


Fibronectin (FN) is an essential glycoprotein of the extracellular matrix; binds integrins, syndecans, collagens, and growth factors; and is assembled by cells into complex fibrillar networks. The RGD motif in FN facilitates cell binding and fibrillogenesis through binding to α5β1 and αv-class integrins. However, whether RGD is the sole binding site for αv-class integrins is unclear. Most notably, substituting aspartate with glutamate (RGE) was shown to eliminate integrin binding in vitro, while mouse genetics revealed that FNRGE preserves αv-class integrin binding and fibrillogenesis. To address this conflict, we employed single-cell force spectroscopy, engineered cells, and RGD motif–deficient mice (Fn1ΔRGD/ΔRGD) to search for additional αv-class integrin–binding sites. Our results demonstrate that α5β1 and αv-class integrins solely recognize the FN-RGD motif and that αv-class, but not α5β1, integrins retain FN-RGE binding. Furthermore, Fn1ΔRGD/ΔRGD tissues and cells assemble abnormal and dysfunctional FNΔRGD fibrils in a syndecan-dependent manner. Our data highlight the central role of FN-RGD and the functionality of FN-RGE for αv-class integrins.
Introduction
Fibronectin (FN) is a glycoprotein of the ECM that is widely expressed in vertebrates throughout all stages of life and assembled by cells into a fibrillar network (McDonald, 1988; George et al., 1993; Mao and Schwarzbauer, 2005). FN fibrils facilitate numerous cellular functions, including cell adhesion, spreading, migration, growth, and survival. The diverse functions of FN are indispensable for mammalian development (George et al., 1993; Georges-Labouesse et al., 1996) and affect diseases such as inflammation, fibrosis, and metastasis (Erdogan et al., 2017). The properties of FN are also used in regenerative medicine, where full-length FN, FN fragments, or FN peptidomimetica serve as substrates for bio-inspired materials (Salmerón-Sánchez et al., 2011; Marín-Pareja et al., 2015). FN is a modular protein composed of three types of folding units (type I, II, and III), which contain interaction motifs for cell adhesion molecules, including integrins and syndecans.
Syndecans consist of four members (syndecan 1–4), bind the heparin II region located within the 12th to 14th FN type III repeats (FNIII12-14; Woods et al., 2000; Mahalingam et al., 2007), colocalize with integrins in focal adhesions (FAs), and cooperate with integrins to promote F-actin assembly, mechanotransduction, and integrin recycling (Morgan et al., 2007; Chronopoulos et al., 2020). Syndecans binding to FN promote the formation of integrin-containing FAs (Woods and Couchman, 1994; Bloom et al., 1999; Roper et al., 2012; Bass et al., 2011). The major FN-binding cell surface receptors are integrins, which bind different sites in FN. A central cell binding site is the Arg-Gly-Asp (RGD) motif in FNIII10 recognized by several integrins, including α5β1, αIIbβ3 (exclusively expressed on platelets), and αv-class integrins (Leiss et al., 2008). Integrin binding to FN-RGD unfolds soluble FN, aligns, and allows cross-links of FN molecules into fibrils, which are further assembled into a complex network (Mao and Schwarzbauer, 2005; Singh et al., 2010). The FN matrices serve as mold for several FN-binding ECM proteins, including collagens, fibrillin, fibulin, latent TGF-β binding protein (LTBP), perlecan (Pln), and tenascin-C, which incorporate into this complex, 3D ECM mesh (Chung and Erickson, 1997; Twal et al., 2001; Dallas et al., 2005; Kadler et al., 2008; Sottile and Hocking, 2002). Genetic experiments in mice revealed that loss of either α5 or αv integrin expression still allowed the assembly of FN fibrils to proceed in vivo (Yang et al., 1993; Yang and Hynes, 1996; Bader et al., 1998), while adhesion dynamics, integrin signaling, actin morphology, and mechanotransduction were impaired (Wennerberg et al., 1996; Schiller et al., 2013; Strohmeyer et al., 2017). The simultaneous disruption of Itga5 (encoding integrin α5) and Itgav (encoding integrin αv) genes in mice markedly reduced FN expression as well as FN fibril formation and caused lethality at approximately embryonic day 7.5 (E7.5) to E8.0 (Yang et al., 1999). In an attempt to test whether similar defects arise in mice carrying a dysfunctional RGD motif in FN, the aspartate residue of the RGD motif was substituted with a glutamate to produce an RGE (FNRGE), which has been extensively used as an α5β1 and αv-class integrin binding–deficient motif in in vitro experiments (Greenspoon et al., 1993; Beumer et al., 2000; Shiokawa et al., 1999; Leyton et al., 2001; Lee et al., 2019; Niederlechner et al., 2012). Contrary to expectations, these mutant mice produced an apparently normal fibrillar FN network in tissues and developed the same defects as α5-null mice (Takahashi et al., 2007; Yang et al., 1993; Girós et al., 2011), which was regarded as evidence for the existence of a so far unrecognized αv-class integrin binding site in FN. Cellular and biochemical experiments assigned this new αv-class integrin binding to an isoDGR motif in FNI5 (Curnis et al., 2006; Takahashi et al., 2007), which was later shown, however, to lack the capacity to mediate cell adhesion and fibrillogenesis (Xu et al., 2010). Therefore, how αv-class integrin bind and enable cell adhesion to FNRGE both in vitro and in vivo is still an open question.
Here, we use atomic force microscopy–based single-cell force spectroscopy (SCFS), mouse genetics, cell biology, and protein engineering to show that the elimination of the entire RGD motif in FN (FNΔRGD) disrupts FN binding to both α5β1 and αv-class integrins in vitroand causes defects in vivo that are more severe than in Fn1RGE/RGE mice and closely resemble those reported for mice lacking the Fn1 gene. Our experiments also reveal that the aspartate substitution in the RGD motif of FN with a glutamate (RGD>RGE) abolishes α5β1 integrin binding but does not impair αv-class integrin–mediated cell adhesion and spreading. Our results underscore the extraordinary importance of the RGD motif in FN and, unexpectedly, unveil that the RGD>RGE mutation can direct cellular behavior by restricting integrin choice.
