


Mitochondrial function is integrated with cellular status through the regulation of opposing mitochondrial fusion and division events. Here we uncover a link between mitochondrial dynamics and lipid metabolism by examining the cellular role of mitochondrial carrier homologue 2 (MTCH2). MTCH2 is a modified outer mitochondrial membrane carrier protein implicated in intrinsic cell death and in the in vivo regulation of fatty acid metabolism. Our data indicate that MTCH2 is a selective effector of starvation-induced mitochondrial hyperfusion, a cytoprotective response to nutrient deprivation. We find that MTCH2 stimulates mitochondrial fusion in a manner dependent on the bioactive lipogenesis intermediate lysophosphatidic acid. We propose that MTCH2 monitors flux through the lipogenesis pathway and transmits this information to the mitochondrial fusion machinery to promote mitochondrial elongation, enhanced energy production, and cellular survival under homeostatic and starvation conditions. These findings will help resolve the roles of MTCH2 and mitochondria in tissue-specific lipid metabolism in animals.
Introduction
Mitochondria are essential cellular metabolic and signaling hubs (Chandel, 2014). They form highly dynamic networks by the concerted action of fusion and division events, which are linked to mitochondrial and cellular status (Labbé et al., 2014). Mitochondrial division is essential for mitochondrial transport and distribution within cells (Mishra and Chan, 2014), for cellular quality control via the elimination of damaged mitochondria (Twig et al., 2008; Youle and Narendra, 2011), and for organelle inheritance during cytokinesis (Mishra and Chan, 2014; Taguchi et al., 2007). Mitochondria fusion promotes mitochondrial mixing (Chen et al., 2003, 2005; Legros et al., 2002), increases cellular energy production via oxidative phosphorylation (Yao et al., 2019), and facilitates mitochondrial import of fatty acids (Rambold et al., 2015).
Mitochondrial fusion and division are mediated by evolutionarily conserved dynamin-like GTPases (DRPs). In mammalian cells, division depends on the activity of cytosolic dynamin-related protein 1 (DRP1), which is recruited to the mitochondrial outer membrane where it oligomerizes into helical structures that catalyze membrane fission (Kraus and Ryan, 2017). Fusion requires the successive actions of the integral mitochondrial outer membrane DRPs, mitofusin 1 and 2 (MFN1 and MFN2), and the inner membrane DRP, optic atrophy 1 (OPA1; Hoppins and Nunnari, 2006). These core DRP division and fusion machines are regulated by accessory proteins and posttranslational modifications, which modulate their recruitment, activation, and/or degradation in a context-specific manner, connecting cellular status to mitochondrial behavior (Wai and Langer, 2016).
Regulation of mitochondrial dynamics is evident during cellular stresses (Shutt and McBride, 2013). Mitochondrial fragmentation is linked to pathogen infection (Castanier et al., 2010; Stavru et al., 2011; Yang et al., 2020), excess nutrients (Molina et al., 2009; Wikstrom et al., 2007), and cell death (Arnoult, 2007). Conversely, stress-induced mitochondrial hyperfusion (SIMH) is linked to DNA damage (Tondera et al., 2009), ER stress (Lebeau et al., 2018), inhibition of translation (Tondera et al., 2009), and nutrient starvation (Gomes et al., 2011; Rambold et al., 2011). SIMH preserves cellular integrity by protecting mitochondria from autophagic degradation and by improving mitochondrial ATP production and requires MFN1, OPA1, and the mitochondrial inner membrane stomatin-like protein 2 (SLP2; Tondera et al., 2009). However, SIMH regulatory networks upstream of these mitochondrial components have not been fully elucidated.
Mitochondrial carrier homologue 2 (MTCH2) is an outer mitochondrial membrane protein that functions in intrinsic cell death and in the regulation of fatty acid metabolism in vivo (Bahat et al., 2018; Bar-Lev et al., 2016; Buzaglo-Azriel et al., 2017; Raemy et al., 2016; Rottiers et al., 2017; Ruggiero et al., 2017; Zaltsman et al., 2010). In humans, MTCH2 and the related proteins MTCH1 and SLC25A46 are members of a group of divergent mitochondrial carrier proteins localized to the mitochondrial outer membrane (Ruprecht and Kunji, 2020), with no known substrates or transporter functions (Robinson et al., 2012; Ruprecht and Kunji, 2020). A fungal-specific member of this group, Ugo1, plays an essential role in mitochondrial fusion, likely by regulating and coordinating membrane fusion DRPs (Coonrod et al., 2007; Hoppins et al., 2009; Palmieri et al., 2011; Sesaki and Jensen, 2001). MTCH2 was recently found to promote mitochondrial elongation in stem cells (Bahat et al., 2018), suggesting a role in the regulation of mitochondrial dynamics. However, how MTCH2 modulates mitochondrial morphology has not been resolved.
Here we report that MTCH2 is a selective mediator of starvation-induced mitochondrial hyperfusion that also functions under homeostatic conditions to maintain elongated mitochondria. Our data indicate that MTCH2 promotes mitochondrial elongation by stimulating mitochondrial fusion in a manner dependent on the bioactive lipid, lysophophatidic acid (LPA), generated during de novo lipogenesis. Our data suggest a model in which MTCH2 functions to connect flux through the de novo lipogenesis pathway to the mitochondrial fusion machinery to coordinate mitochondrial behavior with metabolic status.
Results and discussion
MTCH2 regulates mitochondrial fusion
MTCH2 is an outer mitochondrial membrane carrier protein related to the fungal mitochondrial fusion component, Ugo1 (Coonrod et al., 2007; Hoppins et al., 2009; Palmieri et al., 2011; Sesaki and Jensen, 2001). We examined the effects of MTCH2 overexpression and loss of function on mitochondrial morphology. Consistent with its known outer membrane localization (Grinberg et al., 2005), GFP-tagged MTCH2 overexpressed in COS7 cells localized to the outer rim of mitochondria labeled with matrix-targeted mito-mCherry (Figs. 1 A and S1, A and B). The level of MTCH2-GFP overexpression in cells, assessed by GFP fluorescence intensity, correlated with hyperfused mitochondria, typified by elongated branched networks (Figs. 1 A and S1 A). In contrast, mitochondria were extensively fragmented in two independent HCT116 MTCH2−/− lines generated using CRISPR (Fig. 1, B and C; and Fig. S1 C). Stable expression of MTCH2-GFP in MTCH2−/− cells restored WT mitochondrial morphology, indicating that mitochondrial fragmentation was due to loss of MTCH2 and that MTCH2-GFP is functional (Fig. 1, B–E; and Fig. S1 D). These observations are consistent with published work suggesting a role for MTCH2 in mitochondrial elongation (Bahat et al., 2018).
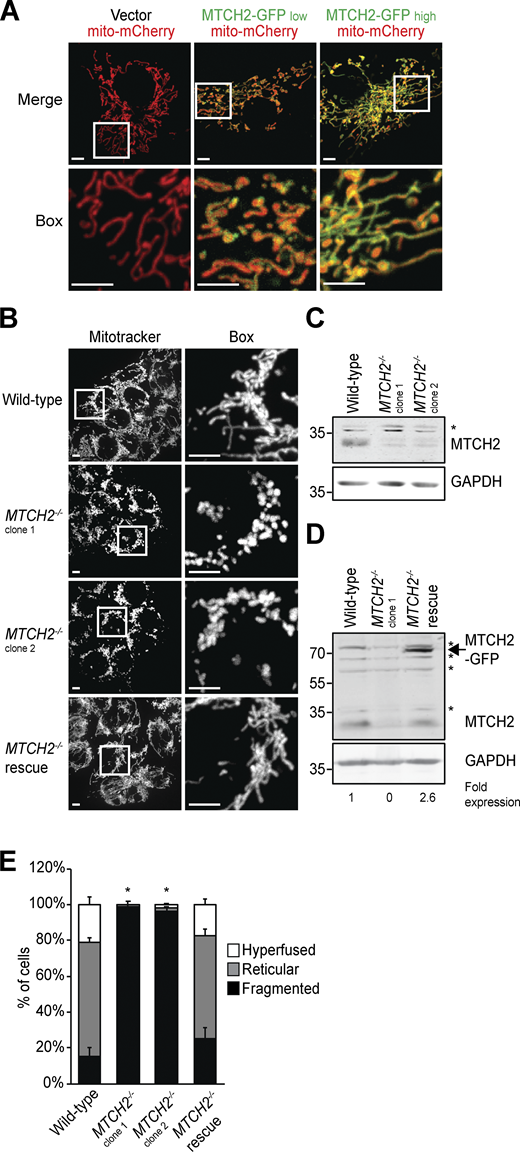
MTCH2 regulates mitochondrial fusion.(A) COS7 cells transiently transfected with matrix-targeted mito-mCherry and a low (25 ng) or high (250 ng) amount of MTCH2-GFP. Scale bars = 5 µm. (B) MitoTracker red staining of WT HCT116 cells, two independently generated MTCH2−/− CRISPR cell lines, and a MTCH2−/− line that stably expresses MTCH2-GFP (MTCH2−/− rescue). Scale bars = 5 µm. (C and D) Immunoblot for MTCH2 expression in lysates from indicated cell lines. Fold expression was determined by relative density analysis. *, Nonspecific bands. (E) Quantification of mitochondrial morphology in cell lines described in B. Error bars show mean + SEM of at least three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. Source data are available for this figure: SourceData F1.
MTCH2 regulates mitochondrial fusion.(A) COS7 cells transiently transfected with matrix-targeted mito-mCherry and a low (25 ng) or high (250 ng) amount of MTCH2-GFP. Scale bars = 5 µm. (B) MitoTracker red staining of WT HCT116 cells, two independently generated MTCH2−/− CRISPR cell lines, and a MTCH2−/− line that stably expresses MTCH2-GFP (MTCH2−/− rescue). Scale bars = 5 µm. (C and D) Immunoblot for MTCH2 expression in lysates from indicated cell lines. Fold expression was determined by relative density analysis. *, Nonspecific bands. (E) Quantification of mitochondrial morphology in cell lines described in B. Error bars show mean + SEM of at least three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. Source data are available for this figure: SourceData F1.
