a combination of two siRNAs and scrambled siRNA as a control. RT-qPCR and immunoblot analyses confirmed the efficient SUV39H2 knockdown at day 0 and day 3 after calcium switch (Fig. 4 A and Fig. S3). In general, after 3 d, the SUV39H2 knockdown cells exhibited a similar phenotype to HNPK cells; that is, the cells prematurely detached from the culture dish and an increase in multinucleated cells was noted (Fig. 4, B–D). Between days 5 and 12, the effect of the SUV39H2 knockdown waned at the mRNA level (Fig. 4 A), and the cells gradually recovered, however without completely returning to control levels. This may suggest that faulty SUV39H2 function at cell cycle exit irreversibly impacts the subsequent differentiation process.

SUV39H2 knockdown mimics the HNPK phenotype. (A) RT-qPCR on SUV39H2 knockdown of control compared with HNPK nasal keratinocytes during calcium-induced differentiation at passage 15. Values represent relative mRNA expression of SUV39H2, SUV39H1, and selected deregulated genes identified in RNA-seq and signaling pathway analyses. Data are calculated relative to control keratinocytes transfected with scrambled siRNA at day 0 set to 1 (n = 3 independent experiments on nasal keratinocytes from three different dogs in duplicate). (B) Quantification of attached cells during differentiation and SUV39H2 knockdown per micrographs at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (C) Quantification of cells in supernatant during differentiation and SUV39H2 knockdown at passage 15, indicating that HNPK and SUV39H2 knockdown keratinocytes detach earlier than control keratinocytes (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (D) Quantification of multinucleated cells in keratinocytes at passage 15 at onset of differentiation (900–1,000 cells/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (E) Immunodetection of SUV39H2, H3K9me3, β-catenin, and plakoglobin (PG) in control, SUV39H2 knockdown, and HNPK nasal keratinocytes at passage 15 and at days 0 and 4 after calcium-induced differentiation (n = 3 independent experiments on nasal keratinocytes from three different dogs in duplicate). Pictures are representative of three independent experiments. Scale bars, 5 µm. (F) SUV39H2, H3K9me3, β-catenin, and plakoglobin protein intensity quantification normalized to nonspecific rabbit IgG and quantification of Ki67-positive cells on >10 micrographs per antibody generated in E. (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated with Kruskal-Wallis test with Dunn post hoc test.
SUV39H2 knockdown mimics the HNPK phenotype. (A) RT-qPCR on SUV39H2 knockdown of control compared with HNPK nasal keratinocytes during calcium-induced differentiation at passage 15. Values represent relative mRNA expression of SUV39H2, SUV39H1, and selected deregulated genes identified in RNA-seq and signaling pathway analyses. Data are calculated relative to control keratinocytes transfected with scrambled siRNA at day 0 set to 1 (n = 3 independent experiments on nasal keratinocytes from three different dogs in duplicate). (B) Quantification of attached cells during differentiation and SUV39H2 knockdown per micrographs at passage 15 (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (C) Quantification of cells in supernatant during differentiation and SUV39H2 knockdown at passage 15, indicating that HNPK and SUV39H2 knockdown keratinocytes detach earlier than control keratinocytes (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (D) Quantification of multinucleated cells in keratinocytes at passage 15 at onset of differentiation (900–1,000 cells/dog, n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). (E) Immunodetection of SUV39H2, H3K9me3, β-catenin, and plakoglobin (PG) in control, SUV39H2 knockdown, and HNPK nasal keratinocytes at passage 15 and at days 0 and 4 after calcium-induced differentiation (n = 3 independent experiments on nasal keratinocytes from three different dogs in duplicate). Pictures are representative of three independent experiments. Scale bars, 5 µm. (F) SUV39H2, H3K9me3, β-catenin, and plakoglobin protein intensity quantification normalized to nonspecific rabbit IgG and quantification of Ki67-positive cells on >10 micrographs per antibody generated in E. (n = 3 independent experiments on nasal keratinocytes from three different dogs in triplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated with Kruskal-Wallis test with Dunn post hoc test.

siRNA experimental design and efficiency and Ki67-positive cells in control and HNPK nasal planum. (A) RT-qPCR of SUV39H2 expression 24 h after SUV39H2 siRNA transfection. Three SUV39H2-specific siRNAs (1, 2, and 3) were designed and tested separately or in combination in a dose response compared with negative scrambled siRNA set to 1 (n = 1 experiment on one control dog in duplicate). (B) Second siRNA setup of mixed SUV39H2 siRNA 2 + 3 efficiency during time course. Data are calculated relative to 50 nM scrambled siRNA at day 0 set to 1. (n = 1 experiment on one control dog in duplicate). (C) Immunoblot analyses of Triton X-100 soluble protein levels after SUV39H2 siRNA knockdown. Left panel: Membrane scanning for indicated proteins with the molecular weight marker (MW) apparent in the scan shown on the left. Note that isoform 2 corresponds to the main protein band identified in human samples. Right panel: Protein band from quantifications. Protein data were normalized to tubulin and calculated relative to scrambled siRNA control set to 1 (n = 1 experiment on one dog). (D) Quantification of the percentile of counted Ki67-positive cells per number of basal layer keratinocytes in control and HNPK nasal epidermis (250–300 Ki67-positive cells/dog, n = 1 independent experiment on three different dogs in triplicate). Data are mean ± SEM. **P < 0.01 for Ki67-positive cell counting. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. Pvalues for D were calculated on pooled numbers with Wilcoxon signed-rank test.
siRNA experimental design and efficiency and Ki67-positive cells in control and HNPK nasal planum. (A) RT-qPCR of SUV39H2 expression 24 h after SUV39H2 siRNA transfection. Three SUV39H2-specific siRNAs (1, 2, and 3) were designed and tested separately or in combination in a dose response compared with negative scrambled siRNA set to 1 (n = 1 experiment on one control dog in duplicate). (B) Second siRNA setup of mixed SUV39H2 siRNA 2 + 3 efficiency during time course. Data are calculated relative to 50 nM scrambled siRNA at day 0 set to 1. (n = 1 experiment on one control dog in duplicate). (C) Immunoblot analyses of Triton X-100 soluble protein levels after SUV39H2 siRNA knockdown. Left panel: Membrane scanning for indicated proteins with the molecular weight marker (MW) apparent in the scan shown on the left. Note that isoform 2 corresponds to the main protein band identified in human samples. Right panel: Protein band from quantifications. Protein data were normalized to tubulin and calculated relative to scrambled siRNA control set to 1 (n = 1 experiment on one dog). (D) Quantification of the percentile of counted Ki67-positive cells per number of basal layer keratinocytes in control and HNPK nasal epidermis (250–300 Ki67-positive cells/dog, n = 1 independent experiment on three different dogs in triplicate). Data are mean ± SEM. **P < 0.01 for Ki67-positive cell counting. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. Pvalues for D were calculated on pooled numbers with Wilcoxon signed-rank test.
Significant effects of the SUV39H2 knockdown were further measured for gene products in the identified altered signaling pathways (Fig. 2). In general, the SUV39H2 knockdown shifted expression of genes of interest in normal keratinocytes toward the expression levels of HNPK nasal keratinocytes (Fig. 4 A). This shift was, for example, marked for two targets and two effectors of Wnt signaling identified in skin keratinocytes, axin2 and c-Myc, as well as β-catenin and plakoglobin (ɣ-catenin; He et al., 1998; Lustig et al., 2002; Williamson et al., 2006). Furthermore, Notch target gene expression and signaling were decreased. As H3K9 trimethylation is a repressive mark, its loss is expected to increase gene expression. The decrease in Notch signaling could therefore be assumed to represent a consequence of overactivation of upstream effectors such as p63, known to inhibit Notch (Nowell and Radtke, 2013). As suggested by the expression profile of specific target genes in vivo and cultured nasal keratinocytes (Fig. 2 and Fig. 3 J), ΔNp63 and TAp63 mRNA levels were increased in HNPK and followed the same trajectory upon SUV39H2 knockdown while remarkably maintaining their inverse expression pattern (Fig. 3 J). Compatible with higher p63 activity, Notch signaling was reduced and desmoglein 3 and desmocollin 1, 2, and 3 as well as E-cadherin mRNA levels were significantly up-regulated in cultured HNPK nasal keratinocytes and SUV39H2 knockdown control cells (Fig. 4 A; Nowell and Radtke, 2013; Soares and Zhou, 2018; Truong et al., 2006). The phenotypic change in SUV39H2 knockdown cells indicated that epigenetic SUV39H2 activity is critically required between day 0 and day 3 to temper the fate conversion of proliferating progenitor cells toward epidermal lineage specification.