Results
The RGD motif in FN is the sole binding site for αv-class integrins
To investigate whether the RGD motif is essential for integrins to bind FN, we used full-length and recombinant fragments of FN with and without mutations in the RGD motif for SCFS experiments with engineered fibroblasts expressing distinct FN-binding integrins. The cell-derived FN (cFN) was purified from the culture medium of wild-type (producing cFNRGD) or Fn1ΔRGD/ΔRGD (producing RGD-deleted cFN [cFNΔRGD]) fibroblasts (see Materials and methods) and recombinantly expressed FN fragments containing the 7th to 10th type III repeats (FNIII7-10), which minimized integrin-unspecific interactions in single-cell adhesion experiments. The following FNIII7-10 fragments were expressed: an RGD-deleted fragment (FNIII7-10ΔRGD), a fragment carrying an aspartate-to-glutamate substitution (FNIII7-10RGE), and the unmodified fragment (FNIII7-10RGD) as control. To quantify ligand binding properties of αv-class and α5β1 integrins to cFNs and FNIII7-10 variants by SCFS, we attached single pan-integrin–deficient fibroblasts (pan-integrin KO [pKO]; Schiller et al., 2013) reconstituted with either αv-class (pKO-αv; expressing αvβ3 and αvβ5 integrins) or β1 (pKO-β1; expressing α5β1 integrins) or both classes of integrins (pKO-αv/β1) to concanavalin A–coated cantilevers and performed SCFS with single-molecule sensitivity. Thereto, we lowered cantilever-bound pKO, pKO-αv, pKO-β1, or pKO-αv/β1 fibroblasts onto different cFNs or FNIII7-10 fragments until a contact force of 200 pN was reached and instantly retracted fibroblasts from the substrate, which resulted in a total contact time of ∼50 ms (Materials and methods and Fig. S1, a–d). For each cantilever-bound fibroblast, this experiment was repeated multiple times, and the binding probability was calculated as ratio of force–distance (FD) curves showing single unbinding events and all attempts. As expected, pKO fibroblasts showed nonspecific binding probabilities to all tested substrates (Fig. 1, a and b). In line with earlier reports (Benito-Jardón et al., 2017; Bharadwaj et al., 2017), pKO-β1 fibroblasts showed a slightly but nonsignificantly higher binding probability to cFNRGD and FNIII7-10RGD compared with cFNΔRGD and FNIII7-10ΔRGD and nonspecific binding probabilities to FNIII7-10RGE (Fig. 1, a and b). On the contrary, pKO-αv fibroblasts showed comparably high binding probabilities to cFNRGD, FNIII7-10RGD, and FNIII7-10RGE and nonspecific binding probabilities to cFNΔRGD and FNIII7-10ΔRGD (Fig. 1, a and b). Expectedly, pKO-αv/β1 fibroblasts showed the same binding probabilities as pKO-αv fibroblasts to all FNIII7-10 fragment variants (Fig. 1, a and b). Importantly, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts showed the same nonspecific binding probabilities to cFNΔRGD and FNIII7-10ΔRGD as pKO fibroblasts, indicating that neither α5β1 nor αv-class integrins bind to RGD-deleted FN within the short contact time (Fig. 1, a and b). To test whether low integrin binding probabilities to FN account for lack of FN binding, we increased the contact time between cantilever-bound cells and FNIII7-10 fragments in binding probability experiments to ∼300 ms (Fig. 1 c). The binding probabilities of all pKO-fibroblast lines to FNIII7-10ΔRGD were not affected by the increased contact time. However, pKO-αv and pKO-αv/β1 fibroblasts displayed an increased binding probability to FNIII7-10RGD and FNIII7-10RGE, whereas the binding probability of pKO-β1 fibroblasts increased only for FNIII7-10RGD and remained at nonspecific levels for FNIII7-10RGE similar to FNIII7-10ΔRGD (Fig. 1 c). Additionally, experiments with specific integrin–FN interactions revealed in ∼25% of all binding probability experiments multiple unbinding events in one experiment, indicating that the ∼300-ms time span is sufficiently long for multiple integrins to bind their ligand (Fig. 1 d and Fig. S1 e). Altogether, our experiments show that the RGD motif is essential for αv-class and α5β1 integrins to bind FN and that binding of αv-class integrins to FNRGE is not compromised, while binding of α5β1 integrin to FNRGE is abrogated.
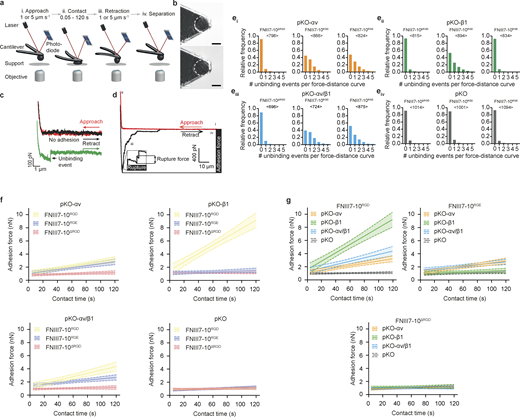
AFM-based SCFS setup to quantify adhesion properties of fibroblasts to different full-length cFNs and FNIII7-10 fragments.(a) SCFS setup. (i) Single fibroblasts are incubated for ∼7–15 min on a concanavalin A–coated cantilever to assure firm attachment. (ii) Cantilever-attached fibroblasts are approached to cFN- or FN fragment–coated supports. (iii and iv) After 0–120 s contact time, the cantilever-bound fibroblast is retracted vertically until the fibroblast is fully detached from substrate to either quantify binding probabilities or measure adhesion force between fibroblast and support. During adhesion experiments, cantilever deflections are recorded and displayed in FD curves. (b) A single round fibroblast attached to the cantilever tip used to measurement adhesion forces (top) until the fibroblast’s morphology changed (i.e., spreading; bottom). Scale bars, 10 µm. (c) FD curves representing binding of single integrins on fibroblasts to FN- or FNIII7-10–coated substrate. To record single integrin unbinding events, fibroblasts are approached to the substrate (red curve) until they contact the substrate at minimal contact force (200 pN) and contact time (∼50 or ∼300 ms) and are then retracted (black/green curve). The green FD curve shows a single adhesion (unbinding) event, while the black FD curve shows no adhesion event. (d) A representative FD curve after longer contact times (5–120 s) show different features; the retraction FD curve (black) records adhesion forces of the fibroblast, which represent the maximum downward deflection of the cantilever and thus the force required to separate the fibroblast from the substrate. During detachment of the fibroblast from the substrate, single receptor unbinding events are observed (ruptures). Ruptures are recorded when bonds formed between cytoskeleton-linked integrins and substrate fail. Tethers (longer force plateaus) are recorded when a membrane tether extrudes from the cell body with a single integrin or multiple integrins at its tip and occur when the integrin linkage to the actomyosin cytoskeleton is either too weak to resist the mechanical load applied or nonexistent. (e) The relative frequency distribution of unbinding events in single FD curves recorded for binding probability experiments with a contact time of ∼300 ms for pKO-αv (i), pKO-β1 (ii), pKO-αv/β1 (iii), or pKO (iv) fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, or FNIII7-10RGE (FD curves taken from experiments from Fig. 1 c). <n> denotes the number of FD curves analyzed per condition. (f and g) Adhesion-strengthening dynamics for pKO-αv, pKO-β1, pKO-αv/β1, or pKO fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, and FNIII7-10RGE are shown. For each condition, a linear fit was used to fit all adhesion forces for all contact times (data taken from Fig 1, h–k). Lines depict the fit and dashed lines the 95% confidence interval of the fit. Adhesion-strengthening dynamics are quantified by the slope of the fit and are composed for each fibroblasts line (f) or FNIII7-10 fragments (g). Statistical tests for slope differences are listed in Table S1.