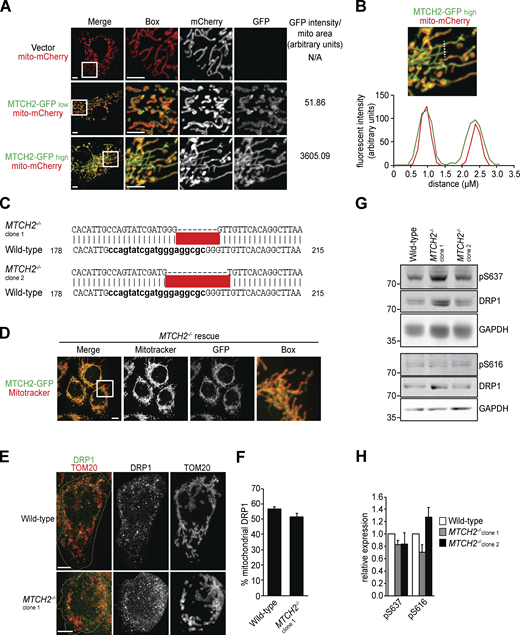
MTCH2 regulates mitochondrial fusion.(A) Individual channels and GFP pixel intensity of fluorescent images shown in Fig. 1 A. Scale bars = 5 µm. (B) Pixel intensity of a line scan drawn across mitochondrial tubules of cell expressing MTCH2-GFP and matrix-targeted mito-mCherry. (C) DNA sequence of CRISPR-generated mutations in HCT116 MTCH2−/− clonal cell lines. (D)MTCH2−/− rescue line stably expressing MTCH2-GFP stained with MitoTracker red. Scale bars = 5 µm. (E) Immunofluorescence for endogenous DRP1 and TOM20 in WT and MTCH2−/− cells. Yellow line demarcates the cell periphery. Scale bars = 5 µm. (F) Quantification of the percent of total cellular DRP1 signal localized to mitochondria in images described in A. Error bars show mean + SEM of 30 cells. Statistical significance was evaluated by two-sided unpaired t test. (G) Immunoblot for total, phospho-S637, or phospho-S616 DRP1 in lysates from WT and MTCH2−/− cells. (H) Quantification of relative density of phospho/total normalized to WT. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. Source data are available for this figure: SourceData FS1.
MTCH2 regulates mitochondrial fusion.(A) Individual channels and GFP pixel intensity of fluorescent images shown in Fig. 1 A. Scale bars = 5 µm. (B) Pixel intensity of a line scan drawn across mitochondrial tubules of cell expressing MTCH2-GFP and matrix-targeted mito-mCherry. (C) DNA sequence of CRISPR-generated mutations in HCT116 MTCH2−/− clonal cell lines. (D)MTCH2−/− rescue line stably expressing MTCH2-GFP stained with MitoTracker red. Scale bars = 5 µm. (E) Immunofluorescence for endogenous DRP1 and TOM20 in WT and MTCH2−/− cells. Yellow line demarcates the cell periphery. Scale bars = 5 µm. (F) Quantification of the percent of total cellular DRP1 signal localized to mitochondria in images described in A. Error bars show mean + SEM of 30 cells. Statistical significance was evaluated by two-sided unpaired t test. (G) Immunoblot for total, phospho-S637, or phospho-S616 DRP1 in lysates from WT and MTCH2−/− cells. (H) Quantification of relative density of phospho/total normalized to WT. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. Source data are available for this figure: SourceData FS1.
We considered whether MTCH2 promotes mitochondrial elongation by inhibiting division, promoting fusion, or a combination of both. Loss of MTCH2 did not alter the expression of core mitochondrial dynamics proteins as assessed by Western blot analysis, as only one of the two MTCH2−/− cell lines showed moderate changes in dynamics protein levels. This finding suggests that MTCH2-dependent regulation of mitochondrial dynamics is posttranslational (Fig. 2, A and B). To assess the contribution of mitochondrial division, we asked if mitochondrial fragmentation in MTCH2−/− cells could be rescued by expressing the dominant-negative DRP1K38A GTPase mutant (Zhu et al., 2004). Transient expression of mCherry-DRP1K38A in WT cells caused mitochondrial hyperfusion, consistent with inhibition of division (Fig. 2, C and D). In MTCH2−/− cells expressing mCherry-DRP1K38A, however, mitochondria remained primarily fragmented (Fig. 2, C and D). In addition, there was no significant difference in DRP1 localization to mitochondria between WT and MTCH2−/− cells, as assessed by indirect immunofluorescence using validated antibodies against DRP1 and the mitochondrial outer membrane protein TOM20 (Fig. S1, E and F). Furthermore, Western blot analysis using phospho-specific antibodies showed that levels of DRP1 phosphorylation at S637 or S616, which inhibit and promote DRP1 translocation to mitochondria, respectively, were similar in WT and MTCH2−/− cells (Liesa and Shirihai, 2013; Fig. S1, G and H). Together, our results indicate that mitochondrial fragmentation in MTCH2−/− cells is not due to an increase in mitochondrial fission and suggest that MTCH2 functions in the positive regulation of mitochondrial fusion.
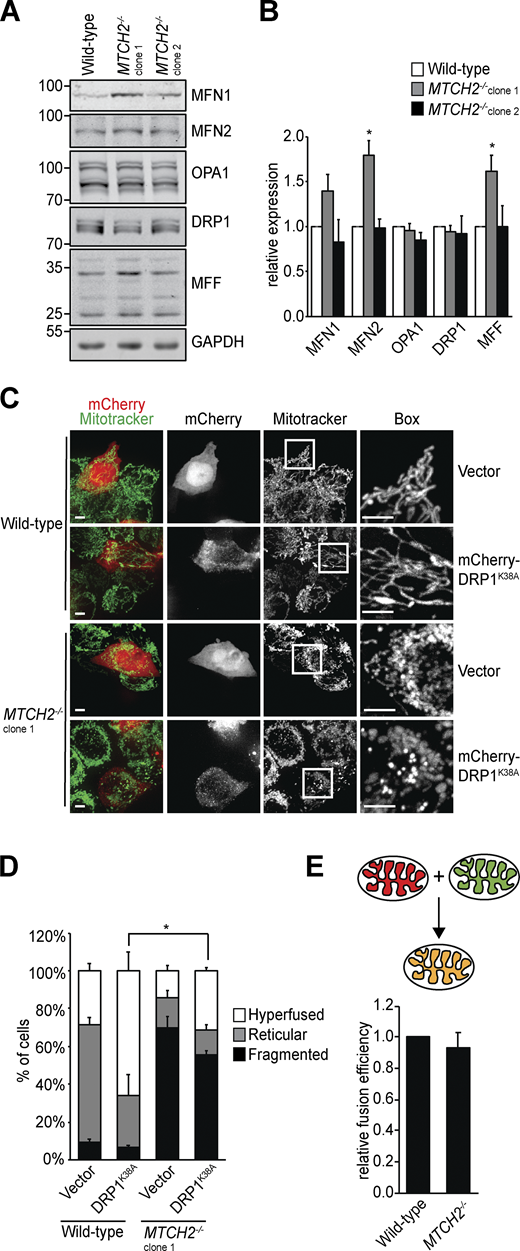
The mitochondrial dynamics machinery is functional in MTCH2−/− cells.(A) Immunoblot for expression of the indicated mitochondrial dynamics proteins in lysates of WT and MTCH2−/− cells. (B) Quantification of the relative density normalized to GAPDH. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C) WT and MTCH2−/− cells transfected with dominant-negative mCherry-DRP1K38A or control empty mCherry vector and stained with MitoTracker green. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) Top: Schematic representation of content mixing in vitro mitochondrial fusion assay. Bottom: In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells. Data are presented as fusion efficiency normalized to the WT reaction. Error bars show mean + SEM of seven independent experiments. Statistical significance was evaluated between fragmented values by two-sided nonparametric t test. Source data are available for this figure: SourceData F2.
The mitochondrial dynamics machinery is functional in MTCH2−/− cells.(A) Immunoblot for expression of the indicated mitochondrial dynamics proteins in lysates of WT and MTCH2−/− cells. (B) Quantification of the relative density normalized to GAPDH. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C) WT and MTCH2−/− cells transfected with dominant-negative mCherry-DRP1K38A or control empty mCherry vector and stained with MitoTracker green. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) Top: Schematic representation of content mixing in vitro mitochondrial fusion assay. Bottom: In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells. Data are presented as fusion efficiency normalized to the WT reaction. Error bars show mean + SEM of seven independent experiments. Statistical significance was evaluated between fragmented values by two-sided nonparametric t test. Source data are available for this figure: SourceData F2.
We assessed a role for MTCH2 in mitochondrial fusion using an established in vitro mitochondrial fusion assay, which uses matrix content-mixing to measure fusion efficiency (Hoppins et al., 2011; Meeusen et al., 2004; Fig. 2 E, schematic). Mitochondria were harvested from WT or MTCH2−/− cells engineered to stably express matrix-mCherry or matrix-GFP. Matrix-mCherry (red) and matrix-GFP (green) mitochondria of the same genotype were mixed in equal amounts in the presence of an exogenous energy regeneration system, and the proportion of fused and unfused mitochondria was quantified. The relative fusion efficiency of the MTCH2−/− mitochondria was similar to that of WT, indicating that, in contrast to Ugo1, MTCH2 is not essential for fusion (Fig. 2 E).