Immunofluorescence microscopy confirmed corresponding changes on the protein level (Fig. 4, E and F). In SUV39H2 knockdown cells, SUV39H2 protein was significantly reduced at day 0, with a slow return toward normal levels observed at day 4 after calcium switch. SUV39H2 levels were not changed in HNPK nasal keratinocytes, compatible with stable RNA levels despite the inactivating mutation (Fig. 3 J and Fig. 4 A; Jagannathan et al., 2013; Schuhmacher et al., 2015). However, H3K9me3 marks were largely absent at day 0 in SUV39H2 knockdown and at days 0 and 4 in HNPK nuclei, similar to the nasal epidermis (Fig. 1). Furthermore, a few nuclei faintly stained for the H3K9me3 mark in HNPK keratinocytes. β-Catenin and plakoglobin were expressed at moderate levels in control cells and up-regulated at all time points in SUV39H2 knockdown and HNPK nasal keratinocytes. This set of data demonstrates first that increased transcripts can be swiftly translated into altered protein levels and second that these proteins reflect a relieved epigenetic mark beyond recovery of SUV39H2 protein levels seen at day 4 in knockdown cells (Fig. 4 F). Moreover, compatible with premature cell cycle exit and quantification in basal keratinocytes of the nasal planum (Fig. S3 D), the relative number of Ki67-positive cells, marking proliferative keratinocytes, was on average lower in HNPK cells but not affected in the short period of siRNA treatment in HNPK knockdown cultures (Fig. 4 F).
Finally, the unexpectedly rapid loss of H3K9me3 marks upon SUV39H2 knockdown (within 24 h; Fig. 4 E) pointed toward the possibility that the relief from SUV39H2-mediated gene repression might be accompanied by collateral de-repression of H3K9 demethylases, erasing preexisting histone marks. Indeed, mRNA expression levels of the two H3K9 demethylases KDM4C and KDM7A were significantly up-regulated in the RNA-seq analysis in vivo (Table S1) as well as in HNPK nasal keratinocytes and SUV39H2 knockdown cells (Fig. 4 A, top lane). This suggests potentiation of the loss of SUV39H2 activity through de-repression of demethylases and erasure of preexisting H3K9me3 marks.
In conclusion, through siRNA knockdown experiments, our results demonstrate that the HNPK phenotype is caused by the functionally inactive SUV39H2 variant (Schuhmacher et al., 2015), resulting in a failure to slow the fate conversion of progenitor cells and temper the speed of cell cycle exit. The concomitant unscheduled activation of signaling pathways known from studies on normal epidermal homeostasis (Choi et al., 2013; Klein and Andersen, 2015; Koster et al., 2004; Lim and Nusse, 2013; Miroshnikova et al., 2018; Negri et al., 2019; Nowell and Radtke, 2013; Truong et al., 2006; Williamson et al., 2006), such as Wnt/p63/adhesion paired with Notch inhibition and deregulated expression of effectors in the epidermal differentiation complex, supports that SUV39H2 is a major gatekeeper of the progenitor pool in epidermal nasal planum keratinocytes through epigenetic control of progenitor-promoting signaling pathways.
SUV39H2 epigenetically targets Wnt/p63/adhesion signaling pathways
We next addressed by comparative chromatin immunoprecipitation (ChIP)-qPCR whether SUV39H2 is directly regulating H3K9 trimethylation on specific target genes of the here-identified affected pathways. Chromatin was precipitated from keratinocytes of three control and two HNPK dogs at day 0 and day 5 after calcium switch using H3K9me3 antibodies (Wiencke et al., 2008) or, as a negative control, rabbit IgG (Fig. 5 A). DNA was amplified by qPCR using “positive primers” designed to recognize the area around the transcriptional start site of genes of interest, where H3K9me3 marks were shown to be predominantly localized (Amabile et al., 2016). Irrelevant “negative primers” were designed roughly 2.5 kb upstream of the transcriptional start site. The gene encoding the telomerase reverse transcription (telomerase) is known to permanently carry H3K9me3 repressive marks in adult tissue (Zhang et al., 2014) and was thus used as a positive control. The ribosomal protein S5, encoding a housekeeping gene free of repressive marks, served as negative control. Notably, while chromatin at the telomerase promoter contained a high fraction of H3K9me3 in cells from control dogs, as judged from the amount of DNA collected, H3K9 marks were significantly lower in cells from both HNPK dogs (Fig. 5 B). This is compatible with the involvement of SUV39H1 and SUV39H2 in controlling the length of telomeres (García-Cao et al., 2004) and suggests that this control can occur through modulation of telomerase activity. Furthermore, in HNPK cells the lack of SUV39H2 activity could not be fully compensated by other H3K9 trimethyltransferases.

ChIP-qPCR reveals that SUV39H2 selectively promotes H3K9me3 marks on gene promoters encoding cell cycle, Wnt/p63/adhesion, and epidermal differentiation complex components. (A) Schematic representation of ChIP primer design for H3K9me3–ChIP-qPCR on transcriptional start site (TSS; positive primers) and negative control (negative primers). Arrows indicate the qPCR amplicon localization on the gene. (B) H3K9me3 ChIP-qPCR on target gene promoters selected from RNA-seq and signaling pathway analyses. All experimental values were normalized to input DNA * 100. Mean values of keratinocytes during calcium-induced differentiation from three control (12 values) and two HNPK (eight values) dogs per data point from n = 2 independent ChIP experiments in duplicate are presented. Dotted lines represent the mean of nonspecific rabbit IgG amplifications (background threshold). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated on pooled values from different dogs per data point using the Kruskal-Wallis test with Dunn post hoc test.
ChIP-qPCR reveals that SUV39H2 selectively promotes H3K9me3 marks on gene promoters encoding cell cycle, Wnt/p63/adhesion, and epidermal differentiation complex components. (A) Schematic representation of ChIP primer design for H3K9me3–ChIP-qPCR on transcriptional start site (TSS; positive primers) and negative control (negative primers). Arrows indicate the qPCR amplicon localization on the gene. (B) H3K9me3 ChIP-qPCR on target gene promoters selected from RNA-seq and signaling pathway analyses. All experimental values were normalized to input DNA * 100. Mean values of keratinocytes during calcium-induced differentiation from three control (12 values) and two HNPK (eight values) dogs per data point from n = 2 independent ChIP experiments in duplicate are presented. Dotted lines represent the mean of nonspecific rabbit IgG amplifications (background threshold). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated on pooled values from different dogs per data point using the Kruskal-Wallis test with Dunn post hoc test.
Consistent with increased mRNA levels (Fig. 4 A), the demethylase promoters KDM4A and KDM7A were H3K9 trimethylated in control but not HNPK cells (Fig. 5 B). Unlike in control keratinocytes, H3K9me3 repressive marks were also absent from promoters of Wnt pathway components and Wnt target genes, β-catenin, plakoglobin, c-Myc, and cyclin D1 as well as p63 isoforms, adhesion molecules, and late terminal differentiation markers, compatible with their unscheduled activation in HNPK cells (Fig. 2 and Fig. 3). H3K9 trimethylation of axin1/2, CDC25A, and Hes5 promoters was not observed in control keratinocytes at the time points investigated.
Collectively, the significant loss of H3K9me3 repressive marks, shown by ChIP-qPCR in HNPK keratinocytes, which affects key signaling pathways of progenitor fate conversion and onset of differentiation (Choi et al., 2013; Klein and Andersen, 2015; Koster et al., 2004; Lim and Nusse, 2013; Miroshnikova et al., 2018; Negri et al., 2019; Nowell and Radtke, 2013; Truong et al., 2006; Williamson et al., 2006), reveals a central role for SUV39H2 in epigenetic silencing of Wnt/p63/adhesion pathways in nasal epidermal progenitor cells.
Elevated Wnt signaling is a mediator of cell fate conversion at cell cycle exit in epidermal keratinocytes and a major cause of HNPK
Wnt signaling has a dominant, albeit controversial, role in epidermal homeostasis, with reported activities ranging from stem cell maintenance to proliferation, lineage selection, and differentiation (Choi et al., 2013; Lim and Nusse, 2013; Williamson et al., 2006). Accordingly, in mice many Wnt signaling mutations display an epidermal phenotype (Lim and Nusse, 2013).
We questioned whether Wnt might be a key driver of progenitor cell fate conversion and the remarkable up-regulation of Wnt signaling in HNPK (Fig. 2, Fig. 3, Fig. 4, and Fig. 5), a major cause of premature cell cycle exit and compromised differentiation. To this aim, we inhibited Wnt signaling in HNPK keratinocytes using IWR1 (Inhibitor of Wnt Response-1; Lu et al., 2009) and activated Wnt in control cells by BIO (Meijer et al., 2003) 1 d before calcium switch (Fig. 6). While IWR1 and BIO strongly modulated Wnt signaling, confirmed by down- and up-regulation of axin1 and axin2 mRNA levels, respectively, they did not affect SUV39H2 expression, positioning SUV39H2 upstream of Wnt (Fig. 6 D and Table S2). In contrast to SUV39H2 knockdown (Fig. 4), Wnt modulation failed to alter the nuclear phenotype in both control and HNPK keratinocytes, in line with direct SUV39H-mediated chromosomal stabilization (Peters et al., 2001; Fig. 6 C). Strikingly, however, Wnt activation shifted the fate of control keratinocytes toward the HNPK phenotype, and Wnt inhibition shifted HNPK keratinocytes toward control cells. Characteristically, BIO-treated control keratinocytes prematurely lifted up from the culture dish while IWR1-treated HNPK keratinocytes converted toward the more adherent status of vehicle-treated control cells (Fig. 6, A and B). These changes were accompanied by differential modulation of mRNA levels in all representative pathways identified in this study toward the HNPK (BIO) or control phenotype (IWR1). This suggested an antagonistic relationship between Wnt and SUV39H2, with differential longitudinal modulation of specific gene products by Wnt effectors and H3K9me3 repressive marks.