AFM-based SCFS setup to quantify adhesion properties of fibroblasts to different full-length cFNs and FNIII7-10 fragments.(a) SCFS setup. (i) Single fibroblasts are incubated for ∼7–15 min on a concanavalin A–coated cantilever to assure firm attachment. (ii) Cantilever-attached fibroblasts are approached to cFN- or FN fragment–coated supports. (iii and iv) After 0–120 s contact time, the cantilever-bound fibroblast is retracted vertically until the fibroblast is fully detached from substrate to either quantify binding probabilities or measure adhesion force between fibroblast and support. During adhesion experiments, cantilever deflections are recorded and displayed in FD curves. (b) A single round fibroblast attached to the cantilever tip used to measurement adhesion forces (top) until the fibroblast’s morphology changed (i.e., spreading; bottom). Scale bars, 10 µm. (c) FD curves representing binding of single integrins on fibroblasts to FN- or FNIII7-10–coated substrate. To record single integrin unbinding events, fibroblasts are approached to the substrate (red curve) until they contact the substrate at minimal contact force (200 pN) and contact time (∼50 or ∼300 ms) and are then retracted (black/green curve). The green FD curve shows a single adhesion (unbinding) event, while the black FD curve shows no adhesion event. (d) A representative FD curve after longer contact times (5–120 s) show different features; the retraction FD curve (black) records adhesion forces of the fibroblast, which represent the maximum downward deflection of the cantilever and thus the force required to separate the fibroblast from the substrate. During detachment of the fibroblast from the substrate, single receptor unbinding events are observed (ruptures). Ruptures are recorded when bonds formed between cytoskeleton-linked integrins and substrate fail. Tethers (longer force plateaus) are recorded when a membrane tether extrudes from the cell body with a single integrin or multiple integrins at its tip and occur when the integrin linkage to the actomyosin cytoskeleton is either too weak to resist the mechanical load applied or nonexistent. (e) The relative frequency distribution of unbinding events in single FD curves recorded for binding probability experiments with a contact time of ∼300 ms for pKO-αv (i), pKO-β1 (ii), pKO-αv/β1 (iii), or pKO (iv) fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, or FNIII7-10RGE (FD curves taken from experiments from Fig. 1 c). <n> denotes the number of FD curves analyzed per condition. (f and g) Adhesion-strengthening dynamics for pKO-αv, pKO-β1, pKO-αv/β1, or pKO fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, and FNIII7-10RGE are shown. For each condition, a linear fit was used to fit all adhesion forces for all contact times (data taken from Fig 1, h–k). Lines depict the fit and dashed lines the 95% confidence interval of the fit. Adhesion-strengthening dynamics are quantified by the slope of the fit and are composed for each fibroblasts line (f) or FNIII7-10 fragments (g). Statistical tests for slope differences are listed in Table S1.

The FN-RGD motif is the exclusive binding site for α5β1 and αv-class integrins. SCFS measurements of pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts adhering to different FNIII7-10 fragments and full-length cFN. (a) Binding probabilities determined after pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblast contact with FNIII7-10RGD, FNIII7-10RGE, or FNIII7-10ΔRGD fragments for ∼50 ms with a contact force of 200 pN. Adhesion probability was calculated as the ratio of FD curves showing unbinding events and total number of recorded FD curves. n in bars give the numbers of individual fibroblasts tested and <n> the total number of recorded FD curves in each condition. Bars depict mean ± SEM of binding probabilities. P values are given on top of lines that indicate compared conditions. (b) Binding probabilities of pKO, pKO-αv, or pKO-β1 fibroblasts to cFNRGD or cFNΔRGD as described for panel a. (c) Binding probability of pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts determined after fibroblasts contacted FNIII7-10RGD, FNIII7-10RGE, or FNIII7-10ΔRGD fragments for ∼300 ms with a contact force of 200 pN. Data representation as described for panel a. (d) Average number of unbinding events per FD curve acquired for c. Only FD curves showing unbinding events were considered. Data representation as described for panel a. (e–g) Adhesion forces of single pKO-αv (e), pKO-β1 (f), or pKO (g) fibroblasts to cFNΔRGD or cFNRGD after a given contact time are shown as dots and their median as a red bar. P values are given for statistical significance between adhesion forces to cFNRGD and cFNΔRGD at the indicated contact times. n (cells) gives the number of fibroblasts tested for each condition. (h–k) Adhesion forces of single pKO-αv (h), pKO-β1 (i), pKO-αv/β1 (j), or pKO (k) fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, or FNIII7-10RGE after given contact times shown as dots and their median as a red bar. Top P values are given for statistical significance between adhesion forces to FNIII7-10RGD and FNIII7-10ΔRGD, and bottom P values are given for comparison of FNIII7-10RGD and FNIII7-10RGE at the indicated contact times. n (cells) gives the number of fibroblasts tested for each condition. (l and m) Rupture forces of single ruptures extracted from FD curves after 5-s (l) or 120-s (m) contact times (FD curves are taken from data presented in h, i, and k). Bars are medians and error bars with 95% confidential intervals. All statistical significances were calculated using two-tailed Mann-Whitney test.