MTCH2 is a specific mediator of starvation stress–induced mitochondrial hyperfusion
Based on our data, we considered the possibility that MTCH2 plays a nonessential positive regulatory role in mitochondrial fusion. Inhibition of translation, ER stress, and nutrient starvation promote a protective SIMH response (Gomes et al., 2011; Lebeau et al., 2018; Rambold et al., 2011; Tondera et al., 2009). In MTCH2−/− cells treated with a translational inhibitor, cycloheximide, mitochondria were predominantly hyperfused (Fig. 3, A and B; and Fig. S2 A), similar to mitochondria in WT cells and consistent with our in vitro data indicating that the fusion machinery is functional in MTCH2−/− cells. However, mitochondria in MTCH2−/− cells remained fragmented under conditions of nutrient starvation (Fig. 3, A and B; and Fig. S2 A), in contrast to the robust mitochondrial hyperfusion observed in WT cells and MTCH2−/− cells rescued by MTCH2-GFP expression (Fig. S2 B). Consistent with previous work, glutamine-deficient culture medium was sufficient to induce starvation-dependent SIMH in WT cells (Fig. S2 C; Rambold et al., 2011). Together, our results indicate that MTCH2 is a selective component of the starvation-dependent SIMH pathway. Recent work identified the protein kinase R–like ER kinase (PERK) arm of the unfolded protein response as required for ER stress–induced SIMH caused by thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+ ATPase (Lebeau et al., 2018). However, PERK was not required for cycloheximide-induced SIMH. Thus, our data showing that MTCH2 functions specifically in starvation-induced hyperfusion indicate that the SIMH response encompasses several distinct pathways with different upstream and downstream mediators.
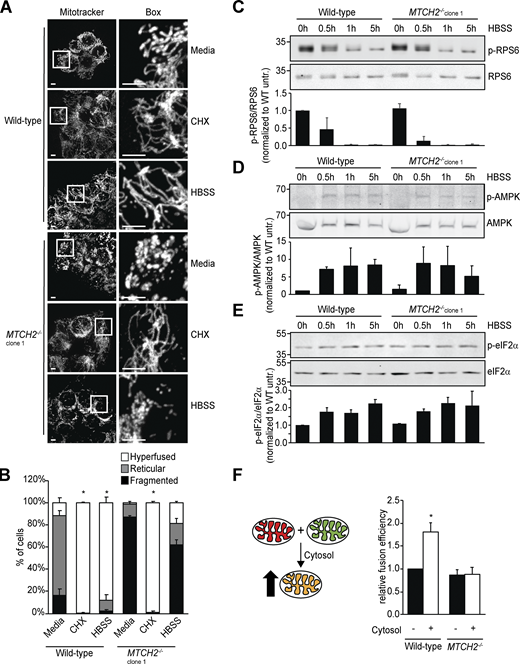
MTCH2 is essential for and specific to starvation stress–induced mitochondrial hyperfusion.(A) MitoTracker red staining of WT and MTCH2−/− cells treated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h. Scale bars = 5 µm. (B) Quantification of mitochondrial morphology in cells described in A. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C–E) Top: Western blot for phospho- and total levels of the indicated proteins in lysates from WT and MTCH2−/− cells starved with HBSS for the specified amount of time. Bottom: Quantification of relative density of phospho/total normalized to WT 0 h. Error bars show mean + SD of two independent experiments. (F) Left: Schematic representation of content mixing in vitro mitochondrial fusion assay. Addition of cytosol to the reaction enhances fusion efficiency in WT cells. Right: In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of cytosol from WT cells. Data are presented as fusion efficiency normalized to the WT no-cytosol reaction. Error bars show mean + SEM of five independent experiments. Statistical significance was evaluated by two-sided unpaired t test. *, P < 0.05. Source data are available for this figure: SourceData F3.
MTCH2 is essential for and specific to starvation stress–induced mitochondrial hyperfusion.(A) MitoTracker red staining of WT and MTCH2−/− cells treated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h. Scale bars = 5 µm. (B) Quantification of mitochondrial morphology in cells described in A. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C–E) Top: Western blot for phospho- and total levels of the indicated proteins in lysates from WT and MTCH2−/− cells starved with HBSS for the specified amount of time. Bottom: Quantification of relative density of phospho/total normalized to WT 0 h. Error bars show mean + SD of two independent experiments. (F) Left: Schematic representation of content mixing in vitro mitochondrial fusion assay. Addition of cytosol to the reaction enhances fusion efficiency in WT cells. Right: In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of cytosol from WT cells. Data are presented as fusion efficiency normalized to the WT no-cytosol reaction. Error bars show mean + SEM of five independent experiments. Statistical significance was evaluated by two-sided unpaired t test. *, P < 0.05. Source data are available for this figure: SourceData F3.
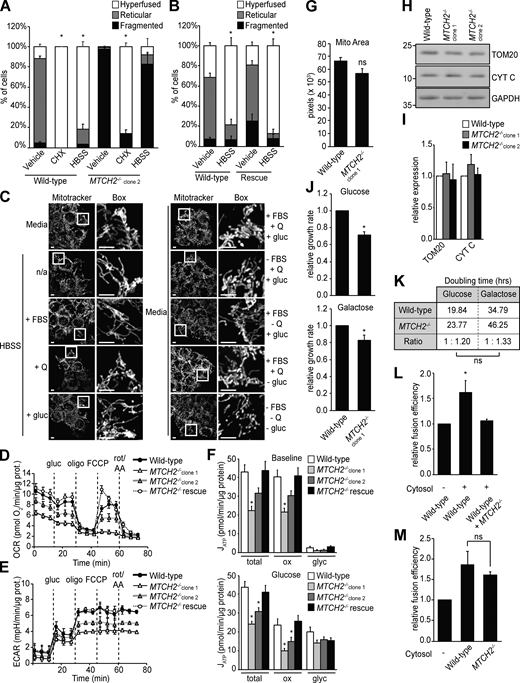
MTCH2 is essential for and specific to starvation stress–induced mitochondrial hyperfusion. (A) Quantification of mitochondrial morphology in WT and MTCH2−/− clone 2 cells treated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (B) Quantification of mitochondrial morphology in WT and MTCH2−/− rescue line starved in HBSS for 5 h. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. *, P < 0.05. (C) MitoTracker red staining of WT cells incubated for 5 h in HBSS (left) or medium (right) with no supplements (n/a) or supplemented with Serum (FBS), glutamine (Q), or glucose (gluc), as indicated. Scale bars = 5 µm. (D and E) Traces of oxygen consumption rates (OCR; D) and extracellular acidification rate (ECAR; E) of WT, MTCH2−/−, and rescue cell lines sequentially treated with glucose (gluc), oligomycin (oligo), FCCP, and rotenone with antimycin A (rot/AA) to assess respiratory states. Error bars show mean + SEM of 18 replicates. (F) Total, oxidative phosphorylation-derived (ox), and glycolysis-derived (glyc) ATP synthesis rates of WT, MTCH2−/−, and MTCH2−/− rescue cell lines as calculated from measurements shown in A and B at baseline, before addition of glucose (top) and after the addition of glucose (bottom). Error bars show mean + SEM of 18 replicates. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (G) Quantification of total mitochondrial pixel area in WT and MTCH2−/− cells stained with MitoTracker red. Error bars show mean + SEM of 15 cells. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. (H) Immunoblot for TOM20 and cytochrome C (CYT C) expression in lysates from indicated cell lines. (I) Quantification of the relative density normalized to GAPDH. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. (J and K) Relative growth rate (J) and doubling time (K) of WT and MTCH2−/− cells grown in media containing glucose or galactose. Error bars show mean + SEM of seven time points measured over two independent experiments. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. *, P < 0.05. (L) In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol from WT or MTCH2−/− cells. Data are presented as fusion efficiency normalized to the no-cytosol reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (M) In vitro fusion efficiency of mitochondria derived from WT cells, MTCH2−/− cells, or a mix of both with or without addition of cytosol from WT cells. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. Source data are available for this figure: SourceData FS2.
MTCH2 is essential for and specific to starvation stress–induced mitochondrial hyperfusion. (A) Quantification of mitochondrial morphology in WT and MTCH2−/− clone 2 cells treated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (B) Quantification of mitochondrial morphology in WT and MTCH2−/− rescue line starved in HBSS for 5 h. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. *, P < 0.05. (C) MitoTracker red staining of WT cells incubated for 5 h in HBSS (left) or medium (right) with no supplements (n/a) or supplemented with Serum (FBS), glutamine (Q), or glucose (gluc), as indicated. Scale bars = 5 µm. (D and E) Traces of oxygen consumption rates (OCR; D) and extracellular acidification rate (ECAR; E) of WT, MTCH2−/−, and rescue cell lines sequentially treated with glucose (gluc), oligomycin (oligo), FCCP, and rotenone with antimycin A (rot/AA) to assess respiratory states. Error bars show mean + SEM of 18 replicates. (F) Total, oxidative phosphorylation-derived (ox), and glycolysis-derived (glyc) ATP synthesis rates of WT, MTCH2−/−, and MTCH2−/− rescue cell lines as calculated from measurements shown in A and B at baseline, before addition of glucose (top) and after the addition of glucose (bottom). Error bars show mean + SEM of 18 replicates. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (G) Quantification of total mitochondrial pixel area in WT and MTCH2−/− cells stained with MitoTracker red. Error bars show mean + SEM of 15 cells. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. (H) Immunoblot for TOM20 and cytochrome C (CYT C) expression in lysates from indicated cell lines. (I) Quantification of the relative density normalized to GAPDH. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. (J and K) Relative growth rate (J) and doubling time (K) of WT and MTCH2−/− cells grown in media containing glucose or galactose. Error bars show mean + SEM of seven time points measured over two independent experiments. Statistical significance was evaluated between hyperfused values by two-tailed unpaired t test. *, P < 0.05. (L) In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol from WT or MTCH2−/− cells. Data are presented as fusion efficiency normalized to the no-cytosol reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (M) In vitro fusion efficiency of mitochondria derived from WT cells, MTCH2−/− cells, or a mix of both with or without addition of cytosol from WT cells. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA. Source data are available for this figure: SourceData FS2.
SIMH and mitochondrial elongation are associated with promoting ATP synthesis (Benador et al., 2018; Gomes et al., 2011; Liesa and Shirihai, 2013). We assessed the bioenergetic capacity of WT and MTCH2−/− cells using the Seahorse XF96 extracellular flux analyzer (Mookerjee et al., 2017). ATP synthesis rate was lower in MTCH2−/− cells as compared with WT or MTCH2−/− rescue cells in both starved and glucose-fed conditions (Fig. S2, D–F). Total respiratory capacity, as induced by uncoupling with carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), was also decreased in MTCH2−/− lines (Fig. S2 D). Mitochondrial content, as assessed in live cells (Fig. S2 G) and by Western blot analysis of TOM20 and cytochrome C in whole-cell lysates (Fig. S2, H and I), was similar between WT and MTCH2−/−, indicating that the lower bioenergetic capacity of the MTCH2−/− is likely not the result of loss of mitochondrial mass. Consistent with a decreased rate of ATP synthesis, MTCH2−/− cells grew at ∼70% the rate of WT in both glucose- and galactose-based media (Fig. S2, J and K). These results suggest a role for MTCH2-dependent mitochondrial elongation in maintaining cellular bioenergetics.