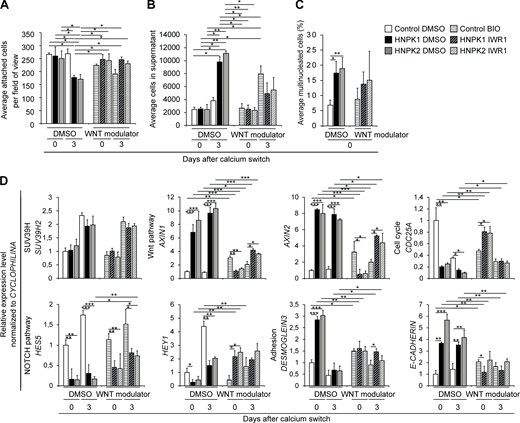
Increased Wnt signaling is a pathomechanism in HNPK.(A) Quantification of attached cells with Wnt modulation (BIO: Wnt activation in control cells; IWR1: Wnt suppression on HNPK cells) during differentiation (n = 2 independent experiments on three different nasal dog cell isolates in triplicate). (B) Quantification of cells in supernatant of cultures in A. (C) Quantification of multinucleated cells of cultures in A (900–1,000 cells/dog). (D) RT-qPCR of selected gene products relative to control DMSO set to 1 (n = 2 independent experiments on three different dog cell isolates in triplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated on pooled values per data point and cell isolate using with Kruskal-Wallis test with Dunn post hoc test. See Table S2 for a compilation of P values.
Increased Wnt signaling is a pathomechanism in HNPK.(A) Quantification of attached cells with Wnt modulation (BIO: Wnt activation in control cells; IWR1: Wnt suppression on HNPK cells) during differentiation (n = 2 independent experiments on three different nasal dog cell isolates in triplicate). (B) Quantification of cells in supernatant of cultures in A. (C) Quantification of multinucleated cells of cultures in A (900–1,000 cells/dog). (D) RT-qPCR of selected gene products relative to control DMSO set to 1 (n = 2 independent experiments on three different dog cell isolates in triplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. P values were calculated on pooled values per data point and cell isolate using with Kruskal-Wallis test with Dunn post hoc test. See Table S2 for a compilation of P values.
To confirm that Wnt signaling also impacts fate conversion in human and mouse epidermal keratinocytes, we treated primary skin keratinocytes of both species with the Wnt activator BIO. Notably, as seen under the same treatment regimen applied to dog control keratinocytes (Fig. 6), BIO increased delamination of keratinocytes from the culture dish compatible with premature cell cycle exit. As expected, nuclear integrity and SUV39H2 mRNA levels remained unaffected (Fig. S5, A–C; and Fig. S5, E–G). Further supporting the contribution of Wnt to fate conversion in normal human and mouse epidermal keratinocytes, axin1 and axin2 levels significantly increased at the onset of differentiation in control cells as well as upon BIO treatment (Fig. S5, D and H). BIO further strongly reduced the mRNA levels of CDC25A, consistent with premature cell cycle exit, but had a more moderate impact on adhesion molecules and no effect on mRNA levels of the Notch gene targets Hey1 and Hes5.
In conclusion, BIO- and IWR1-mediated Wnt modulation in HNPK and control keratinocytes combined with RNA-seq, RT-qPCR, and ChIP-qPCR data identified the premature increase in Wnt signaling as a major cause in the molecular pathogenesis of HNPK and a prominent target of transcriptional repression by SUV39H2 trimethylation in the fate conversion of normal progenitor cells. Epidermal keratinocytes from mouse and human similarly responded with fate conversion to ectopic Wnt activation, warranting further in-depth investigations.
Lack of an HNPK phenotype in skin correlates with low SUV39H2 and high SUV39H1 levels
Finally, we addressed the conundrum of why the SUV39H2 variant severely affects homeostasis of the glabrous nasal epidermis but spares hairy skin (Bannoehr et al., 2020; Jagannathan et al., 2013). We first questioned whether repressive H3K9me3 marks are present in hairy skin biopsies of one of the HNPK dogs used in the nose study (HNPK 1; Fig. 1). Strikingly, the distribution pattern and abundance of H3K9me3 marks was similar in HNPK and control dog skin (Fig. 7 A). With the 77-times-higher mRNA levels of SUV39H2 than of SUV39H1 in control nose epidermal keratinocytes in mind (Fig. 3 J and Fig. 7 B), we isolated haired skin keratinocytes from the same dog (HNPK1; Fig. S4) and compared the relative expression levels of these two enzymes between nose and hairy skin (Fig. 7 B). Interestingly, in keratinocytes isolated from control hairy skin, the ratio of the transcripts was inverted, with 11 times higher SUV39H1 than SUV39H2 mRNA levels at day 0. Furthermore, the relative steady-state levels of SUV39H1 in hairy skin were in a range similar to that of SUV39H2 levels in nose epidermis. However, the expression pattern was different. The predominant SUV39H1 expression in hairy skin further correlated with the absence of lesions in hairy skin of HNPK affected dogs, along with a normal H3K9me3 pattern in hairy skin biopsies (Fig. 7 A). Hairy skin keratinocytes from the HNPK dog exhibited the same morphology as normal dog cells, did not age significantly faster over 14 passages, and did not exhibit signs of premature exit from the cell cycle in a differentiation study (Fig. S4; and Fig. 7, A–G). Moreover, the mRNA expression pattern of pathway effectors, as identified in nose epidermis, did not exhibit significant differences between control and HNPK hairy skin keratinocytes (Fig. 7 H). Finally, we also confirmed on biopsies from one control and one HNPK dog that markers of the Wnt/p63/adhesion axis were increased in HNPK nose tissue but, with the exception of some increase in Dsg3, were not deregulated in hairy skin (Fig. S1).

SUV39H2 loss of function has no phenotypic consequences for haired skin. (A) Immunodetection of H3K9me3 in control and HNPK haired skin. Note that H3K9me3 labeling is comparable between genotypes (n = 3 independent experiments on three different dog biopsies in triplicate; shown are two dogs). See also Fig. S1. Scale bars, 100 µm. Dashed line indicates the basement membrane zone. (B) RT-qPCR comparison of relative mRNA expression levels of SUV39H2/H1and AXIN2 in nasal and haired skin keratinocyte cultures during differentiation at passage 15. Data are calculated relative to SUV39H2 and axin2 levels, respectively, in control nose keratinocytes at day 0 set to 1. Note that SUV39H1 and SUV39H2 mRNA expression patterns are tissue dependent (n = 3 independent experiments on keratinocytes from two control dogs (one nose, one skin) in triplicate). (C) Cumulative population doubling during expansion of proliferative keratinocytes along passages. Note that control and HNPK population doubling is not significantly different (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (D) Cell viability of proliferating hairy skin keratinocytes along passages. Graph depicts no differences in apoptotic rate (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (E) Quantification of attached cells at passage 15 during differentiation per micrographs, indicating that hairy skin control and HNPK keratinocytes exhibit similar cell detachment (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (F) Quantification of cells in supernatant during differentiation at passage 15 (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (G) Multinucleated cell quantification in keratinocytes at passage 15 at onset of differentiation (900–1,000 cells/dog, n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (H) RT-qPCR during differentiation of haired skin keratinocytes at passage 15 of SUV39H2, SUV39H1, and selected deregulated genes identified in nose keratinocytes. Data are calculated relative to control keratinocytes at day 0 set to 1 (n = 3 independent experiments on skin keratinocytes from two different dogs in duplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. Pvalues were calculated with Wilcoxon signed-rank test.
SUV39H2 loss of function has no phenotypic consequences for haired skin. (A) Immunodetection of H3K9me3 in control and HNPK haired skin. Note that H3K9me3 labeling is comparable between genotypes (n = 3 independent experiments on three different dog biopsies in triplicate; shown are two dogs). See also Fig. S1. Scale bars, 100 µm. Dashed line indicates the basement membrane zone. (B) RT-qPCR comparison of relative mRNA expression levels of SUV39H2/H1and AXIN2 in nasal and haired skin keratinocyte cultures during differentiation at passage 15. Data are calculated relative to SUV39H2 and axin2 levels, respectively, in control nose keratinocytes at day 0 set to 1. Note that SUV39H1 and SUV39H2 mRNA expression patterns are tissue dependent (n = 3 independent experiments on keratinocytes from two control dogs (one nose, one skin) in triplicate). (C) Cumulative population doubling during expansion of proliferative keratinocytes along passages. Note that control and HNPK population doubling is not significantly different (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (D) Cell viability of proliferating hairy skin keratinocytes along passages. Graph depicts no differences in apoptotic rate (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (E) Quantification of attached cells at passage 15 during differentiation per micrographs, indicating that hairy skin control and HNPK keratinocytes exhibit similar cell detachment (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (F) Quantification of cells in supernatant during differentiation at passage 15 (n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (G) Multinucleated cell quantification in keratinocytes at passage 15 at onset of differentiation (900–1,000 cells/dog, n = 3 independent experiments on skin keratinocytes from two different dogs in triplicate). (H) RT-qPCR during differentiation of haired skin keratinocytes at passage 15 of SUV39H2, SUV39H1, and selected deregulated genes identified in nose keratinocytes. Data are calculated relative to control keratinocytes at day 0 set to 1 (n = 3 independent experiments on skin keratinocytes from two different dogs in duplicate). Data are mean ± SEM. *P < 0.05; **P < 0.01. Normal distribution was excluded using the Anderson-Darling P value normality distribution with P < 0.05. Pvalues were calculated with Wilcoxon signed-rank test.