The FN-RGD motif is the exclusive binding site for α5β1 and αv-class integrins. SCFS measurements of pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts adhering to different FNIII7-10 fragments and full-length cFN. (a) Binding probabilities determined after pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblast contact with FNIII7-10RGD, FNIII7-10RGE, or FNIII7-10ΔRGD fragments for ∼50 ms with a contact force of 200 pN. Adhesion probability was calculated as the ratio of FD curves showing unbinding events and total number of recorded FD curves. n in bars give the numbers of individual fibroblasts tested and <n> the total number of recorded FD curves in each condition. Bars depict mean ± SEM of binding probabilities. P values are given on top of lines that indicate compared conditions. (b) Binding probabilities of pKO, pKO-αv, or pKO-β1 fibroblasts to cFNRGD or cFNΔRGD as described for panel a. (c) Binding probability of pKO, pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts determined after fibroblasts contacted FNIII7-10RGD, FNIII7-10RGE, or FNIII7-10ΔRGD fragments for ∼300 ms with a contact force of 200 pN. Data representation as described for panel a. (d) Average number of unbinding events per FD curve acquired for c. Only FD curves showing unbinding events were considered. Data representation as described for panel a. (e–g) Adhesion forces of single pKO-αv (e), pKO-β1 (f), or pKO (g) fibroblasts to cFNΔRGD or cFNRGD after a given contact time are shown as dots and their median as a red bar. P values are given for statistical significance between adhesion forces to cFNRGD and cFNΔRGD at the indicated contact times. n (cells) gives the number of fibroblasts tested for each condition. (h–k) Adhesion forces of single pKO-αv (h), pKO-β1 (i), pKO-αv/β1 (j), or pKO (k) fibroblasts to FNIII7-10ΔRGD, FNIII7-10RGD, or FNIII7-10RGE after given contact times shown as dots and their median as a red bar. Top P values are given for statistical significance between adhesion forces to FNIII7-10RGD and FNIII7-10ΔRGD, and bottom P values are given for comparison of FNIII7-10RGD and FNIII7-10RGE at the indicated contact times. n (cells) gives the number of fibroblasts tested for each condition. (l and m) Rupture forces of single ruptures extracted from FD curves after 5-s (l) or 120-s (m) contact times (FD curves are taken from data presented in h, i, and k). Bars are medians and error bars with 95% confidential intervals. All statistical significances were calculated using two-tailed Mann-Whitney test.
Adhesion strengthening of αv-class integrins remains unaffected by RGE substitution
To test how the loss of the RGD motif or the D>E substitution influences the adhesion strengthening of αv-class and α5β1 integrins to FN, we quantified adhesion forces of the pKO fibroblast lines to cFNs and FNIII7-10 variants by increasing contact times stepwise from 5 s to 120 s (Fig. 1, e–k). In line with previous reports (Spoerri et al., 2020; Strohmeyer et al., 2017; Bharadwaj et al., 2017), pKO-αv, pKO-β1, or pKO-αv/β1 fibroblast lines readily initiated and strengthened adhesion to cFNRGD and FNIII7-10RGD with characteristic integrin-specific dynamics (Fig. 1, e–k; Fig. S1, f and g; and Table S1). Thereby, α5β1 integrins, when expressed alone, strengthened adhesion faster than αv-class integrins alone or when coexpressed with αv-class integrins, which, in the latter case, is due to the out-competition of α5β1 integrins by αv-class integrins (Bharadwaj et al., 2017). Importantly, adhesion forces of pKO-αv, pKO-β1, or pKO-αv/β1 fibroblasts to cFNΔRGD and FNIII7-10ΔRGD were reduced to nonspecific levels and did not strengthen with increasing contact times, confirming that the RGD motif is essential for integrin-mediated adhesion initiation and strengthening to FN (Fig. 1, e–k; Fig. S1, f and g; and Table S1). Further, pKO-αv and pKO-αv/β1 fibroblasts showed integrin-dependent adhesion forces on FNIII7-10RGE that were similar to those on FNIII7-10RGD, while pKO-β1 fibroblasts exhibited nonspecific adhesion at all contact times (Fig. 1, h–k). Interestingly, the adhesion-strengthening dynamics of pKO-αv and pKO-αv/β1 fibroblasts were not affected on FNIII7-10RGE (Fig. S1 g).
From FD curves, we extracted rupture forces of single unbinding events after 5 s (Fig. 1 l and Fig. S1 d) or 120 s (Fig. 1 m) contact times. We found αv-class integrins showed similar rupture forces during detachment from FNIII7-10RGD and FNIII7-10RGE fragments at either contact time that were significantly higher than those from FNIII7-10ΔRGD. The rupture forces of α5β1 integrins on pKO-β1 fibroblasts, however, were significantly lower during detachment from FNIII7-10RGE compared with FNIII7-10RGD at both 5 s and 120 s contact times and similar to those recorded for FNIII7-10ΔRGD. Altogether, these results show that (1) α5β1 and/or αv-class integrins require the RGD motif to strengthen cell adhesion, (2) αv-class integrins strengthen adhesion to FNRGE similar to FNRGD, and (3) α5β1 integrins do not bind or strengthen adhesion to FNRGE.
Cell spreading is altered by RGD mutations
To test whether full-length cFNRGE and cFNΔRGD allows α5β1 and αv-class integrin–mediated adhesion maturation and spreading, we plated pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts in serum replacement medium (SRM) on purified cFN variants and determined cell spreading as well as morphology and distribution of FAs (Fig. 2). At 60 min after seeding, the great majority of pKO-αv (95.8 ± 4.2%), pKO-β1 (93.1 ± 4.6%), and pKO-αv/β1 (98.0 ± 2.0%) fibroblasts spread on cFNRGD; a slightly lower percentage of pKO-αv (85.0 ± 5.6%) and pKO-αv/β1 fibroblasts (79.6 ± 7.9%) spread on cFNRGE; and a neglectable percentage of pKO-β1 fibroblasts spread on cFNRGE. On cFNΔRGD, the numbers of spread pKO-αv and pKO-αv/β1 fibroblasts dropped to 22.2 ± 15% and 10.4 ± 7.0%, respectively, and of pKO-β1 fibroblasts to <1% (Fig. 2 a). Moreover, pKO-αv fibroblasts covered less spreading area than pKO-β1 or pKO-αv/β1 fibroblasts on cFNRGD (744 ± 92 µm2 versus 897 ± 72 µm2, P = 0.022; and 906 ± 72 µm2, P = 0.018), similar to pKO-αv (583 ± 42 µm2) and pKO-αv/β1 fibroblasts (514 ± 49 µm2) on cFNRGE. The few adherent pKO-β1 fibroblasts covered a reduced area of 419 ± 36 µm2 (P < 0.05 compared to pKO-αv cells) on cFNRGE. In Fig. 2 b, statistical calculations change the significance of pKO-β1 compared to pKO-αv fibroblasts on cFNRGE cells from P < 0.001 to P < 0.05. On cFNΔRGD, the adherent pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts covered significantly smaller spreading areas (253 ± 42 µm2, 136 ± 8 µm2, and 227 ± 27 µm2, respectively) when compared with the same cell lines seeded on cFNRGD (Fig. 2 b).