We assessed whether MTCH2 promotes nutrient-dependent SIMH through modulation of nutrient-sensing mTORC1, AMP kinase (AMPK), or integrated stress response pathways (Costa-Mattioli and Walter, 2020; Efeyan et al., 2015). We monitored the activity of these respective pathways via phosphorylation of their substrates, RPS6, AMPK, and eIF2a, by immunoblotting at fixed time points during starvation. We observed expected and comparable changes in both starved WT cells and MTCH2−/− cells (Fig. 3, C–E), suggesting that MTCH2 functions in SIMH downstream from these signaling pathways.
Addition of cytosol has previously been shown to increase fusion efficiency of WT mitochondria in vitro up to twofold, indicating the presence of regulatory pro-fusion factors (Hoppins et al., 2011; Schauss et al., 2010). We examined the effect of cytosol on mitochondrial fusion efficiency of WT cells and MTCH2−/− mitochondria in vitro. As expected, WT cytosol increased fusion efficiency of WT mitochondria approximately twofold (Fig. 3 F). In contrast, WT cytosol had no significant effect on the fusion efficiency of MTCH2−/− mitochondria (Fig. 3 F) or on the fusion efficiency of heteroallelic WT and MTCH2−/− mitochondria (Fig. S2 L), indicating that MTCH2 is required on both mitochondrial partners to promote cytosol-dependent fusion. Cytosol isolated from MTCH2−/− cells stimulated fusion efficiency of WT mitochondria in vitro to the same extent as WT cytosol (Fig. S2 M), indicating that MTCH2−/− cells are not deficient in cytosolic pro-fusion effectors. Taken together, our results suggest that MTCH2 functions to regulate mitochondrial fusion downstream of and in response to pro-fusion cytosolic factors.
Disruption of de novo fatty acid synthesis phenocopies loss of MTCH2
To gain insight into the mechanism of MTCH2-regulated mitochondrial fusion, we analyzed the MTCH2 interactome. Mass spectrometry analysis of cross-linked MTCH2-GFP immunoprecipitated from MTCH2−/− rescue cell extracts identified MFN2 and OPA1 as significant interactors (Fig. 4 A and Table S1). This suggests that, similar to fungal Ugo1, MTCH2 interacts with core outer and inner mitochondrial fusion proteins (Sesaki and Jensen, 2004; Wong et al., 2003) and may play a direct regulatory role in fusion.

Disruption of de novo fatty acid synthesis phenocopies loss of MTCH2. (A) Volcano plot of significant interactors identified by mass spectrometry analysis of MTCH2-GFP immunoprecipitated from cross-linked MTCH2−/− rescue cells. Significant interactors with a log2 fold-change (Log2FC) >1.0 are shown in black. Significant interactors involved in fatty acid/TAG de novo synthesis or catabolism are indicated in red and listed in B. (C) MitoTracker red staining of WT cells treated with vehicle or with the CPT1a inhibitor etomoxir (CPT1i; 100 nM) for 16 h. CPT1i-treated cells were then incubated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) MitoTracker red staining of WT cells treated with vehicle or with the ACC inhibitor TOFA (ACCi; 2 µg/ml) for 16 h. ACCi-treated cells were then incubated with CHX (10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (F) Quantification of mitochondrial morphology in cells described in E. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05.
Disruption of de novo fatty acid synthesis phenocopies loss of MTCH2. (A) Volcano plot of significant interactors identified by mass spectrometry analysis of MTCH2-GFP immunoprecipitated from cross-linked MTCH2−/− rescue cells. Significant interactors with a log2 fold-change (Log2FC) >1.0 are shown in black. Significant interactors involved in fatty acid/TAG de novo synthesis or catabolism are indicated in red and listed in B. (C) MitoTracker red staining of WT cells treated with vehicle or with the CPT1a inhibitor etomoxir (CPT1i; 100 nM) for 16 h. CPT1i-treated cells were then incubated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) MitoTracker red staining of WT cells treated with vehicle or with the ACC inhibitor TOFA (ACCi; 2 µg/ml) for 16 h. ACCi-treated cells were then incubated with CHX (10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (F) Quantification of mitochondrial morphology in cells described in E. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05.
The MTCH2 interactome was also enriched in enzymes involved in de novo fatty acid synthesis, storage and use (Fig. 4, A and B; and Fig. S3 A, in red), consistent with the proposed in vivo role of MTCH2 in fat storage (Bar-Lev et al., 2016; Buzaglo-Azriel et al., 2017; Rottiers et al., 2017). These include carnitine palmitoyltransferase (CPT1A) and the voltage-dependent anion channel (VDAC), which together are required for import of long chain fatty acids destined for β-oxidation (Eaton, 2002); 1-acylglycerol-3-phosphate O-acyltransferase 5 (AGPAT5), which functions in fatty acid esterification to convert LPA to phosphatidic acid (PA; Prasad et al., 2011); perilipin-3 (PLIN3), which modulates the storage of esterified fatty acids in triacylglycerol (TAG) in lipid droplets (Kimmel and Sztalryd, 2016); fatty acid synthase and acyl-CoA thioesterase (ACOT7), which catalyze the formation of palmitate, the first fatty acid synthesized in the de novo pathway (Currie et al., 2013; Kirkby et al., 2010); and hexokinase (HK1), aldolase A (ALDOA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which catalyze steps in glycolysis that generate critical substrates needed for lipogenesis (Patra and Hay, 2014).
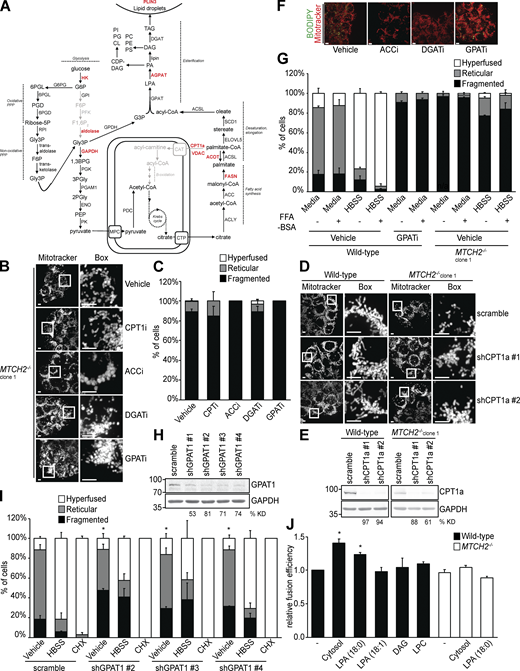
LPA promotes mitochondrial fusion via MTCH2.(A) Schematic of the de novo fatty acid and TAG synthesis pathway. Enzymes identified as interactors in the mass-spectrometry analysis of MTCH2-GFP immunoisolation are shown in red. (B) MitoTracker red staining of MTCH2−/− cells treated with vehicle, CPT1i (100nM), ACCi (2 µg/ml), DGATi (20 µM), or GPATi (75 µM) for 16 h. Scale bars = 5 µm. (C) Quantification of mitochondrial morphology in cells described in B. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. (D) MitoTracker red staining of WT and MTCH2−/− cells stably expressing scramble shRNA or one of two different shRNAs targeting CPT1a. Scale bars = 5 µm. (E) Immunoblot for CPT1a expression in lysates from indicated cell lines. The percentage knockdown efficiency (% KD) was determined by relative density analysis. (F) MitoTracker red and BODIPY 493/503 staining of WT cells treated with vehicle, ACCi (2 µg/ml), DGATi (20 µM), or GPATi (75 µM) for 16 h. Scale bars = 5 µm. (G) Quantification of mitochondrial morphology in WT and MTCH2−/− clone 1 cells treated with vehicle or GPATi (75 µM) for 16 h and starved in HBSS for 5 h with or without BSA (0.1% wt/vol) in the presence of vehicle or inhibitor. Error bars show mean + SEM of three independent experiments. (H) Immunoblot for GPAT1 expression in lysates from indicated cell lines. The % KD was determined by relative density analysis. (I) Quantification of mitochondrial morphology in WT cells stably expressing scramble shRNA or one of three different shRNAs targeting mitochondrial GPAT1. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. *, P < 0.05. (J) In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of LPA 18:0 (400 µM), LPA 18:1 (400 µM), DAG (400 µM), or LPC (400 µM). Data are presented as fusion efficiency normalized to the WT control reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. *, P < 0.05. Source data are available for this figure: SourceData FS3.
LPA promotes mitochondrial fusion via MTCH2.(A) Schematic of the de novo fatty acid and TAG synthesis pathway. Enzymes identified as interactors in the mass-spectrometry analysis of MTCH2-GFP immunoisolation are shown in red. (B) MitoTracker red staining of MTCH2−/− cells treated with vehicle, CPT1i (100nM), ACCi (2 µg/ml), DGATi (20 µM), or GPATi (75 µM) for 16 h. Scale bars = 5 µm. (C) Quantification of mitochondrial morphology in cells described in B. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. (D) MitoTracker red staining of WT and MTCH2−/− cells stably expressing scramble shRNA or one of two different shRNAs targeting CPT1a. Scale bars = 5 µm. (E) Immunoblot for CPT1a expression in lysates from indicated cell lines. The percentage knockdown efficiency (% KD) was determined by relative density analysis. (F) MitoTracker red and BODIPY 493/503 staining of WT cells treated with vehicle, ACCi (2 µg/ml), DGATi (20 µM), or GPATi (75 µM) for 16 h. Scale bars = 5 µm. (G) Quantification of mitochondrial morphology in WT and MTCH2−/− clone 1 cells treated with vehicle or GPATi (75 µM) for 16 h and starved in HBSS for 5 h with or without BSA (0.1% wt/vol) in the presence of vehicle or inhibitor. Error bars show mean + SEM of three independent experiments. (H) Immunoblot for GPAT1 expression in lysates from indicated cell lines. The % KD was determined by relative density analysis. (I) Quantification of mitochondrial morphology in WT cells stably expressing scramble shRNA or one of three different shRNAs targeting mitochondrial GPAT1. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. *, P < 0.05. (J) In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of LPA 18:0 (400 µM), LPA 18:1 (400 µM), DAG (400 µM), or LPC (400 µM). Data are presented as fusion efficiency normalized to the WT control reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between fragmented values by one-way ANOVA. *, P < 0.05. Source data are available for this figure: SourceData FS3.