Haired skin keratinocyte morphology. Haired skin keratinocyte morphology during differentiation at passage 15. From top to bottom: Representative pictures of cells at day 0 before calcium switch and at 3, 5, and 12 days after calcium switch (three independent experiments on skin keratinocytes from three different dogs in triplicate). Scale bars, 10 µm.
Haired skin keratinocyte morphology. Haired skin keratinocyte morphology during differentiation at passage 15. From top to bottom: Representative pictures of cells at day 0 before calcium switch and at 3, 5, and 12 days after calcium switch (three independent experiments on skin keratinocytes from three different dogs in triplicate). Scale bars, 10 µm.
In summary, comparison of control haired skin and nose keratinocytes concerning the expression profile of key effectors during progenitor fate conversion revealed two noticeable aspects; first, differentially regulated signaling effectors were similar than in nose keratinocytes, implicating Wnt/p63/adhesion and Notch pathway modulation. Second, their expression profiles as well as those of SUV39H1 and SUV39H2 were different, and resembled those observed in human and mouse skin keratinocytes (Fig. S5). mRNA expression of key effectors such as axin2 and Hey1 increased over the inspected time course in canine (Fig. 7 H) and also human and mouse hairy skin keratinocytes (Fig. S5). In contrast, in nose keratinocytes expression returned to basal levels at day 5, following the SUV39H2 expression profile. This points toward site-specific regulation of similar pathways in the epidermal homeostatic process.

Modulation of Wnt signaling in human and mouse skin keratinocytes. (A–H) P(passage)15 normal epidermal human skin keratinocytes (A–D) and P15 normal epidermal mouse skin keratinocytes (E–H), both treated with DMSO (vehicle control, white bars) or the Wnt pathway activator BIO (black bars) 24 h after seeding. (n = 2 independent experiments on two conditions and two time points for either human or mouse keratinocytes in triplicates). (A and E) Quantification of attached cells during early differentiation counted on random micrographs. (B and F) Quantification of keratinocytes in supernatant. Note that increased Wnt signaling reduces the average of attached cells and increases delaminated cells over time in line with results of premature cell cycle exit in HNPK nasal keratinocytes (Fig. 2 and Fig. 3). (C and G) Quantification of multinucleated cells (n = 900–1,000 cells/mouse). (D and H) Graphs of results from RT-qPCR on total RNA isolated from cultured keratinocytes at indicated time points. Relative mRNA levels of SUV39H2 and representative genes of major deregulated pathways identified in canine keratinocytes. DMSO-treated cells set to 1. Note that Wnt activation affects the phenotype as in canine nose keratinocytes (see Fig. 6). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. P values were calculated with Wilcoxon signed-rank test with six pooled values obtained from the two independent experiments done in triplicates.
Modulation of Wnt signaling in human and mouse skin keratinocytes. (A–H) P(passage)15 normal epidermal human skin keratinocytes (A–D) and P15 normal epidermal mouse skin keratinocytes (E–H), both treated with DMSO (vehicle control, white bars) or the Wnt pathway activator BIO (black bars) 24 h after seeding. (n = 2 independent experiments on two conditions and two time points for either human or mouse keratinocytes in triplicates). (A and E) Quantification of attached cells during early differentiation counted on random micrographs. (B and F) Quantification of keratinocytes in supernatant. Note that increased Wnt signaling reduces the average of attached cells and increases delaminated cells over time in line with results of premature cell cycle exit in HNPK nasal keratinocytes (Fig. 2 and Fig. 3). (C and G) Quantification of multinucleated cells (n = 900–1,000 cells/mouse). (D and H) Graphs of results from RT-qPCR on total RNA isolated from cultured keratinocytes at indicated time points. Relative mRNA levels of SUV39H2 and representative genes of major deregulated pathways identified in canine keratinocytes. DMSO-treated cells set to 1. Note that Wnt activation affects the phenotype as in canine nose keratinocytes (see Fig. 6). Data are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. P values were calculated with Wilcoxon signed-rank test with six pooled values obtained from the two independent experiments done in triplicates.
Loss of SUV39H2 function affects stem cell potential in the absence of significant SUV39H1 levels
Our data obtained from nasal and skin keratinocytes identify increased Wnt signaling as a major mediator in fate conversion of epidermal progenitor cells toward keratinocytes committed to differentiation. Conversely, low-level Wnt signaling is required for stem cell maintenance in epidermis (Habib et al., 2013) and hair follicles (Blanpain and Fuchs, 2006). Hence, we expected that sustained Wnt activity upon loss of SUV39H2 function would exhaust stem cells in nasal but not hairy skin keratinocyte cultures. We confirmed this hypothesis by colony-forming efficiency assays with keratinocytes from the two different anatomical locations (nose vs. hairy skin). As shown in Fig. 8 A, HNPK nose keratinocytes grew only sparse and very small colonies. In contrast, control nose keratinocytes yielded many colonies but fewer than those from normal hairy skin. We did not observe a difference in colony-forming efficiency between HNPK and control keratinocytes of hairy skin. Thus, while stem cell growth potential was dramatically reduced in HNPK nose keratinocytes, it was not affected in those from haired skin. Nevertheless, keratinocytes from an epidermis with only a few layers had higher growth potential.
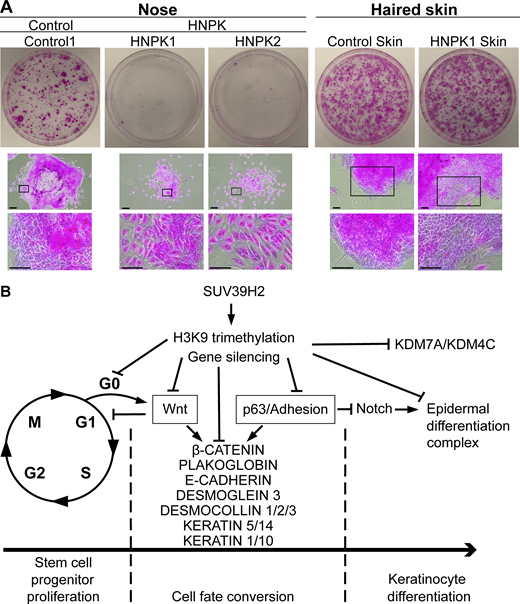
SUV39H2 loss of function reduces stem cell potential and proposed SUV39H2 epigenetic silencing scheme in nose keratinocytes.(A) Colony-forming efficiency (CFE) assay on control and HNPK nose and haired skin keratinocytes at passage 15 after 12 d in culture. Top panel: Overview of representative CFE plates stained with Rhodamine B. Bottom panel: Representative closeup pictures of CFE colonies. Note that HNPK nasal keratinocytes grow in a dispersed manner and fail to form colonies like control cells or HNPK skin keratinocytes. (n = 1 experiment on nasal keratinocytes from three different dogs and skin keratinocytes from two different dogs in triplicate). Scale bars, 25 µm. (B) Summary of the targeted signaling pathways epigenetically repressed by SUV39H2 in nasal epidermis revealed by ChIP-qPCR on representative genes (Fig. 5) and aligning with functional analyses of cultured nasal keratinocytes (Fig. 3, Fig. 6, and Fig. 8 A) and RNA-seq results from nasal planum biopsies (Fig. 2 and Table S1). Summary shows that SUV39H2 epigenetic gene silencing tampers fate conversion of proliferating progenitor cells toward epidermal differentiation through rate-limiting transcriptional repression of key effectors of the cell cycle (CDC25A, cyclin D1), the Wnt/p63/adhesion axis, and epidermal differentiation complex (see Discussion for complementary information). Progenitor-promoting Wnt signaling and downstream genes thereby represent core targets of SUV39H2 repression. If repression is relieved due to SUV39H2 inactivation, as shown in this study, H3K9me3 marks are erased, and overactivated Wnt signaling promotes the premature exit of progenitor cells from the cell cycle in concert with elevated expression of p63 and downstream targets, such as cell–cell adhesion components and keratin intermediate filaments. Consequences are exhaustion of stem and progenitor cells through enhanced mobilization while Notch signaling and terminal differentiation as well as genome stability are compromised. Results obtained in haired epidermis further suggest that SUV39H1 can compensate for loss of SUV39H2.