The FN-RGD motif determines α5β1 and αv -class integrin–mediated fibroblast adhesion and spreading. (a) Percentage of pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts spread on cFNRGD, cFNRGE, or cFNΔRGD after 60-min culture. Bars indicate mean and SD (n = 20–30 cells from three independent experiments). (b) Cell area of pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts seeded on cFNRGD, cFNRGE, or cFNΔRGD. Lines indicate mean (n = 20–30 cells assessed from three independent experiments). (c) Immunofluorescence staining of paxillin (white), F-actin (with phalloidin, red), and nuclei (with DAPI, blue) in pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts seeded for 90 min on cFNRGD, cFNRGE, or cFNΔRGD. Statistical significance was calculated using a two-tailed unpaired Student t test; *, P < 0.05; ***, P < 0.001. Scale bars, 10 µm.
The FN-RGD motif determines α5β1 and αv -class integrin–mediated fibroblast adhesion and spreading. (a) Percentage of pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts spread on cFNRGD, cFNRGE, or cFNΔRGD after 60-min culture. Bars indicate mean and SD (n = 20–30 cells from three independent experiments). (b) Cell area of pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts seeded on cFNRGD, cFNRGE, or cFNΔRGD. Lines indicate mean (n = 20–30 cells assessed from three independent experiments). (c) Immunofluorescence staining of paxillin (white), F-actin (with phalloidin, red), and nuclei (with DAPI, blue) in pKO-αv, pKO-β1, and pKO-αv/β1 fibroblasts seeded for 90 min on cFNRGD, cFNRGE, or cFNΔRGD. Statistical significance was calculated using a two-tailed unpaired Student t test; *, P < 0.05; ***, P < 0.001. Scale bars, 10 µm.
FA and cell morphological parameters were analyzed 90 min after seeding by staining for paxillin and F-actin (Fig. 2 c). Whereas pKO-αv fibroblasts seeded on cFNRGD formed large paxillin-positive FAs at the edges of cellular protrusions, the pKO-β1 and pKO-αv/β1 fibroblasts formed, in addition to the protrusion-located FAs, abundant and small fibrillar adhesions (FBs) that aligned along stress fibers inside the cells. While pKO-αv and pKO-αv/β1 fibroblasts seeded on cFNRGE behaved similarly and developed large FAs at the tips of cellular protrusions but no FBs along stress fibers, clearly resembling the phenotype of pKO-αv fibroblasts seeded on cFNRGD, the few adherent pKO-β1 fibroblasts on cFNRGE developed neither FAs nor FBs (Fig. 2 c). These findings indicate that the RGE motif abolishes α5β1- but does not compromise αv-class integrin–mediated cellular functions such as FA assembly and spreading.
Lack of the FN-RGD motif arrests development before gastrulation
The findings so far indicate that the absence of the RGD motif is sufficient to abrogate αv-class integrin functions in vitro. To test whether this observation also holds true in vivo, we generated a Fn1ΔRGD allele by site-directed mutagenesis in embryonic stem (ES) cells and mice (Fig. S2). Heterozygous Fn1ΔRGD/+ mice were viable, fertile, and without an obvious phenotype. Intercrosses of Fn1ΔRGD/+ mice produced no Fn1ΔRGD/ΔRGD mice at postnatal day 21. Time-mated heterozygous intercrosses revealed a normal Mendelian distribution of Fn1ΔRGD/ΔRGD embryos at E7.5, a reduction from the expected 25% to 20% at E8.5 and 15% at E9.5 (Table 1). The Fn1ΔRGD/ΔRGD embryos appeared normal at E7.5, whereas at E8.5, they displayed several pronounced defects, including a shortened anterior–posterior axis, lack of somites, small and distorted headfolds, a small primitive heart bulge, and failure to undergo inside-out turning (Fig. 3 a). At E9.5, the Fn1ΔRGD/ΔRGD embryos had still not turned, were severely underdeveloped, lacked somites, and had small heart bulges, and 30% of Fn1ΔRGD/ΔRGD embryos showed also dilated and misshaped head folds (arrowhead in Fig. 3 b). Similar phenotypic defects were also described in embryos with paired null mutations in Itgav and Itga5 genes or with a null mutation in the Fn1 gene (Yang et al., 1999; George et al., 1993; Georges-Labouesse et al., 1996). Moreover, Fn1ΔRGD/ΔRGD embryos were phenotypically different from Fn1RGE/RGE embryos (Fig. 3 c), which develop the same defects as α5 integrin–deficient mice, including shortened posterior trunk at E9.5, cardiovascular defects, and death at E10–E11 (Takahashi et al., 2007; Girós et al., 2011; Yang et al., 1993). Altogether, these observations indicate that in line with the in vitro experiments, α5β1 as well as αv-class integrins mediate their in vivo functions through binding to the FNIII10-RGD motif.

Information to Fn1ΔRGD/ΔRGD mouse generation and FN distribution in BMs and yolk sac from Fn1ΔRGD/ΔRGD embryos. (a) Strategy to generate Fn1ΔRGD/ΔRGD mice. (b) PCR genotypes with primers indicated in panel a by arrows. (c and d) Nucleotide sequence of wild-type Fn1 (c) and Fn1ΔRGD allele (d) confirms RGD deletion. (e) Immunofluorescence of FN (green), laminin (LM; red), and collagen type IV (Col IV; red) in E9.5 Fn1+/+ and Fn1ΔRGD/ΔRGD embryos. (f and g) Immunofluorescence of FN (green) and nidogen (red) in Fn1−/− embryos. Arrow indicates FN in the yolk sac (Ys). Scale bars, 50 µm (e) and 200 µm (f and g).
Information to Fn1ΔRGD/ΔRGD mouse generation and FN distribution in BMs and yolk sac from Fn1ΔRGD/ΔRGD embryos. (a) Strategy to generate Fn1ΔRGD/ΔRGD mice. (b) PCR genotypes with primers indicated in panel a by arrows. (c and d) Nucleotide sequence of wild-type Fn1 (c) and Fn1ΔRGD allele (d) confirms RGD deletion. (e) Immunofluorescence of FN (green), laminin (LM; red), and collagen type IV (Col IV; red) in E9.5 Fn1+/+ and Fn1ΔRGD/ΔRGD embryos. (f and g) Immunofluorescence of FN (green) and nidogen (red) in Fn1−/− embryos. Arrow indicates FN in the yolk sac (Ys). Scale bars, 50 µm (e) and 200 µm (f and g).