Based on this MTCH2 interactome, we investigated whether fatty acid metabolism was linked to MTCH2-dependent mitochondrial fusion. Specifically, we tested whether inhibiting enzymes involved in fatty acid consumption, synthesis, and/or storage phenocopied MTCH2−/− mitochondrial morphology defects. WT cells were treated with a CPT1 inhibitor, etomoxir (CPT1i), to inhibit mitochondrial import of fatty acids for β-oxidation. CPT1i treatment caused mitochondrial fragmentation in WT cells; however, the phenotype was not as penetrant as the morphological fragmentation defect of MTCH2−/− cells (Fig. 4, C and D), and CPT1i had no effect on MTCH2−/− mitochondrial morphology (Fig. S3, B and C). In addition, CPT1i had no significant effect on either starvation- or cycloheximide-induced SIMH (Fig. 4, C and D). Given the reported off-target effects of CPT1i (Divakaruni et al., 2018; Yao et al., 2018), we also generated efficient stable knockdowns of CPT1A by two different shRNAs and observed no effect of CPT1A silencing on mitochondrial morphology in either WT or MTCH2−/− cells (Fig. S3, D and E). These data indicate that defects in mitochondrial fusion in MTCH2−/− cells are likely not the result of defective fatty acid β-oxidation.
Treatment of WT cells with 5-(tetradecyloxy)-2-furoic acid (TOFA [ACCi]), an inhibitor of acetyl-CoA carboxylase (ACC) that functions early in lipogenesis in the formation of malonyl-CoA, caused mitochondrial fragmentation similar to the mitochondrial morphology defect observed in MTCH2−/− cells, but did not prevent cycloheximide-induced SIMH (Fig. 4, E and F). However, upon starvation, mitochondria remained fragmented in ACCi-treated cells, similar to MTCH2−/− cells (Fig. 4, E and F). ACCi also had no effect on mitochondrial morphology in MTCH2−/− cells (Fig. S3, B and C). Thus, inhibition of de novo fatty acid synthesis phenocopied the mitochondrial morphological defects observed in MTCH2−/− cells.
Consistent with inhibition of TAG production, ACCi treatment also caused loss of cellular lipid droplets, as visualized by BODIPY 493/503 staining (Fig. S3 F). To test whether the generation of TAG was responsible for mitochondrial fragmentation observed in WT cells treated with ACCi, we inhibited diglyceride acyltransferases (DGATs) using T863 and PG-06424439, which target DGAT1 and DGAT2, respectively (together, DGATi; Nguyen et al., 2017). As expected, DGATi treatment caused loss of lipid droplets in cells (Fig. S3 F); however, mitochondrial morphology was unaffected in WT and MTCH2−/− cells (Fig. 5, A and B; and Fig. S3, B and C), suggesting that defects in fatty acid storage are not the cause of mitochondrial fragmentation in ACCi-treated cells. Instead, our findings raise the possibility that an intermediate in the de novo fatty acid synthesis pathway upstream of TAG generation regulates mitochondrial fusion in a MTCH2-dependent manner.

LPA promotes mitochondrial fusion via MTCH2. (A) MitoTracker red staining of WT cells treated with vehicle or the DGAT1 inhibitor T863 (20 µM) and DGAT2 inhibitor PF-06424439 (20 µM; together, DGATi) for 16 h. DGATi-treated cells were then incubated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (B) Quantification of mitochondrial morphology in cells described in A. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C) MitoTracker red staining of WT cells treated with vehicle or the GPAT inhibitor FSG67 (GPATi; 75 µM) for 16 h. GPATi-treated cells were incubated with CHX (10 µM) or starved in HBSS for 5 h with or without BSA (0.1% wt/vol) in the presence of inhibitor. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) Top: Schematic representation of content-mixing in vitro mitochondrial fusion assay with addition of LPA. Bottom: In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol (cyto) or the indicated amount of LPA 16:0. Data are presented as fusion efficiency normalized to the control reaction. Error bars show mean + SEM of three or four independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (F) In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of LPA 16:0 (400 µM) or PA (400 µM). Data are presented as fusion efficiency normalized to the WT control reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (G and H) In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol from cells treated with vehicle, ACCi (2 µg/ml; G), or GPATi (75 µM; H) for 16 h. Data are presented as fusion efficiency normalized to the no-cytosol reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (I) Schematic representation of the TAG synthesis pathway and the generation of LPA stimulating MTCH2-dependent mitochondrial fusion.
LPA promotes mitochondrial fusion via MTCH2. (A) MitoTracker red staining of WT cells treated with vehicle or the DGAT1 inhibitor T863 (20 µM) and DGAT2 inhibitor PF-06424439 (20 µM; together, DGATi) for 16 h. DGATi-treated cells were then incubated with cycloheximide (CHX; 10 µM) or starved in HBSS for 5 h in the presence of inhibitor. Scale bars = 5 µm. (B) Quantification of mitochondrial morphology in cells described in A. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (C) MitoTracker red staining of WT cells treated with vehicle or the GPAT inhibitor FSG67 (GPATi; 75 µM) for 16 h. GPATi-treated cells were incubated with CHX (10 µM) or starved in HBSS for 5 h with or without BSA (0.1% wt/vol) in the presence of inhibitor. Scale bars = 5 µm. (D) Quantification of mitochondrial morphology in cells described in C. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated between hyperfused values by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (E) Top: Schematic representation of content-mixing in vitro mitochondrial fusion assay with addition of LPA. Bottom: In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol (cyto) or the indicated amount of LPA 16:0. Data are presented as fusion efficiency normalized to the control reaction. Error bars show mean + SEM of three or four independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (F) In vitro fusion efficiency of mitochondria isolated from WT or MTCH2−/− cells with or without addition of LPA 16:0 (400 µM) or PA (400 µM). Data are presented as fusion efficiency normalized to the WT control reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (G and H) In vitro fusion efficiency of mitochondria isolated from WT cells with or without addition of cytosol from cells treated with vehicle, ACCi (2 µg/ml; G), or GPATi (75 µM; H) for 16 h. Data are presented as fusion efficiency normalized to the no-cytosol reaction. Error bars show mean + SEM of three independent experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s HSD test. *, P < 0.05. (I) Schematic representation of the TAG synthesis pathway and the generation of LPA stimulating MTCH2-dependent mitochondrial fusion.
In the de novo lipogenesis pathway, a family of four glycerol-3-phosphate acyltransferases (GPATs) catalyze production of LPA via fatty acid esterification of glycerol-3-phosphate (Fig. S3 A; Wendel et al., 2009; Yu et al., 2018). GPAT3 and GPAT4 are localized at the ER, whereas GPAT1 and GPAT2 reside on the mitochondrial outer membrane. GPAT1, the predominant mitochondrial GPAT, has been reported to promote mitochondrial elongation in Caenorhabditis elegans and mammalian cells (Ohba et al., 2013). Treatment of cells with the pan GPAT inhibitor FSG67 (GPATi; Kuhajda et al., 2011) caused mitochondrial fragmentation (Fig. 5, C and D), consistent with these previous findings. Additionally, treatment with GPATi had no effect on cycloheximide-induced mitochondrial hyperfusion (Fig. 5, C and D), similar to the phenotype of MTCH2−/− cells. However, under starvation conditions, cells treated with GPATi lost mitochondrial membrane potential, had abnormal cell morphology, and underwent cell death (Fig. 5, C and D), suggesting that GPAT is required for cell survival under nutrient deprivation conditions. During starvation, fatty acids are mobilized, and lipotoxicity is prevented via their esterification to yield TAG, which is stored in lipid droplets (Listenberger et al., 2003; Nguyen et al., 2017; Walther and Farese, 2012). Thus, we tested whether lipotoxicity was responsible for the cell death observed in starved cells treated with GPATi by buffering fatty acid accumulation with fatty acid–free BSA. Addition of fatty acid–free BSA to starvation medium rescued the viability of GPATi-treated cells (Fig. 5, C and D), consistent with a role for GPAT activity in preventing lipotoxicity during nutrient deprivation. In addition, similar to MTCH2−/− cells, mitochondria in WT cells treated with GPATi and fatty acid–free BSA were fragmented under starvation conditions (Fig. 5, C and D), indicating that GPAT activity is required for starvation-dependent SIMH. In contrast, fatty acid–free BSA treatment had no effect on mitochondrial morphology in WT cells under nutrient-replete conditions in the absence or presence of GPATi or in starved WT and MTCH2−/− cells (Fig. S3 G). To further test a role for GPAT in starvation-dependent SIMH, we created stable knockdown cell lines of GPAT1, which encodes a mitochondrial GPAT, using three different shRNAs (Fig. S3 H). Consistent with published work (Ohba et al., 2013), knockdown of GPAT1 caused mitochondrial fragmentation as compared with control cells (Fig. S3 I). GPAT1 deficiency had no effect on cycloheximide-induced SIMH, but starvation-dependent SIMH was attenuated as compared with WT cells (Fig. S3 I), similar to MTCH2−/− cells. Together, these data suggest that GPAT activity is required for MTCH2-dependent mitochondrial fusion.