SUV39H2 loss of function reduces stem cell potential and proposed SUV39H2 epigenetic silencing scheme in nose keratinocytes.(A) Colony-forming efficiency (CFE) assay on control and HNPK nose and haired skin keratinocytes at passage 15 after 12 d in culture. Top panel: Overview of representative CFE plates stained with Rhodamine B. Bottom panel: Representative closeup pictures of CFE colonies. Note that HNPK nasal keratinocytes grow in a dispersed manner and fail to form colonies like control cells or HNPK skin keratinocytes. (n = 1 experiment on nasal keratinocytes from three different dogs and skin keratinocytes from two different dogs in triplicate). Scale bars, 25 µm. (B) Summary of the targeted signaling pathways epigenetically repressed by SUV39H2 in nasal epidermis revealed by ChIP-qPCR on representative genes (Fig. 5) and aligning with functional analyses of cultured nasal keratinocytes (Fig. 3, Fig. 6, and Fig. 8 A) and RNA-seq results from nasal planum biopsies (Fig. 2 and Table S1). Summary shows that SUV39H2 epigenetic gene silencing tampers fate conversion of proliferating progenitor cells toward epidermal differentiation through rate-limiting transcriptional repression of key effectors of the cell cycle (CDC25A, cyclin D1), the Wnt/p63/adhesion axis, and epidermal differentiation complex (see Discussion for complementary information). Progenitor-promoting Wnt signaling and downstream genes thereby represent core targets of SUV39H2 repression. If repression is relieved due to SUV39H2 inactivation, as shown in this study, H3K9me3 marks are erased, and overactivated Wnt signaling promotes the premature exit of progenitor cells from the cell cycle in concert with elevated expression of p63 and downstream targets, such as cell–cell adhesion components and keratin intermediate filaments. Consequences are exhaustion of stem and progenitor cells through enhanced mobilization while Notch signaling and terminal differentiation as well as genome stability are compromised. Results obtained in haired epidermis further suggest that SUV39H1 can compensate for loss of SUV39H2.
Collectively, this set of experiments supports that, as in the hair follicle (Blanpain and Fuchs, 2006), high-level Wnt has progenitor-promoting activity implicating stem cell activation and progenitor fate conversion of epidermal keratinocytes. SUV39H2 thereby plays a critical and major role in tempering this process through repression of key signaling pathways of the Wnt/p63/adhesion axis (Fig. 8 B).
Discussion
The role of SUV39H1- and SUV39H2-catalyzed H3K9me3 marks in epidermal homeostasis has so far remained elusive. However, differentially expressed SUV39H1 or SUV39H2 have been identified in global screens of human progenitor versus differentiating keratinocytes (Lopez-Pajares et al., 2015; Sen et al., 2010), and epigenetic mechanisms have been implicated in epidermal homeostasis of human and mouse epidermis (Frye and Benitah, 2012; Kang et al., 2019; Kang et al., 2020).
Here, we performed comparative analyses of epidermal homeostasis in the canine monogenic HNPK disorder with an N324K loss-of-function variant in an evolutionary highly conserved amino acid of the SUV39H2 catalytic domain. Using a combination of analyses on skin biopsies and cultured keratinocytes from HNPK patients involving RNA-seq or SUV39H2 knockdown as well as ChIP-qPCR, our results now demonstrate that the H3K9 methyltransferase SUV39H2 plays a critical role in the epigenetic maintenance of genome stability and the stem and progenitor state (Fig. 2, Fig. 4, Fig. 5, and Fig. 8 B). We show that SUV39H2 prevents progenitor cells from prematurely exiting the cell cycle by H3K9 trimethylation and transcriptional repression of genes of the Wnt/p63/adhesion axis. Thereby, timely modulation of progenitor-promoting Wnt signaling is primordial as demonstrated by pharmacological intervention on nasal keratinocytes from dogs and skin keratinocytes from humans and mice (Fig. 6 and Fig. S5), as discussed in more detail below. While the loss of SUV39H2 function in the dog nose exposes these levels of SUV39H2 involvement in the conversion process, both in vivo and in cultured keratinocytes, it also shows that impaired progenitor cell maintenance due to loss of SUV39H2 function exhausts the stem cell pool, impedes the differentiation process, and results in a senescence-like state. These changes are exemplified by reduced stem cell growth potential in cultured HNPK (Fig. 8 A), impaired Notch signaling, and faulty expression of the epidermal differentiation markers in nasal tissue and cultured keratinocytes (Fig. 2, Fig. 3, and Fig. 4; Bannoehr et al., 2020) coupled with aberrant and disintegrating nuclei along with increased p53 signaling (Fig. 2;,Fig. 3, D–J; and Fig. 4 D). These findings illustrate that the precise spatiotemporal activation of fate conversion in basal layer keratinocytes (including genomic stabilization) is a prerequisite to proper buildup of a functional stratified epithelium.
The comparative investigation of nasal and hairy skin keratinocytes from the dog further suggests that SUV39H1 and SUV39H2 exhibit functional redundancy (Fig. 7). Redundancy is stipulated by the observation that loss of SUV39H2 function correlates with a severe decrease in repressive H3K9me3 marks paired with aberrant keratinocyte differentiation in nasal epidermis with low SUV39H1 expression. In contrast, H3K9me3 marks and differentiation (likely requiring H3K9 trimethylation on similar gene promoters) are unaltered in haired skin with high SUV39H1 expression levels. Although it cannot be excluded that H3K9 methyltransferases other than SUV39H1 compensate for SUV39H2 in haired skin, our data suggest for the first time that SUV39H2, and potentially also SUV39H1, restricts a master switch during epidermal homeostasis, which consists in fate conversion of progenitor cells and commitment to keratinocyte differentiation.
To prevent unscheduled fate conversion, SUV39H2 also suppresses the expression of H3K9-specific demethylases KDM4C and KDM7A, as indicated by loss of H3K9me3 marks on these promoters in HNPK nasal keratinocytes (Fig. 5). This grip on maintaining H3K9me3 marks wanes in control nasal keratinocytes around day 5, when H3K9me3 marks on KDM4C and KDM7A promoters decrease and their mRNA steady-state levels increase (Fig. 4 A and Fig. 5 B). The concomitant decrease in SUV39H2 and also SUV39H1 mRNA expression levels (Fig. 7 B) raises the possibility that their reduced expression is a prerequisite to allow for swift demethylation of promoters in progenitor cells to enter the orderly spatiotemporal keratinocyte differentiation program. The epigenetic maintenance of a progenitor state by SUV39H2 is compatible with reduced expression of SUV39H2 in human skin keratinocytes forced to exit the progenitor cell compartment through DNA methyltransferase 1 depletion (Sen et al., 2010). Although it is currently unknown how SUV39H2 transcription is regulated, the latter supports cross-talk between DNA methylation and SUV39H2 expression in human epidermis (Gasser, 2016; Rose and Klose, 2014).
The critical role of SUV39H2 in slowing the release of progenitor cells from the proliferative compartment in nasal epidermis is congruent with the contrasting reduction of proliferative Ki67-positive progenitor cells in HNPK nasal keratinocytes as well as in biopsies of the nasal planum from dogs with SUV39H2 loss of function (Fig. 4 F and Fig. S3 D). In these dogs, this further matches the paucity of typical growth-promoting and transforming Wnt coactivators, contrasting with enhanced progenitor-promoting Wnt signaling, identified as a major driver of progenitor fate conversion at cell cycle exit in this study (Fig. 2, Fig. 6, and Table S1) These observations paint a picture whereby SUV39H2 tempers fate conversion by restricting progenitor-promoting Wnt activity through promoter H3K9 trimethylation of genuine Wnt targets and effectors, such as c-Myc and β-catenin, as well as downstream effectors of Wnt signaling in the p63 pathway, such as keratins and cell–cell adhesion molecules (Truong et al., 2006; Fig. 5 and Fig. 8). Remarkably, some of these SUV39H2 targets were also differentially regulated by Wnt activation/inhibition (Fig. 6 and Fig. S5), adding an additional layer of complexity by suggesting a reciprocal, antagonizing relationship between SUV39H2 and progenitor-promoting Wnt. Deciphering this relationship, including the cross-talk to Notch signaling in a longitudinal fashion along the homeostatic epidermal process, warrants further investigation, for which the HNPK canine model in combination with primary keratinocytes from humans and mice represents an ideal tool.
The role of SUV39H2 in slowing the release of progenitor cells is fully compatible with the correlation of overexpressed SUV39H2 and increased proliferation in a variety of cancer types in humans; these are colorectal, cervical, bladder, breast, esophagus, and prostate as well as some leukemia types and osteosarcoma, the most common primary bone cancer, augmenting the relevance of these findings for applications in human oncology (Mutonga et al., 2015; Shuai et al., 2018; Vougiouklakis et al., 2018; Piao et al., 2019).
Taken together, our findings demonstrate for the first time that SUV39H2, which promotes repressive H3K9me3 marks in constitutive and facultative heterochromatin, epigenetically controls cell fate conversion through rate-limiting H3K9me3 roadblocks in epidermal progenitor cells. The compatibility of these regulatory mechanisms with those in a variety of cancer conditions in humans in combination with the conserved mechanistic functions of H3K9me3 marks from worms to men now suggests that the here-described SUV39H2-mediated suppression of progenitor-promoting Wnt signaling and the cross-talk with other pathways of general importance in tissue homeostasis warrants further investigations in other tissues and pathological conditions in humans and mice. Furthermore, the concerted action of SUV39H2 and SUV39H1, as well as their relationship with other trimethylases and epigenetic marks such as H3K27me3 in mammalian epidermis (Frye and Benitah, 2012; Kang et al., 2019; Kang et al., 2020), represent an important step to be investigated in future studies; these marks may, to a certain degree, be controlled by H3K9me3 modifications, as suggested from analyses on heterochromatin (Gasser, 2016; Rose and Klose, 2014).