Progeny of Fn1+/ΔRGD intercrosses

Development of Fn1ΔRGD/ΔRGD embryos arrests at gastrulation. (a–c) Phenotype of Fn1+/+ and Fn1ΔRGD/ΔRGD littermate embryos at E8.5 (a) and E9.5 (b) and for comparison of Fn1RGE/RGE embryo at E9.5 (c). Arrowhead in b points the abnormally shaped telencephalic vesicles. (d) FN (green) distribution in E9.5 Fn1+/+ and Fn1ΔRGD/ΔRGD embryos. (e) Higher magnification of insets depicted in d showing FN and nidogen (red) signals in the telencephalic vesicle and adjacent mesenchyme. Arrowheads in e indicate interstitial tissue and arrow indicates FN in BM of Fn1ΔRGD/ΔRGD embryo. Hf, head folds; Hr, heart; Tb, tail bud. Scale bars, 125 µm (E8.5), 250 µm (E9.5), 200 µm (d), and 50 µm (e).
Development of Fn1ΔRGD/ΔRGD embryos arrests at gastrulation. (a–c) Phenotype of Fn1+/+ and Fn1ΔRGD/ΔRGD littermate embryos at E8.5 (a) and E9.5 (b) and for comparison of Fn1RGE/RGE embryo at E9.5 (c). Arrowhead in b points the abnormally shaped telencephalic vesicles. (d) FN (green) distribution in E9.5 Fn1+/+ and Fn1ΔRGD/ΔRGD embryos. (e) Higher magnification of insets depicted in d showing FN and nidogen (red) signals in the telencephalic vesicle and adjacent mesenchyme. Arrowheads in e indicate interstitial tissue and arrow indicates FN in BM of Fn1ΔRGD/ΔRGD embryo. Hf, head folds; Hr, heart; Tb, tail bud. Scale bars, 125 µm (E8.5), 250 µm (E9.5), 200 µm (d), and 50 µm (e).
Fn1ΔRGD/ΔRGD embryos assemble an aberrant FNΔRGD matrix
Since Fn1ΔRGD/ΔRGD and Fn1−/− mice develop similar defects and arrest development before E8.5, we hypothesized that not only integrin signaling but also the assembly of a FNΔRGD matrix are compromised. To test the hypothesis, we immunostained FN in E9.5 embryos and found that FN was widely distributed in both Fn1+/+ and Fn1ΔRGD/ΔRGD embryos (Fig. 3 d). A close inspection of consecutively immunostained sections of the telencephalic vesicle for the basement membrane (BM) markers nidogen, laminin, and collagen type IV (Fig. 3 e and Fig. S2 e) revealed that FN was present in BMs as well as interstitial ECM of the Fn1+/+ head mesenchyme (arrowhead in Fig. 3 e). In Fn1ΔRGD/ΔRGD embryos, the FN was mostly restricted to BMs, where irregular deposits were apparent (arrow in Fig. 3 e). Interestingly, laminin, nidogen, and collagen type IV showed the same irregular distribution as the FNΔRGD (Fig. S2 e). In addition to BMs, tiny, puncta-like FNΔRGD deposits were visible in the poorly developed mesenchyme of Fn1ΔRGD/ΔRGD embryos (arrowhead in Fig. 3 e). To exclude that diffusion of maternally derived FN contributes to FN fibril formation in Fn1ΔRGD/ΔRGD embryos, we generated E8.5 Fn1−/− embryos by intercrossing the Fn1-floxed allele (Sakai et al., 2001) with a deletor-Cre transgene, prepared sections, and found, in line with a previous report (George et al., 1993), that FN deposits were absent from embryonic BMs and interstitial tissues (Fig. S2, f and g), whereas maternal FN deposits were present in the external layer of the yolk sac (arrow in Fig. S2 f). These findings indicate that the aberrant and short, puncta-like deposits in tissues of Fn1ΔRGD/ΔRGD embryos were not maternally derived.
FNΔRGD forms short fibrils in vitro
Although FN fibril formation is thought to be an integrin-dependent process, the lack of α5β1 and αv-class integrin binding to FNΔRGD indicates that the fibrils observed in Fn1ΔRGD/ΔRGD embryos are formed integrin independently. To further investigate this conundrum, we established Fn1ΔRGD/ΔRGD ES cells and fibroblasts to confirm and characterize FNΔRGD fibril formation in vitro. As shown in Fig. 4 a, wild-type as well as Fn1ΔRGD/ΔRGD ES cell aggregates grown in FN-depleted serum clearly contained FN fibrils, although the FNΔRGD fibrils appeared thicker and shorter. The similar expression levels of α5, αv, β1, and β3 and absence of α4 integrin expression on wild-type and Fn1ΔRGD/ΔRGD ES cells (Fig. S3 a) excluded an involvement of α4β1 integrins in FNΔRGD fibril formation (Sechler et al., 2000).

FNΔRGD is assembled into short and thick fibrils. (a) Immunofluorescence of FN (green) assembled by Fn1+/+ and Fn1ΔRGD/ΔRGD ES cell aggregates. (b–d)Fn1+/+, Fn1ΔRGD/ΔRGD, and Fn1RGE/RGE fibroblasts seeded on laminin and stained for F-actin (phalloidin; red), paxillin (white), and FN (green) at indicated time points after cell seeding. (e) Representative WBs of FN content in whole-cell lysates, including DOC-insoluble and DOC-soluble fractions, and in the conditioned medium. Tub, tubulin. (f) Quantification of DOC-insoluble and DOC-soluble FN from cell extracts and FN in the conditioned medium at indicated time points after seeding. FN levels are shown relative to tubulin in the cell extract. au, arbitrary units. Bars indicate mean and SEM of three independent experiments (n = 3). Statistical significance was calculated using two-tailed unpaired Student t tests and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; n.s., not significant. (g) Pln (green) deposition in matrices assembled by Fn1+/+,Fn1RGE/RGE, and Fn1ΔRGD/ΔRGD fibroblasts. Scale bars, 10 µm.
FNΔRGD is assembled into short and thick fibrils. (a) Immunofluorescence of FN (green) assembled by Fn1+/+ and Fn1ΔRGD/ΔRGD ES cell aggregates. (b–d)Fn1+/+, Fn1ΔRGD/ΔRGD, and Fn1RGE/RGE fibroblasts seeded on laminin and stained for F-actin (phalloidin; red), paxillin (white), and FN (green) at indicated time points after cell seeding. (e) Representative WBs of FN content in whole-cell lysates, including DOC-insoluble and DOC-soluble fractions, and in the conditioned medium. Tub, tubulin. (f) Quantification of DOC-insoluble and DOC-soluble FN from cell extracts and FN in the conditioned medium at indicated time points after seeding. FN levels are shown relative to tubulin in the cell extract. au, arbitrary units. Bars indicate mean and SEM of three independent experiments (n = 3). Statistical significance was calculated using two-tailed unpaired Student t tests and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; n.s., not significant. (g) Pln (green) deposition in matrices assembled by Fn1+/+,Fn1RGE/RGE, and Fn1ΔRGD/ΔRGD fibroblasts. Scale bars, 10 µm.