LPA promotes mitochondrial fusion via MTCH2
Based on our data, we tested whether the product of GPAT activity, LPA, stimulates mitochondrial fusion in a MTCH2-dependent manner. LPA is a potent bioactive lipid previously implicated in mitochondrial fusion (Ohba et al., 2013). We directly assessed whether exogenous LPA (16:0) regulates fusion in vitro. Addition of LPA increased fusion efficiency of WT mitochondria in a dose-dependent manner to levels similar to those measured with the addition of cytosol (Fig. 5 E). Addition of LPA with a longer carbon tail (18:0) also increased in vitro fusion efficiency (Fig. S3 J). However, addition of unsaturated LPA (18:1), PA, diacylglycerol (DAG), or lysophosphatidylcholine (LPC) had no effect on WT mitochondrial fusion efficiency (Figs. 5 F and S3 J). These negative data are difficult to interpret given the potential impact of the differential solubility of these lipid species. LPA-stimulated fusion was dependent on MTCH2, as addition of LPA did not enhance the in vitro fusion efficiency of MTCH2−/− mitochondria (Figs. 5 F and S3 J). We also tested cytosol isolated from cells treated with ACCi or GPATi on in vitro fusion efficiency. In contrast to the cytosol from vehicle-treated cells, addition of either inhibitor-treated cytosol to WT mitochondria did not significantly increase fusion efficiency (Fig. 5, G and H), consistent with endogenous LPA being a pro-fusion factor in cytosolic extracts.
Taken together, our data demonstrate that the mitochondrial outer membrane protein MTCH2 regulates mitochondrial fusion in response to LPA, a lipogenesis pathway intermediate whose levels change in response to nutritional status (Zhang et al., 2013). During starvation, cells shift their mode of energy production from the utilization of exogenous glucose to endogenous substrates, such as fatty acids (Cabodevilla et al., 2013; Finn and Dice, 2006). Fatty acids are mobilized by lipolysis of TAG stored in lipid droplets and metabolized via β-oxidation in mitochondria (Olzmann and Carvalho, 2019; Rambold et al., 2015,Cabodevilla et al., 2013). Paradoxically, starvation also stimulates a burst of fatty acid esterification, resulting in the biogenesis of new lipid droplets (Cabodevilla et al., 2013; Nguyen et al., 2017; Rambold et al., 2015) that serve to store and buffer newly available fatty acids (Nguyen et al., 2017; Fig. 5, C and D). We propose that, during starvation, this increased flux through the fatty acid esterification pathway is sensed via LPA by MTCH2, which stimulates mitochondrial fusion to produce elongated mitochondrial networks that are protected from mitophagy, more productively generate energy via oxidative phosphorylation, and more efficiently accumulate fatty acids for either β-oxidation or esterification and TAG production (Benador et al., 2018; Rambold et al., 2015; Fig. 5 I).
Although MTCH2 is predicted to have the structural fold that characterizes the carrier family, it is divergent in regions that play a mechanistic role in transport, making it unlikely that it possesses canonical solute transport activity (Robinson et al., 2012). Thus, it is possible that MTCH2 evolved to function as a receptor to monitor and link cellular status with mitochondrial dynamics. Whether MTCH2 directly senses LPA via binding or through alterations in the membrane environment requires further studies. However, our interactome analysis suggests that MTCH2 is in close proximity to AGPAT5, an outer mitochondrial membrane protein that catalyzes the esterification of LPA to PA (Prasad et al., 2011). Mitochondrial AGPAT5 functions in parallel to ER-localized AGPATs (Takeuchi and Reue, 2009). In this context, it is significant that the upstream formation of LPA by GPATs also occurs in parallel on the mitochondrial outer membrane (GPAT1 and GPAT2) and the ER (GPAT3 and GPAT4; Wendel et al., 2009; Yu et al., 2018). The existence of a mitochondrial-localized fatty acid esterification pathway suggests that MTCH2 may sense LPA proximal to its synthesis and consumption to facilitate a direct and rapid response of mitochondrial fusion to lipogenesis status. Consistent with this idea and our findings, deletion of mitochondrial GPATs in C. elegans or knockdown in HeLa cells causes mitochondrial fragmentation, similar to loss of MTCH2 function, which is reversed in C. elegans by injection of LPA (Ohba et al., 2013). Our observations also raise the possibility that the related modified outer membrane carrier SLC25A46, which promotes mitochondrial shortening and has been implicated in lipid transfer from the ER to the mitochondria (Abrams et al., 2015; Janer et al., 2016; Steffen et al., 2017), functions in a similar but antagonistic manner to MTCH2 to regulate mitochondrial morphology in response to nutrient availability.
Mitochondria in MTCH2−/− cells are fragmented even under nutrient-replete growth conditions, suggesting that flux through de novo lipogenesis regulates mitochondrial fusion via MTCH2 under homeostatic conditions. This is similar to a number of cell stress pathways whose effectors have also been implicated in promoting mitochondrial function under homeostatic conditions, such as mitophagy (Exner et al., 2007; Yang et al., 2008; Yu et al., 2019), inflammation (Laforge et al., 2016), and apoptosis (Hoppins and Nunnari, 2012). For example, mitochondrial elongation in healthy cells requires the soluble form of the BCL-2 family member BAX, which, during apoptosis, inserts into the mitochondrial outer membrane to facilitate its permeabilization (Cleland et al., 2011; Hoppins et al., 2011; Karbowski et al., 2006). In this context, MTCH2 was first described as a proapoptotic factor that interacts with truncated BH3-interacting domain death agonist (tBID; Grinberg et al., 2005), a BCL-2 protein that is activated by caspase cleavage and triggers the recruitment and activation or proapoptotic BAX at the outer membrane. MTCH2 is thought to promote apoptosis by directly recruiting tBID to the mitochondrial outer membrane (Grinberg et al., 2005; Zaltsman et al., 2010). Our MTCH2 interactome analysis identifies uncleaved BID as a MTCH2 interactor in healthy cells. Thus, further studies are warranted to explore the roles and potential crosstalk of the MTCH2–BID interactions in LPA-dependent mitochondrial fusion in healthy cells and during cell death.
Our findings are consistent with the reported conserved role of MTCH2 in the regulation of lipid homeostasis and mitochondrial metabolism (Bahat et al., 2018; Bar-Lev et al., 2016; Buzaglo-Azriel et al., 2017; Maryanovich et al., 2015; Rottiers et al., 2017; Ruggiero et al., 2017). Overexpression or depletion of MTCH2 was reported to be sufficient to increase or deplete lipid accumulation, respectively, in animal models (Bar-Lev et al., 2016; Rottiers et al., 2017), and genome-wide association studies have linked a MTCH2 single polymorphism to high body mass index and obesity in humans (Willer et al., 2009). Recent work revealed the complex relationship of mitochondria and lipogenesis by identifying a distinct population of mitochondria in brown fat cells that is associated with lipid droplets. These elongated mitochondria have reduced β-oxidation activity and instead fuel the production of TAG for storage (Benador et al., 2018). Our findings demonstrating that MTCH2 functions to stimulate mitochondrial fusion in response to lipogenesis via LPA will help to resolve the mechanistic basis of the roles of both MTCH2 and mitochondria in the tissue-specific regulation of lipid metabolism in animals.
Materials and methods
Reagents
Reagents, suppliers, and catalog numbers are listed in Table S2.
Plasmids and cloning
All cloning was done using Gibson Assembly Master Mix (NEB). Plasmids pAC-GFP, mito-mCherry, and mito-GFP were generous gifts from G. Voeltz (University of Colorado Boulder, Boulder, CO; Friedman et al., 2011). MTCH2-GFP was generated by PCR amplifying the mammalian MTCH2 cDNA sequence (Mammalian Gene Collection, Horizon Discovery; clone ID 3461469) and ligating into the vector pAC-GFP-N1 linearized with Kpn1 and HindIII. mCherry-DRP1K38A was constructed by PCR amplifying the DRP1K38A sequence of the pcDNA3-HA-DRP1K38A plasmid previously described (Smirnova et al., 2001) and subcloning into pAC-mCherry-C1. pBABE-CMV-mito-mCherry and pBABE-CMV-mito-GFP were generated by subcloning the CMV-mito-GFP or CMV-mito-mCherry sequences described elsewhere (Friedman et al., 2011) into pBABE-puro (Addgene; 1764; Morgenstern and Land, 1990) linearized with EcoRI and SalI.
Cell culture and transfections
HCT116 parental cell line was obtained from ATCC. COS7 cells were a generous gift from G. Voeltz. COS7 cells were grown in DMEM (Gibco), and HCT116 cells were grown in McCoy’s modified medium (Gibco). All culture media were supplemented with 10% FBS and 100 mg/ml penicillin/streptomycin and maintained in 5% CO2 at 37°C. All cell lines were verified as free of mycoplasma contamination by a PCR-based method.
Growth rate was measured by plating 0.106 cells per well, in duplicate, in a six-well plate in DMEM or DMEM without glucose supplemented with 10 mM galactose, counting total cell numbers 72 h later. Cells were then repeatedly replated at 0.106 cells per well and counted every 72 h for a total of 12 d.
For imaging experiments, 0.08 × 106 cells were seeded in 35-mm poly-d-lysine glass-bottom dishes (MatTek Corp) and allowed to adhere for 24 h (COS7) or 48 h (HCT116). For SIMH experiments, cells were treated with 100 µM cycloheximide in complete medium or washed three times in HBSS (Gibco) to remove any residual medium, then kept in HBSS. Where indicated, cells were incubated with 100 nM etomoxir (Cayman Chemical), 2 µg/ml TOFA (Sigma-Aldrich), 20 µM T863 (Sigma-Aldrich), and 20 µM PF-06424439 (Sigma-Aldrich) or 2 µg/ml FSG-67 (Focus Biomolecules) for 16 h and included throughout further manipulations and imaging.
Transfections were performed for 4 h in serum- and antibiotic-free medium with 4 µl Lipofectamine 2000 (Thermo Fisher Scientific) per dish, following the manufacturer’s protocol. Cells were imaged the next day in complete medium.
Fluorescence microscopy
All fluorescent imaging was performed on live cells in the culture/treatment medium described in Materials and methods (DMEM, McCoy’s, or HBSS) unless fixed for immunofluorescence and imaged in PBS. For all fluorescence images, 0.25 µm step size z-series of cells were collected at room temperature using the spinning-disk module of a Marianas SDC Real Time 3D Confocal-TIRF microscope (Intelligent Imaging Innovations) fitted with a 100×, 1.46-NA objective and a Photometrics QuantEM electron-multiplying charge-coupled device camera. For immunofluorescence experiments, a Hamamatsu Orca Flash 4.0 scientific complementary metal–oxide–semiconductor camera was used. Images were captured with SlideBook (Intelligent Imaging Innovations), and settings were kept identical for all images within the same experiment.