Materials and methods
Ethics statement
Biopsies were taken with informed owner consent. All animal experiments were performed according to local regulations, and the work conducted was approved by the cantonal committee on animal experimentation (permits BE22/07 and BE23/10).
Dog biopsies
Biopsies from the nasal planum of anesthetized or euthanized dogs were collected, based on availability of tissue and compatible history, from three HNPK Labrador retriever patients (a 2.5-yr-old male [HNPK I] and two females, 5 and 14.8 yr old [HNPK II and III]) and three Labrador retriever controls (three males, 10, 11, and 12 yr old [Controls I, II, and III]); biopsies were used for RNA-seq analyses (European Nucleotide Archive and GenBank accession no. PRJEB32103; Bannoehr et al., 2020) and immunofluorescence microscopy.
Biopsies for the establishment of nose keratinocyte cultures were collected from the nasal planum of two HNPK Labrador retriever patients (a 2-yr-old female [HNPK 1] and a 2.5-yr-old male [HNPK 2, the same as in HNPK I]) and three control dogs (a 12-yr-old male Labrador retriever [Control 1]; one 10-yr-old female Spitz [Control 2]; and one 10-yr-old female Basset des Alpes [Control 3]). Samples for the establishment of haired skin keratinocyte cultures were collected from HNPK 1 (2-yr-old female [HNPK 1 Skin]) and one control Labrador retriever (5.5-yr-old female [Control Skin]). For immunofluorescence microscopy of skin biopsies, we additionally used one 2-yr-old female beagle.
The genotype ± the SUV39H2:p.N324K variant was confirmed for all dogs by PCR on genomic DNA isolated from EDTA blood or from paraffin-embedded tissue samples followed by Sanger sequencing.
Antibodies
RNA-seq and pathway analysis
RNA-seq data were obtained from nasal planum biopsies of three HNPK-affected dogs and three control dogs (European Nucleotide Archive and GenBank accession no. PRJEB32103; Bannoehr et al., 2020). The nasal epidermis (including the rete ridges) was manually separated from the dermis using a stereo microscope. Total RNA was isolated with the RNeasy fibrous tissue kit (Qiagen), RNA integrity was confirmed on a 2100 Bioanalyzer (Agilent Technologies AG), and the concentration was determined by fluorimetry. 1 µg of total RNA was processed into stranded mRNA libraries using the commercial TruSeq stranded total RNA sample prep kit according to the manufacturer’s instructions (Illumina). Six fragment libraries (three control and three HNPK libraries) with 350-bp insert size were prepared, and two lanes of Illumina HiSeq2000 paired-end reads (2 × 100 bp) were collected, obtaining on average 63,280,070 tags per library. Prior to mapping reads to the dog reference genome, we filtered all sequences to remove adaptor sequences and low-quality bases (sequences with 50 or more bases were kept, and bases with quality value <15 were removed). Quality-filtered reads were mapped to the dog reference genome canFam3.1 using the splice alignment program TopHat2 (version 2.0.4) with default parameters (Kim et al., 2013). HTseq (version 0.5.3p9) was used for counting the number of sequencing reads mapping to genes after the read alignment step. The gene models from Ensembl build 70 were used for read counting. We also included the curated catalog of improved keratin gene annotations for gene counts (Balmer et al., 2017).
On average, 80% of the reads were mapped to the reference genome, of which 90% were uniquely mapped and 76.5% were concordantly aligned (see Table S1 for details). RSeQC (v2.3.3) was used for read distribution over the gene body to check 5′/3′ bias (Wang et al., 2012). Differentially expressed genes between cases and controls were identified with DeSeq2 from Bioconductor (https://bioconductor.org/packages/release/bioc/html/DESeq2.html; Love et al., 2014). Genes that passed the nominal statistical significance threshold of adjusted P value of 0.05 and a log2 fold change threshold of >|2| were considered to be differentially expressed genes (Fig. S2).
EnhancedVolcano v.1.4.0 (https://github.com/kevinblighe/EnhancedVolcano) was used to visualize the RNA-seq results (Fig. S2). Pathway analysis was done with AMIGO (Carbon et al., 2009) and IPA (Qiagen) tools. Briefly, all statistically significant canine genes (P value <0.05) with a log2 fold change threshold of >|2| (and corresponding mouse gene IDs) were used for pathway analysis, and genes from most deregulated pathways (canonical and noncanonical Wnt, cell cycle, Notch, p53, protein ubiquitination) were complemented by manual analysis of literature from online databases (National Center for Biotechnology Information [NCBI], AMIGO, and Wnt homepage [Roel Nusse] for Wnt signaling pathway; Table S1). To establish a protein connection network, protein names of differentially expressed genes were submitted to STRING software (Szklarczyk et al., 2015) on Canis familiaris specific–organism and with default parameters yielding the additional protein networks of deregulated adhesion molecules, intermediate filaments, and epidermal differentiation complex (Fig. 2 C and Table S1).
Primary keratinocyte isolation and subculture
The nasal planum or haired skin from the left thigh of anesthetized or euthanized dogs was washed with 70% EtOH, and at least one 4-mm punch biopsy was collected. Exceeding fat and muscle were removed, and the biopsy was immediately placed in culture medium (1/3 CnT-09 and 2/3 CnT-07; CELLnTEC Advanced Cell Systems AG) supplemented with 12 mg/ml Dispase 2 (CELLnTEC) and 10X antibiotic/antimycotics (CELLnTEC) overnight for 24 h at 4°C. Epidermis was removed from the dermis and placed on 500 µl TrypLE (GIBCO; 12605036) during 20 min at RT. The epidermis was rubbed, and cells were suspended in culture medium with 10X antibiotics/antimycotics (CELLnTEC) and centrifuged at 1,000 rpm for 5 min at RT. Primary keratinocytes were resuspended in 1 ml culture medium, stained with trypan blue, counted, and seeded at 30,000 cells/cm2. Primary keratinocytes from mouse skin were isolated previously as described (Caldelari and Müller, 2010), and pooled primary keratinocytes from the foreskin of three young human donors were purchased from CELLnTEC Advanced Cell Systems AG (human primary epidermal keratinocytes, pooled; HPEKp).
For cell expansion, primary canine keratinocytes were cultured at 8,000 cells/cm2 at 35°C under 5% CO2 in 1/3 CnT-09 and 2/3 CnT-07 (CELLnTEC) culture medium with 1X antibiotic/antimitotic (CELLnTEC). Cells were seeded and grown to 70–80% confluency during approximatively 4–4.5 d for nose keratinocytes and 3 d for haired skin keratinocytes. At 70–80% confluency, cells were detached with accutase (CELLnTEC) at 35°C during 15 min, resuspended in culture medium, and centrifuged 5 min at 1,200 rpm, suspended, and counted with trypan blue staining in order to quantify cell viability and population doubling. Cultured cells were frozen in 1:1 CnT–CRYO-50 (CELLnTEC) and culture medium (1/3 CnT-9 and 2/3 CnT-07; CELLnTEC) with 1X antibiotic/antimitotic (CELLnTEC). Frozen cells were thawed by adding 1 ml 1/3 CnT-09 and 2/3 CnT-07 (CELLnTEC) at RT with 1X antibiotic/antimitotic (CELLnTEC) and entered experiments after one passage.
For experiments, canine keratinocytes were seeded at 30,000 cells/cm2 and human and mouse keratinocytes at 35,000 cells/cm2 in 1/3 CnT-09 and 2/3 CnT-07 (CELLnTEC) with 1X antibiotic/antimitotic (CELLnTEC) and cultured for 2 d. At confluency, three micrographs were taken for each replicate, and attached cells were counted with FIJI software (version 1.52b). At similar density (<5% difference between each replicate), calcium concentration was elevated to a final concentration of 1.8 mM, and 1/3 of the medium was replaced every day until harvest (0, 3, 5, and 12 d after calcium switch). The vessel type was adapted depending on the experimental approach following sample type collection: in 96-well plates (Techno Plastic Products; 92096) for luciferase assay, in 24-well plates (TPP; 92048) for siRNA transfection and RNA collection (including Wnt modulations, see below), in six-well plates (TPP; 92006) for protein collection, and in 75-cm2 flasks (TPP; 90076) for ChIP. At each time point, micrographs of each vessel were taken before and after washing with PBS+ (supplemented with MgCl2 and CaCl2). To quantify the number of cells in supernatants, culture medium was collected and centrifuged 5 min at 1,200 rpm, and the pellet was resuspended in 1 ml culture medium. Cells were stained with trypan blue and counted.
siRNA transfection
Three canine SUV39H2-specific siRNA sequences and a negative control (scrambled siRNA) were designed by Microsynth (Switzerland) on the canine genome canFam3.1 (Table S4). The combination of siRNA 2 + 3 exhibited maximal SUV39H2 knockdown as tested in a dose-response experiment assessing SUV39H2 mRNA and protein levels by immunoblotting (Fig. S3 and see below). siRNA transfection was performed on one control (12-yr-old male Labrador retriever [Control 1]) and two HNPK (HNPK 1 and 2) dog nose keratinocyte isolates at passage 15. Keratinocytes were seeded in 24-well plates (TPP; 92048) at 30,000 cells/cm2 in triplicate, and the experiment was repeated three times. After 24 h, cells were washed with PBS+ and incubated at 35°C during 1 h until transfection in serum-free 1/3 BM2 and 2/3 BM1 (CELLnTEC). 50 nM of scrambled siRNA, used as negative control, or 25 nM each of the two SUV39H2 siRNAs was diluted in 1/3 BM2 and 2/3 BM1 (CELLnTEC) and incubated with 3 µl SilenceMag (OZ Bioscience; SM10500) per well during 20 min at RT. siRNA/medium complex was added to the cells and incubated on a magnetic plate for 20 min at 35°C. The siRNA/medium complex was replaced by normal culture medium after 4 h of incubation, and culture continued until harvesting. Knockdown efficiency was assessed by RT-qPCR and Western blotting (Fig. S3 and see below).