Integrin expression. (a and b) FACS-based measurements of integrin levels on the surface of Fn1+/+ and Fn1ΔRGD/ΔRGD ES cells (a) and integrin and syndecan 4 levels on Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts (b). Intensities obtained with specific antibodies were normalized to isotype controls. Intensity values near 1 indicate that similar values were obtained with specific antibody and the isotype control. Raw cells were used as positive control for α4 integrin expression. Experiments were repeated three times, and 10,000 cells were analyzed per experiment. AU, arbitrary units. (c) Quantitative real-time PCR analysis of Itga8 gene expression in Fn1ΔRGD fibroblasts with mRNA from mouse lung as a positive control. Data are from three independent experiments performed in triplicate (normalized to GAPDH). Bars represent mean ± SD of the average fold induction compared with mouse lung tissue. Statistical significances were calculated using two-tailed unpaired Student t tests; *, P < 0.05; ***, P < 0.001.
Integrin expression. (a and b) FACS-based measurements of integrin levels on the surface of Fn1+/+ and Fn1ΔRGD/ΔRGD ES cells (a) and integrin and syndecan 4 levels on Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts (b). Intensities obtained with specific antibodies were normalized to isotype controls. Intensity values near 1 indicate that similar values were obtained with specific antibody and the isotype control. Raw cells were used as positive control for α4 integrin expression. Experiments were repeated three times, and 10,000 cells were analyzed per experiment. AU, arbitrary units. (c) Quantitative real-time PCR analysis of Itga8 gene expression in Fn1ΔRGD fibroblasts with mRNA from mouse lung as a positive control. Data are from three independent experiments performed in triplicate (normalized to GAPDH). Bars represent mean ± SD of the average fold induction compared with mouse lung tissue. Statistical significances were calculated using two-tailed unpaired Student t tests; *, P < 0.05; ***, P < 0.001.
Next, we differentiated Fn1+/+ and Fn1ΔRGD/ΔRGD ES cells into fibroblasts and established immortalized cell lines that expressed similar levels of α5, αv, β1, and β3 integrins and lacked expression of integrin α4 (Fig. S3 b). We also excluded α8 integrin subunit expression in pKO (Schiller et al., 2013), Fn1+/+, Fn1RGE/RGE (Takahashi et al., 2007), and Fn1ΔRGD/ΔRGD fibroblasts (Fig. S3 c) to ensure a faithful comparison of αv-class and α5β1 integrin–mediated FN fibrillogenesis via wild-type or mutant RGD motifs. First, we compared the FN fibrillar network assembled by Fn1+/+, Fn1ΔRGD/ΔRGD, and Fn1RGE/RGE fibroblasts. To this end, we seeded serum-starved cells on laminin, which mediates cell adhesion via α6β1 integrins and leaves αv-class and α5β1 integrins unoccupied and, hence, available for the assembly of self-expressed and secreted FN (Fig. 4, b and c). 24 h after cell seeding and growing in SRM, the Fn1+/+ fibroblasts assembled short FN fibrils and formed characteristic stress fibers that connected paxillin-positive FAs at the cell periphery and aligned with FBs in the cell interior. At 72 h, Fn1+/+ fibroblasts assembled long and thin FN fibrils that were distributed in the cell center, and at 120 h after seeding, the FN fibrils increased in length, were denser, and aligned with the actin fibers (Fig. 4 b). In sharp contrast, Fn1ΔRGD/ΔRGD fibroblasts harbored most of their FN in intracellular vesicles, and only very few short and thick fibrils were visible at the cell tips 24 h after seeding. Furthermore, the Fn1ΔRGD/ΔRGD fibroblasts contained fewer stress fibers connected to the adhesion sites at the cell periphery, and the majority of paxillin remained diffusely distributed in the cytoplasm. Although at 72 and 120 h after seeding the FNΔRGD matrix increased in complexity, the FN fibrils remained short, thick, and located to peripheral adhesions (Fig. 4 c). Finally, the Fn1RGE/RGE fibroblasts assembled their secreted FNRGE already 24 h after seeding into thin fibrils that were poorly aligned with the stress fibers and mainly located to the cell periphery, forming fibrillar networks that were of lower density than those of Fn1+/+ fibroblasts (Fig. 4 d). To quantify the amount of soluble and of functional FN that is assembled into insoluble cross-linked fibrils, cell cultures were extracted with sodium deoxycholate (DOC), which efficiently separates soluble from insoluble FN. In addition, we also quantified unassembled FN secreted into the conditioned SRM by the cells. Expectedly, Fn1+/+ fibroblasts assembled increasing amounts of insoluble fibrils with increasing culturing time, whereas the unassembled FN in the conditioned medium remained low. Consistently with the poor assembly of FNΔRGD fibrils, the levels of DOC-extracted, insoluble cross-linked FNΔRGD were down to ∼8% in Fn1ΔRGD/ΔRGD cells after 5 d in culture (Fig. 4, e and f), whereas the unassembled FNΔRGD increased in the conditioned medium, reaching ninefold-higher levels when compared with Fn1+/+ fibroblasts. The FNRGE secreted by Fn1RGE/RGE fibroblasts also accumulated in the conditioned medium to significantly higher amounts than in conditioned medium of Fn1+/+ fibroblasts. Importantly, the intracellular, DOC-soluble FN contents in cell extracts were similar in Fn1+/+, Fn1RGE/RGE, and Fn1ΔRGD/ΔRGD fibroblasts at all time points tested (Fig. 4, e and f), indicating that FN expression is not altered in Fn1RGE/RGE or Fn1ΔRGD/ΔRGD cells. To exclude that FNΔRGD fibrils are only assembled when fibroblasts are seeded on laminin, we cultured Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts on vitronectin (VN) or collagen type I and found that Fn1ΔRGD/ΔRGD fibroblasts formed short and thick FNΔRGD fibrils on both substrates (Fig. S4), although not restricted to the periphery tips, as observed with Fn1ΔRGD/ΔRGD fibroblasts seeded on laminin (Fig. 4 c).

FNΔRGD assembly by cells cultured on VN or collagen I. Fluorescence signal of FN (green), paxillin (Pax; white), F-actin (phalloidin; red), and nuclei (DAPI; blue) by Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts seeded on either VN or collagen I for 24 h in SRM. Scale bars, 10 µm.