Mitochondria morphology was visualized by incubating cells with 50 nM MitoTracker Red (Life Technologies) for 10 min at 37°C, then rinsing with medium and allowing the cells to recover for ∼1 h before imaging. Lipid droplets were labeled with 200 ng/ml BODIPY 493/503 (Life Technologies) immediately before imaging.
Image analysis
All image analysis was performed using Fiji (ImageJ) software (Schindelin et al., 2012). Linear adjustments to brightness/contrast were made to equalize image pixel intensity display range. For mitochondrial morphology analysis, 6–15 random fields were selected, and a total of 50–100 cells were scored per condition for each of ≥3 independent experiments. Linescan analysis was performed manually on z-projections using the ImageJ RGB Plot Profile function. Mitochondrial area was determined by thresholding and masking the MitoTracker signal of individual cells in z-projected images. Values were normalized to cell size.
The percentage of mitochondrial DRP1 was measured for 20 individual cells in z-projected images. The fluorescence intensity of DRP1-488 on mitochondria was determined by thresholding and masking the TOM20-561 signal and measuring the fluorescence intensity in the 488-nm channel across the entire mask. This was then divided by the total cellular 488 nm intensity and multiplied by 100.
CRISPR genome editing and rescue
CRISPR genome editing was performed using donor vector pX330 (Addgene; 42230), a human codon–optimized SpCas9 and chimeric gRNA expression plasmid, as previously described (Cong et al., 2013; Ran et al., 2013). Briefly, pairs of annealed oligonucleotides coding for three different guide RNA sequences were ligated into pX330 linearized with BsbI. HCT116 cells were cotransfected with one of these plasmids and a pAC-GFP vector, allowed to recover for 48 h, and harvested for single-cell FACS sorting into 96-well plates. Clonal isolates were expanded, and ∼50 clones were screened by PCR-based genotyping. Candidate clones were reconfirmed by PCR-based genotyping and Western blot. Guide 2 (5′-CCAGTATCGATGGGAGGCGCGGG-3′) generated two clones with a deletion in exon 3 of MTCH2 that resulted in loss of protein expression.
For rescue lines, MTCH2-GFP was cloned into the pULTRA lentiviral vector (Addgene; 24129; Lou et al., 2012) linearized with AgeI and NheI to remove the GFP-P2A-TAV2 sequence. The resulting plasmid was cotransfected with plasmids expressing GAG/POL, pREV, TAT, and VSVG into HEK293T cells plated in a six-well dish. Virus-containing medium was harvested 48 h later, filtered through a 0.45 µm polyethersulfone (PES) membrane, and added for 16 h to MTCH2−/− clone 1 cells along with 8 µg/ml sterile polybrene. GFP-expressing cells were sorted by FACS for the top ∼2% expressing cells.
shRNA
For a target gene, pairs of annealed oligonucleotides coding for different shRNA sequences were ligated into donor vector pLKO.1-Blasti (Addgene; 26655; Bryant et al., 2010) linearized with AgeI and EcoRI. The resulting plasmid was cotransfected with plasmids expressing GAG/POL, pREV, TAT, and VSVG into HEK293T cells plated in a six-well dish. Virus-containing medium was harvested 48 h later, filtered through a 0.45 µm PES membrane, and added for 16 h to target cells along with 8 µg/ml sterile polybrene. After recovering for 48 h, cells underwent selection in complete medium supplemented with 10 µg/ml blasticidin for 10 d.
shRNAs were as follows: CPT1a #1: forward, 5′-CCGGGCCATGAAGCTCTTAGACAAACTCGAGTTTGTCTAAGAGCTTCATGGCTTTTTG-3′; reverse, 5′-AATTCAAAAAGCCATGAAGCTCTTAGACAAACTCGAGTTTGTCTAAGAGCTTCATGGC; CPT1a #2: forward, 5′-CCGGCGATGTTACGACAGGTGGTTTCTCGAGAAACCACCTGTCGTAACATCGTTTTTG-3′; reverse, 5′-AATTCAAAAACGATGTTACGACAGGTGGTTTCTCGAGAAACCACCTGTCGTAACATCG; GPAT1 #1: forward, 5′-CCGGCAAGCGTTGTTACCAGCTATACTCGAGTATAGCTGGTAACAACGCTTGTTTTTG-3′; reverse, 5′-AATTCAAAAACAAGCGTTGTTACCAGCTATACTCGAGTATAGCTGGTAACAACGCTTG; GPAT1 #2: forward, 5′-CCGGCCCAATCTTCAGTACCTTGATCTCGAGATCAAGGTACTGAAGATTGGGTTTTTG-3′; reverse, 5′-AATTCAAAAACCCAATCTTCAGTACCTTGATCTCGAGATCAAGGTACTGAAGATTGGG; GPAT1 #3: forward, 5′-CCGGCTGCTGAATTAAACCCTGATCCTCGAGGATCAGGGTTTAATTCAGCAGTTTTTG-3′; reverse, 5′-AATTCAAAAACTGCTGAATTAAACCCTGATCCTCGAGGATCAGGGTTTAATTCAGCAG; and GPAT1 #4: forward, 5′-CCGGGCAAGCGTTGTTACCAGCTATCTCGAGATAGCTGGTAACAACGCTTGCTTTTTG-3′; reverse, 5′-AATTCAAAAAGCAAGCGTTGTTACCAGCTATCTCGAGATAGCTGGTAACAACGCTTGC-3′.
Western blots
Cells were washed in PBS, harvested by scraping, and lysed on ice for 30 min in B150 lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM KCl, 10% glycerol, 5 mM MgCl2, 0.1% NP-40, and protease inhibitors) before performing three freeze–thaw cycles followed by centrifugation (10,000 rpm, 10 min, 4°C) to isolate the supernatant fraction. For signaling time courses, cells were directly lysed in 2× Laemmli sample buffer (0.125 M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 0.004% bromophenol blue, and 10% 2-mercaptoethanol) and boiled for 10 min.
Lysates were analyzed by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with the following antibodies at the indicated dilutions: anti-MTCH2 (Proteintech; 1:1,000), anti-GAPDH (Sigma-Aldrich; 1:10,000), anti-TOM20 (Proteintech; 1:2,000), anti-cytochrome C (BD Biosciences; 1:1,000), anti-MFN1 (a generous gift from Richard Youle, National Institute of Neurological Disorders and Stroke, National Institute of Health, Bethesda, MD; 1:2,000), anti-MFN2 (Sigma-Aldrich; 1:2,000), anti-OPA1 (BD Biosciences; 1:2,000), anti-DRP1 (BD Biosciences; 1:2,000), anti-DRP1 pS637 (Cell Signaling; 1:1,000), anti-DRP1 pS616 (Cell Signaling; 1:1,000), anti-MFF (Proteintech; 1:2,000), anti-RPS6 (Cell Signaling; 1:1,000), anti-RPS6 pS235/236 (Cell Signaling; 1:1,000), anti-AMPK (Cell Signaling; 1:1,000), anti-AMPK pT172 (Cell Signaling; 1:1,000), anti-eIF2α (Cell Signaling; 1:1,000), anti-eIF2α pS51 (Cell Signaling; 1:1,000), anti-CPT1a (Proteintech; 1:1,000), and anti-GPAT1 (Abcam; 1:1,000). The appropriate secondary antibodies conjugated to DyLight 680 and DyLight 800 (Thermo Fisher Scientific; 1:10,000) were used and visualized with the Odyssey Infrared Imaging System (LI-COR Biosciences). Linear adjustments to images were made using Adobe Photoshop. Quantification of band intensity was performed with Image Studio Lite (LI-COR Biosciences).
Immunofluorescence
HCT116 cells were plated at 0.08 × 106 in 35-mm poly-d-lysine glass-bottom dishes (MatTek Corp.) and allowed to adhere for 48 h before fixing for 20 min at room temperature with 4% PFA in PBS prewarmed to 37°C. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min, blocked in PBS + 20% FBS for 1 h, and incubated overnight at 4°C with anti-DRP1 (BD Biosciences; 1:400) and anti-TOM20 (Proteintech; 1:500) in labeling buffer (PBS, 0.1% Tween 20, and 1% FBS). After five PBS washes, secondary antibody conjugated to Alexa Fluor 488 or Alexa Fluor 568 (1:1,000) were added for 1 h in labeling buffer. All steps were done at room temperature unless otherwise noted, and cells were protected from light throughout.
Immunoprecipitation and proteomics analysis
Cells were grown in complete medium, harvested with trypsin, and washed once with PBS. Cells were cross-linked with 0.5 mM dithiobis(succinimidylpropionate) in PBS for 30 min at room temperature, and the reaction was quenched with 100 mM Tris-HCl (pH 7.5) before pelleting the cells and lysing in RIPA buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholate, and protease inhibitors). Lysates were centrifuged (10,000 rpm, 10 min, 4°C), and the resulting supernatants were incubated with 25 µl GFP-Trap magnetic agarose beads (Chromotek) at 4°C for 3 h. Beads were isolated with μ columns (Miltenyi Biotec) and magnetic separator (Miltenyi Biotec) and washed three times with wash buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.01% SDS, 0.05% deoxycholate, and protease inhibitors) and twice with wash buffer containing no detergents or protease inhibitors. Sample were eluted using on-bead digestion. Briefly, beads were incubated for 30 min at room temperature in 25 µl trypsin solution (2 M urea, 50 mM Tris, pH 7.5, 1 mM DTT, and 5 µg/ml trypsin), followed by two 15-min applications of elution buffer (2 M urea, 50 mM Tris, pH 7.5, and 5 mM chloroacetamide). Eluates were collected, and digestion was allowed to proceed overnight at room temperature until reactions were stopped with 1 µl trifluoroacetic acid and stored at −80°C.