Wnt modulation
Wnt signal modulation was performed on one control (12-yr-old male Labrador retriever [Control 1]) and two HNPK (HNPK 1 and HNPK 2) primary dog nose keratinocyte isolates, as well as on one human and one mouse control primary keratinocyte isolate at passage 15. Two independent experiments per cell type were performed in triplicates (Fig. 6 and Fig. S5). Keratinocytes were seeded in 24-well plates (TPP; 92048) at 30,000 cells/cm2 for dog keratinocytes and at 35,000 cells/cm2 for human and mouse keratinocytes. After 24 h, cells were washed with PBS+, and medium was changed to 1/3 CnT-09 and 2/3 CnT-07 (CELLnTEC) supplemented with Wnt enhancer BIO (Meijer et al., 2003; 100-nM final concentration) for control dog, human, and mouse keratinocytes; Wnt suppressor IWR1 (Lu et al., 2009; 10-µM final concentration) for HNPK dog keratinocytes; and DMSO as negative control (with respective final concentration) for all cell types. Subsequent culture conditions were as described for experimental assays. 1/3 Wnt modulators/medium or DMSO/medium was replaced each day until harvesting.
RNA extraction, reverse transcription, and RT-qPCR of cultured keratinocytes
Total RNA was extracted with the RNeasy Mini kit (QIAGEN; 74104) according to the manufacturer’s protocol from dog nose and hairy skin keratinocytes at passage 15. Total RNA concentration and quality were assessed using Nanodrop and fragment analyzer (DNF-472), respectively. 1 µg total RNA was subjected to reverse transcription with the SuperScript IV Reverse transcription kit (Thermo Fisher Scientific; 18090010), according to the manufacturer’s protocol. Primers for RT-qPCR were designed using the NCBI primer software (primers used for RT-qPCR are summarized in Table S3). Specificity of the primers was tested in silico using the BLASTn software, and only primers with 100% identity and an E-value under 0.05 were selected and synthesized by Microsynth AG. Primer specificity was preexperimentally confirmed by serial dilution of primers from 1/10 to 1/100,000 to produce a dilution curve on a cDNA sample. Only primers with an efficiency (R2) over 0.95 (or 95%) were selected. Furthermore, during RT-qPCR, melting curves for each primer pair were generated to confirm specificity and experimental accuracy. For RT-qPCR, 5 ng of cDNA and 0.5 µM primers were used with 10 µl of PowerUP SYBR green mastermix (BioSystems; A25742) in a final volume of 20 µl. All measurements were performed in duplicates of each sample from two or three independent experiments on an ABI (Applied Biosystems) 7300 real-time PCR cycler, and obtained values were normalized to cyclophilin.
ChIP and ChIP-qPCR
At passage 15, nose keratinocytes from controls and HNPK dogs were seeded at 30,000 cells/cm2 in 4 × 75 cm2 flasks per dog (technical duplicates for each time point) and cultured in 1/3 CnT-09 and 2/3 CnT-07 (CELLnTEC) with 1X antibiotics/antimycotics (CELLnTEC) for 2 d until confluency. Two 75-cm2 flasks per cell isolate were used for each time point (Fig. 5). Proteins were cross-linked with disuccinimidyl glutarate (Fluka; 80424, 2 mM final) in 4 ml PBS+ (supplemented with MgCl2 and CaCl2) and incubated 45 min at RT. Cells were fixed with 1% PFA for 10 min at RT to cross-link DNA and proteins, and cross-linking was stopped with 265 µl of 2 M glycine (0.125 M final) for 10 min at RT. 3 ml TrypLE (GIBCO; 12605036) was added to each flask and incubated 10 min at 37°C to detach cells. Cells were scraped on ice into 3 ml of ice-cold PBS+, supplemented with protease inhibitors (Complete; Roche; 11697498001, 1× final), transferred into a 5-ml conic tube, and centrifuged at 4°C and 1,000 rpm for 10 min. The pellet was resuspended in 500 µl lysis buffer containing 1% SDS, 10 mM EDTA, and 50 mM Tris-HCl, pH 8.0) supplemented with protease inhibitors and incubated 10 min on ice. DNA was fragmented with a Covaris E220 Evolution sonicator with preprogrammed setting for 500-bp DNA fragment length. Sonicated samples were diluted 1:1 with dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl, pH 8.1) supplemented with protease inhibitors and centrifuged 1 min at 4°C and 14,000 rpm. For each sonicated sample, 200 µl was saved as input for qPCR, and DNA length was confirmed by Bioanalyzer (Agilent Technologies; Agilent 2100). For ChIP, 5 µg H3K9me3 antibody or ChIP-grade rabbit IgG (Abcam) as isotype control was added per sample and incubated overnight at 4°C on a rotating wheel. 30 µl transfer RNA preblocked DynaBeads G (Invitrogen; 10009D) was added, and the mix was incubated 3 h at 4°C. Beads were precipitated with DynaMag magnet (Invitrogen; 12321D). Beads were sequentially and slowly washed for 5 min on a rotator at RT with 1 ml of washing buffer I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl, pH 8.1), buffer II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, and 20 mM Tris-HCl, pH 8.1), buffer III (0.25 M LiCl, 1% NP-40, 1% DOC, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1), and twice with Tris EDTA buffer (10 mM Tris-HCl, pH 8.0, and 2 mM EDTA). Each washing buffer was supplemented with protease inhibitors. Complexes were eluted two times by incubating 20 min at 65°C with 50 µl elution buffer (1% SDS and 1 mM NaHCO3). Eluate and input samples were diluted 1:1 with TE buffer containing 20 µl 5 M NaCl and incubated overnight at 65°C for reverse cross-linking. Proteinase K (100 ng/µl final) and RNase A (100 ng/µl final) were added to each sample and incubated for 1 h at 55°C. 10% (vol/vol) 4 M LiCl and 100% (vol/vol) phenol/chloroform/isoamyl alcohol were added, and the sample was mixed by vortex for 1 min and centrifuged 10 min at RT at 14,000 rpm. The upper phase was precipitated with 750 µl of ice-cold 100% EtOH and 2 µl of transfer RNA (1 mg/ml) for 30 min at −80°C, and then centrifuged 25 min at 4°C and 14,000 rpm in an Eppendorf centrifuge. The pellet was washed with 500 µl of ice-cold 70% EtOH, followed by centrifugation during 15 min at 4°C and 14,000 rpm. EtOH was aspirated, and pellets were resuspended in 200 µl nuclease-free water.
Primers for ChIP-qPCR were designed using the NCBI primer software according to published H3K9me3 sites (Amabile et al., 2016; Wiencke et al., 2008; Table S5), and specificity was tested as described under RT-qPCR using purified DNA. Two primer pairs were designed: (i) positive primers matching the promoter sequence close to the transcriptional start site expected to carry H3K9me3 and (ii) negative primers ∼2.5kb upstream of the transcriptional start site. 1 µl ChIP DNA was mixed with 1 µM primers and 10 µl of PowerUP SYBR green master mix (BioSystems; A25742) for a final volume of 20 µl. All measurements were performed in duplicates from two independent experiments. All experimental values were normalized to input DNA multiplied by 100. Numbers presented are the mean of three control and two HNPK ChIP-qPCR values per time point, respectively (Fig. 5).
Immunofluorescence microscopy on paraffin-embedded tissue
Paraffin-embedded nose biopsies used for RNA-seq studies (Control I, II, and III; HNPKI, II, and III) and haired skin biopsies (Control skin; HNPK 1 skin) were processed together for immunofluorescence staining. Tissue was deparaffinized, followed by heat-mediated antigen retrieval in 0.01 M sodium-citrate buffer, pH 6.0, in a microwave (3 × 5 min at 800 W). Slides were blocked in a moisture chamber for 90 min at RT with blocking buffer containing 1% BSA and 10% normal goat serum, 0.1% Triton X-100, and 1% cold fish gelatin in PBS+. The slides were incubated with primary antibodies (Table S6) overnight at 4°C using a moisture chamber, followed by incubation with the suitable fluorescent secondary antibody (Alexa Fluor 488 or 594 conjugate) for 90 min at RT in the dark. Nuclei were counterstained with Hoechst 33258 (Sigma-Aldrich). Slides were mounted with fluorescence mounting medium (DAKO) and stored protected from light at 4°C until analysis. In all experiments, nonspecific rabbit or mouse IgG (Santa Cruz Biotechnology) was used instead of primary antibody as negative control. The complete set of slides for individual antibodies per biological replicate was processed on the same day to ensure identical laboratory conditions. Micrographs of slides were taken at RT with constant settings per antibody using a Nikon Eclipse Ti microscope either dry (objectives: 20×; NA: 0.5) or with oil immersion (40×, NA 1.3; 60× NA 1.4) and a Nikon DS-Ri2/Qi2 camera. Pictures were captured with NIS elements 4.40 software (Nikon), and the brightness and contrast were adjusted equally between control and test groups using FIJI software.
Immunofluorescence microscopy of cultured canine keratinocytes
Nasal control keratinocytes at passage 15 were seeded in three different experiments in duplicates in eight-well Lab-Teks (Thermo Fisher Scientific; 177445) at 30,000 cells/cm2, cultured during 24 h, and transfected with siRNA or scrambled RNA as described above, with the following adaptations: 1 µl of SilenceMag per chamber was used for a final volume of 200 µl. For immunofluorescence staining, these cells, including two HNPK isolates, were fixed with cold methanol, 10 min at −20°C for Ki67 detection or with 4% PFA in PBS+ for all other antibodies, and 20 min at RT (see Table S6 for antibodies). Cells were permeabilized 5 min at RT with 0.5% Triton X-100 in PBS+ and blocked 1 h at RT with 1% BSA (Sigma-Aldrich; A2153). They were incubated with primary antibody diluted in 1% BSA (Sigma-Aldrich; A2153) for 1 h at RT and with secondary antibody (Alexa Fluor 488 conjugate) in 1% BSA for another hour at RT in the dark. Antibodies were discarded, cells were washed three times with PBS+, and nuclei were counterstained with Hoechst 33258 (Sigma-Aldrich) during 1 min. Slides were mounted with fluorescence mounting medium (DAKO) and incubated at 4°C overnight before microscopic inspections. In all experiments, nonspecific rabbit or mouse IgG (Santa Cruz Biotechnology) was used instead of primary antibody as negative control. The complete set of slides was processed on the same day to ensure identical laboratory conditions. Micrographs were taken with a Nikon Eclipse Ti microscope with oil immersion (60× objective; NA 1.4) and Nikon DS-Qi2 camera at RT. Pictures were captured with NIS elements 4.40 software (Nikon) with constant settings at RT. Brightness and contrast of images were adjusted equally between control and test groups using FIJI software. SUV39H2, H3K9me3, β-catenin, and plakoglobin protein intensity was calculated with FIJI software (version 1.52b) on >10 random micrographs per antibody and normalized to nonspecific rabbit or mouse IgG (Santa Cruz Biotechnology). Ki67-positive cell ratios (Ki67-positive basal cells over total basal cells) were calculated manually.
Brightfield pictures of cultured keratinocytes were taken at RT with a Nikon Eclipse TS100 microscope with Nikon 1J5 camera and Nikon FT1 adapter using a 4× objective.
Colony-forming efficiency assay
5,000 nose or skin keratinocytes at passage 15 from three and two different dogs, respectively, were seeded in triplicates in 100-mm dishes (TPP; cat9300) in 1/3 CnT-09 and 2/3 CnT-07 supplemented with 1X antibiotics/antimycotics and incubated during 12 d without changing medium. Colonies were then washed with PBS+, fixed with 4% PFA during 2 h at RT, and stained with 1% Rhodamine B (Baxter) during 24 h.
Reporter gene assay
Reporter gene assays were performed with some adaptations (Williamson et al., 2006) using the Dual Luciferase Reporter Assay System (Promega; E1919) as indicated by the manufacturer. Nose keratinocytes at passage 6 (Control 2; HNPK 1) or 15 (Controls 1–3; HNPK 1 or 2) were seeded in two independent experiments in triplicates at 30,000 cells/cm2 in 96-well plates and cultured in 1/3 CnT-9 and 2/3 CnT-07 with 1X antibiotics/antimycotics (CELLnTEC). 24 h after seeding, cells were transiently transfected (Williamson et al., 2006). Briefly, keratinocytes were transfected with 1.5 µl polyethylenimine (1 mg/ml; linear; Polyscience Inc.; MW-25000), 0.2 µg TOP or FOP reporter plasmid (a kind gift of Prof. Hans Clevers, Hubrecht Institute, KNAW, Utrecht, Netherlands), and 7 ng plasmid encoding the Renilla Luciferase for normalization. Cells were incubated 2 h at 35°C with shaking every 15 min. Transfection medium was replaced by 1/3 CnT-09 and 2/3 CnT-07 with 1X antibiotics/antimycotics (CELLnTEC) and 10% FCS and cultured for 24 h. Measurements were performed with a TEKAN infinite 200, with default parameters for luminescence measurement.
Protein isolation and Western blotting
At passage 15 from the siRNA experiment (see above), nose keratinocytes were harvested with a scraper on ice into 150 µl Triton X-100 lysis buffer (1% Triton-100, 20 mM Tris, pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 10 mM NaF, 10 mM Na3VO4, and 1 mM PMSF) supplemented with protease inhibitor (Complete, Roche; 11697498001, 1× final) to extract the TritonX-100 soluble protein fraction. Lysates were centrifuged 20 min at 4°C and 12,000 rpm. Immunoblotting was done according to standard procedures, and primary antibodies are listed in Table S7. Secondary antibodies were conjugated with IRDyeTM 800CW or IRDye 700 (Rockland), and revealed proteins were imaged using the LI-COR Odyssey instrument and LI-COR Image Studio Lite software for signal quantification. Experimental data are from one experiment done with nasal keratinocytes from one control dog (Control 1) transfected with scrambled siRNA or siRNA 2 + 3 and two HNPK dogs for comparison.
Statistical analysis
All tests applied were nonparametric as defined by the Anderson-Darling P value test (P < 0.05). The Kruskal-Wallis test was used for experiments with three or more groups, while the Wilcoxon signed-rank test was used for experiments with two groups to compare differences between multiple dog genotypes on values of independent experiments. The Kruskal-Wallis test with Dunn post hoc test was performed with the online software Astatsa.com, and the Wilcoxon signed-rank test was performed with the online software scistatcal.html. Asterisks denote statistical significance (P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001).
Online supplemental material
Fig. S1 shows immunofluorescence microscopy micrographs of H3K9me3 and H3K27me3 marks as well as Axin2, p63, and Dsg3 expression in control and HNPK nose epidermis and haired skin biopsies. Fig. S2 shows a Volcano plot comparing RNA-seq data obtained from HNPK and control dog biopsies. Fig. S3 shows siRNA experimental design and efficiency of knockdown while confirming decreased Ki67 levels in nasal epidermis. Fig. S4 shows haired skin keratinocyte morphology. Fig. S5 shows consequences of Wnt activation on human and mouse skin keratinocytes. Table S1 provides a list of differentially regulated gene products obtained from RNA-seq analysis of epidermal nose biopsies from three control and three HNPK dogs (European Nucleotide Archive and GenBank accession no. PRJEB32103; Bannoehr et al., 2020) classified according to signaling pathways. Table S2 compiles P values of relative differences related to Fig. 6. Table S3 gives primary antibodies used for immunofluorescence microscopy on paraffin sections, cell culture, and ChIP-qPCR. Table S4 shows primary antibodies used for Western blot analyses. Table S5 shows siRNA sequences used for knockdown. Table S6 depicts primers used for RT-qPCR. Table S7 shows primers used for ChIP-qPCR.
Acknowledgments
We thank Prof. Luca Borradori, Insel, Bern University Hospital for the possibility to conduct this work, Anina Bauer for performing SUV39H2 variant identification on control and HNPK dogs, and the Microscopy Imaging Center, University of Bern, Switzerland, for technical support.
This work was funded by the Swiss National Science Foundation/Sinergia grant CRSII3_160738.
T. Leeb has commercialized the genetic testing of dogs for the SUV39H2 variant. E.J. Müller is a founder and the president of CELLnTEC, whose media have been used in this study. The remaining authors declare no competing financial interests.
Author contributions: P. Balmer was responsible for investigations and provided the original draft of the manuscript. P. Balmer, V. Jagannathan, and W.V.J. Hariton were dedicated to data curation and formal analysis, and P. Balmer, E.J. Müller, W.V.J. Hariton, A. Galichet, T. Leeb, and B.S. Sayar to methodology. E.J. Müller and P. Roosje were responsible for resources, conceptualization, project administration, validation, supervising, review, and editing; E.J. Müller and P. Roosje for visualization; and E.J. Müller, P. Roosje, and T. Leeb for funding acquisition. All coauthors revised the manuscript.