Liquid chromatography–tandem mass spectrometry analysis was performed at the University of California, Davis, Genome Center Proteomics Core. Samples were run on a Thermo Scientific Fusion Lumos mass spectrometer in data-independent acquisition (DIA) mode. Peptides were separated on an Easy-spray 100 µm × 25 cm C18 column using a Dionex Ultimate 3000 nUPLC with 0.1% formic acid (solvent A) and 100% acetonitrile, 0.1% formic acid (solvent B) and the following gradient conditions: 2–50% solvent B over 60 min, followed by a 50–99% solvent B in 6 min, held for 3 min, and finally 99–2% solvent B in 2 min. The total run time was 90 min. Mass spectra were acquired using a collision energy of 35 eV, resolution of 30,000, maximum inject time of 54 ms, and an automatic gain control target of 50,000. Each individual sample was run using a 2× gas-phase fractionation strategy where each sample was analyzed two times with the first gas-phase fraction being 360–760 m/z, and the second being 760–1,158 m/z. Both gas phase fractions used 4D nonoverlapping windows.
Raw files acquired in DIA mode on the Fusion/Lumos instrument were analyzed with DIA-NN v1.7.10 (Demichev et al., 2020) using the following settings (protease, Trypsin/P; missed cleavages, 2; variable modifications, 1; peptide length range, 7–30; precursor m/z range, 300–1,800; fragment ion m/z range, 200–1800; precursor false discovery rate, 1). The N-terminal M excision, C carbamidomethylation, and retention time profiling options were enabled and all other parameters were set to default. To generate a sample-specific spectral library, we initially used DIA-NN to create a large proteome-scale in silico deep learning–based library from the Uniprot human reference proteome (UP000005640, one protein per gene). This large spectral library was used to create a sample specific spectral library from all raw files from the analytical runs. This optimized spectral library was subsequently used to reprocess the analytical samples. DIA-NN output files were imported and analyzed in R. A false discovery rate cutoff of 1% was applied, and protein intensities were quantified (only proteins and protein groups with q value <1%) from proteotypic peptides only. Proteins identified in less than half the samples were discarded. Missing values were imputed using the MinDet deterministic minimal value approach from the MSnbase package, and relative changes between conditions were measured using a two-sample t test with Welch correction.
In vitro mitochondrial fusion assay
To generate mito-red and mito-green cells, pBABE-CMV-mito-mCherry or pBABE-CMV-mito-GFP was transfected into platinum A cells (Cellbiolab) plated in a six-well dish. Virus-containing medium was harvested 48 h later, filtered through a 0.45 µm PES membrane, and added for 16 h to HCT116 WT or MTCH2−/− clone 1 cells along with 8 µg/ml sterile polybrene. After recovering for 48 h, cells underwent selection in complete medium supplemented with 3 µg/ml puromycin for 10 d before being sorted by FACS for the top ∼10% expressing cells.
Fusion assays were performed as described in Hoppins et al. (2011) and detailed here. To prepare mitochondrial and cytosol-enriched fractions, cells were harvested by scraping, washed in mitochondrial isolation buffer (0.25 M sucrose, 10 mM Tris-MOPS, pH 7.4, and 1 mM EGTA), and resuspended in one pellet volume of cold mitochondrial isolation buffer. Cells were lysed with 15 strokes of a Potter-Elvehjem grinder (Kontes) on ice, and the homogenate was centrifuged (600 g, 10 min, 4°C) to remove nuclei and unbroken cells. The resulting supernatant was centrifuged (7,400 g, 10 min, 4°C) to pellet the crude mitochondrial fraction. The supernatant was the cytosol-enriched fraction. The mitochondrial pellet was collected in resuspension buffer (0.3 M mannitol, 10 mM Tris-MOPS, pH 7.4, and 1 mM EGTA), and protein concentration was determined by Bradford assay (Bio-Rad).
For fusion assays, an equivalent mass (15 µg) of red and green mitochondria were combined, concentrated by centrifugation (7,400 g, 10 min, 4°C), and incubated for 10 min on ice. The supernatant was removed, and the mitochondrial pellet was resuspended in 10 µl S2 fusion buffer (20 mM Pipes-KOH, pH 6.8, 150 mM KOAc, 5 mM Mg(OAc)2, 0.6 M sorbitol, 0.12 mg/ml creatine phosphokinase, 40 mM creatine phosphate, 0.75 mM ATP, and 0.75 mM GTP). Where indicated, S2 fusion buffer was supplemented with 25% cytosol-enriched fraction or with lipid species at the indicated concentration. Lipids stocks were resuspended in a chloroform solution at 10 mg/ml, aliquoted, and stored at −20°C. A fresh lipid aliquot was used for each experiment within 3 mo of preparing the stock solution. Lipids were dried under nitrogen gas, placed under vacuum for ≥1 h, and rehydrated in PBS using a sonicating bath for 10 min to make a 2-mg/ml solution that was then used to supplement S2 buffer.
For quantification, fusion reactions were fixed in 4% PFA in PBS for 30 min on ice. Fixed mitochondria were pelleted (7,400 g, 10 min, 4°C) and resuspended in 20 uL resuspension buffer, and 2 µl was distributed between a glass slide and coverslip. Mitochondria were imaged by confocal microscopy as described above, selecting ≥10 different random fields on the slide. Mitochondrial fusion was assessed by blinded analysis, by scoring the colocalization of red and green fluorophores in three dimensions for ≥350 total mitochondria per condition.
Bioenergetics
Cellular bioenergetics were measured using a Seahorse XF96 Extracellular Flux Analyzer. Cells were plated at 0.01 × 106 per well and allowed to adhere for 48 h. Culture medium was replaced with 37°C Krebs–Ringer phosphate Hepes medium (2 mM Hepes, 136 mM NaCl, 2 mM NaH2PO4, 3.7 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, and 0.1% fatty-acid-free BSA, pH 7.4) before the experiment. Oxygen consumption rate and extracellular acidification rate were measured through the sequential addition through ports of 10 mM glucose, 2 µg/ml oligomycin, 1 µM FCCP, and 1 µM rotenone with 1 µM antimycin A. After the assay, cells were washed in PBS and lysed with 5% NaDOC before measuring protein concentration by BCA assay (Thermo Fisher Scientific) according to the manufacturer’s instructions. Rates of oxygen consumption and extracellular acidification were expressed relative to the protein content of the corresponding well. ATP synthesis rates were calculated according to the method described in Mookerjee et al. (2017).
Statistical analysis
All data analyses were performed using Microsoft Excel or GraphPad Prism. Data are presented as mean ± SEM of replicates and were analyzed using unpaired two-tailed Student’s t test or one-way ANOVA followed by a post hoc Tukey honestly significantly different (HSD) test. Data distribution was assumed to be normal but was not formally tested. For mitochondrial morphology analysis, statistical analysis was done against the “fragmented” values in untreated cells, and against the “hyperfused” values in SIMH conditions. Statistical significance is indicated in all figures by *, P < 0.05.
Online supplemental material
Fig. S1 shows quantification of MTCH2-GFP fluorescence in COS7 cells, sequences of the CRISPR-generated deletions in the MTCH2 gene of HCT116 cells, MTCH2-GFP expression in the MTCH2−/− rescue line, and the cellular distribution and phosphorylation status of DRP1 in MTCH2−/− cells. Fig. S2 shows quantification of SIMH in MTCH2−/− clone 2 and rescue cells; mitochondrial morphology of glucose-, serum-, or glutamine-starved cells; an analysis of MTCH2−/− cellular bioenergetics, mitochondrial content, and cell growth rates; the in vitro fusion efficiency of heteroallelic reactions; and the effects of MTCH2−/− cytosol on mitochondrial in vitro fusion efficiency. Fig. S3 shows a schema of the de novo fatty acid synthesis and esterification pathways, mitochondrial morphology of MTCH2−/− cells treated with fatty acid synthesis inhibitors, mitochondrial morphology of stable CPT1A knockdown cells, lipid droplet content in inhibitor-treated cells, quantification of SIMH in BSA-treated WT and MTCH2−/− cells, quantification of SIMH in stable GPAT1 knockdown cells, and the effects of fatty acid esterification intermediates on mitochondrial fusion efficiency in vitro. Table S1 lists the raw intensity value of the hits identified by spectrometry analysis of immunoprecipitated MTCH2-GFP. Table S2 lists the reagents, suppliers, and catalog numbers used in this study.
Acknowledgments
We thank Suzanne Hoppins at the University of Washington and members of the Nunnari and Brand laboratories for helpful discussions and critical reading of the manuscript. We are grateful for the instrumental contributions of Oliver Fiehn and Megan Showalter of the West Coast Metabolomics Center. We thank Michael Paddy at the University of California Davis Molecular and Cellular Biology–College of Biological Sciences Imaging Facility. Thank you to Brett Phinney and Michelle Salemi of the UC Davis Proteomics Core Facility for their help and expertise in mass spectrometry.
The 3iMarianas spinning-disk confocal microscopes used in this study were purchased using a National Institutes of Health Shared Instrumentation grant (1S10RR024543-01). We are grateful for the assistance of Bridget McLaughlin and Jonathan Van Dyke from the UC Davis Flow Cytometry Shared Resource Center, which is supported by National Cancer Institute grant P30CA093373. Liquid chromatography–mass spectrometry was supported by a National Institutes of Health Shared Instrumentation grant (S10OD021801). K. Labbé is supported by a Canadian Institutes of Health Research postdoctoral fellowship. J. Nunnari is supported by National Institutes of Health grants R37GM097432 and R01GM126081.
J. Nunnari is on the scientific advisory board of Mitobridge, an Astellas Company. The authors declare no further competing financial interests.
Author contributions: K. Labbé conceptualized, performed, analyzed, and interpreted experiments. S. Mookerjee conceptualized, performed, analyzed, interpreted, and provided resources for bioenergetics experiments. E. Gibbs and C. Lerner performed bioenergetics experiments. M. Le Vasseur performed formal analysis of the proteomics data. J. Nunnari acquired funding, supervised, and conceptualized ideas and experiments. K. Labbé and J. Nunnari wrote the manuscript. S. Mookerjee and M. Le Vasseur provided essential feedback and reviewed the manuscript.