FNΔRGD assembly by cells cultured on VN or collagen I. Fluorescence signal of FN (green), paxillin (Pax; white), F-actin (phalloidin; red), and nuclei (DAPI; blue) by Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts seeded on either VN or collagen I for 24 h in SRM. Scale bars, 10 µm.
It has been reported that FN fibrils serve as scaffold for several ECM proteins, including collagens and proteoglycans such as Pln (Kadler et al., 2008; Chung and Erickson, 1997). The Pln matrix assembled by Fn1+/+ or Fn1RGE/RGE fibroblasts cultured in SRM on a laminin-coated substrate for 5 d consisted of a similar and elaborated network (Fig. 4 g). In sharp contrast, the Pln network laid down by Fn1ΔRGD/ΔRGD fibroblasts was less dense, and the majority of Pln was inside the cells. These results indicate that FNΔRGD fibrils are dysfunctional to nucleate complex ECMs.
The heparan sulfate–binding sites in FN facilitate FNΔRGD fibril formation
Our findings demonstrate that αv-class integrins assemble FNRGE exclusively by binding the RGE motif in vitro and in vivo. The assembly of FNΔRGD fibrils was unanticipated as FN fibril formation is believed to be integrin driven. Since α4 and α8 integrins are not expressed on Fn1ΔRGD/ΔRGD ES cells and fibroblasts (Fig. S3, a and b) and during early mouse development (Takahashi et al., 2007), α5β1 and αv-class integrins do not bind FNΔRGD, and an additional αv-class integrin binding site is not available in FN, we hypothesized that the assembly is most likely orchestrated by nonintegrin cell surface proteins. Potential candidates are members of the syndecan family, which are ubiquitously expressed (Morgan et al., 2007), bind the heparan sulfate binding motifs of FN, and were shown to promote integrin-induced formation of FN fibrils in vitro (Stepp et al., 2010). Western blot (WB) analyses revealed the presence of all four syndecans in Fn1+/+ as well as Fn1ΔRGD/ΔRGD fibroblasts with syndecan 1, 2, and 4 expressed at higher levels by Fn1ΔRGD/ΔRGD than by Fn1+/+ fibroblasts (Fig. 5 a). Flow cytometry analysis confirmed the twofold higher surface expression levels of syndecan 4 on Fn1ΔRGD/ΔRGD fibroblasts compared with Fn1+/+ fibroblasts (Fig. S3 b). To investigate whether syndecans mediate FNΔRGD fibrillogenesis, we cultured cells in SRM and characterized the assembly of self-secreted FN in the presence of heparin (Fig. 5, b and c; and Fig. S5 a), which blocks the heparan sulfate binding motifs on FN and, hence, syndecan binding to FN (Peterson et al., 2005). Whereas Fn1+/+ fibroblasts seeded on laminin formed similar FN networks with similar numbers of FN fibril branch points in the presence as well as absence of heparin, Fn1ΔRGD/ΔRGD fibroblasts assembled short and thick FN fibrils with significantly fewer branch points (Fig. S5 a), and in the presence of heparin, FNΔRGD fibrils were absent and FNΔRGD was only visible intracellularly (Fig. 5 c and Fig. S5 a). Fn1ΔRGD/ΔRGD fibroblasts treated with heparin were also less spread and accumulated F-actin at the cell cortex. Consistent with the blocking effect of heparin on FN fibrillogenesis by Fn1ΔRGD/ΔRGD fibroblasts, significantly less DOC-insoluble FNΔRGD was isolated from the heparin-treated cells compared with untreated Fn1ΔRGD/ΔRGD fibroblasts (Fig. 5 d). Heparin treatment affected neither the amount of DOC-insoluble FN isolated from Fn1+/+ fibroblasts nor from Fn1RGE/RGE fibroblasts (Fig. 5 d and Fig. S5, b and c). Addition of MnCl2 to the cell culture medium, which binds to and activates integrins (Beauvais and Rapraeger, 2010), further confirmed that integrins are not involved in the assembly of FNΔRGD fibrils, as heparin treatment blocked FNΔRGD fibril formation irrespective of the presence of MnCl2 (Fig. S5 d). Expectedly, MnCl2 induced a robust assembly of FN fibrils in Fn1+/+ and Fn1RGE/RGE fibroblasts, both in the presence and absence of heparin.

The heparan sulfate–binding site in FN facilitates FNΔRGD fibrillogenesis. (a) Representative WB of syndecans 1–4 in Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts and quantification of three (n) independent experiments adjusted to tubulin (Tub) levels. Bars indicate mean and SEM. Statistical significance was calculated using a two-tailed unpaired Student t test; *, P < 0.05. (b and c) FN assembly by Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts seeded on laminin and treated with heparin (Hep) at the indicated time points. (d) Representative WB and quantification of DOC-insoluble and DOC-soluble FN in cell extracts and FN in the conditioned medium without and with heparin treatment for 24 h. (e) Percentage of syndecan protein expression in Fn1ΔRGD/ΔRGD and Fn1+/+ fibroblasts transfected with 150 nM siRNA. (f) Fluorescence of FN (green), F-actin (red), and nuclei (DAPI) by indicated fibroblast lines. (g) Representative WB and quantification of DOC-insoluble and DOC-soluble FN in cell extracts and FN in the conditioned medium of cells treated with either scr RNA or siRNA. FN levels are shown relative to tubulin in the cell extract. Bars indicate mean SEM of three (n) independent experiments. Statistical significance was calculated using a two-tailed unpaired Student t test and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant. Scale bars in b, c, and f, 10 µm. au, arbitrary units; KD, knockdown.
The heparan sulfate–binding site in FN facilitates FNΔRGD fibrillogenesis. (a) Representative WB of syndecans 1–4 in Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts and quantification of three (n) independent experiments adjusted to tubulin (Tub) levels. Bars indicate mean and SEM. Statistical significance was calculated using a two-tailed unpaired Student t test; *, P < 0.05. (b and c) FN assembly by Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts seeded on laminin and treated with heparin (Hep) at the indicated time points. (d) Representative WB and quantification of DOC-insoluble and DOC-soluble FN in cell extracts and FN in the conditioned medium without and with heparin treatment for 24 h. (e) Percentage of syndecan protein expression in Fn1ΔRGD/ΔRGD and Fn1+/+ fibroblasts transfected with 150 nM siRNA. (f) Fluorescence of FN (green), F-actin (red), and nuclei (DAPI) by indicated fibroblast lines. (g) Representative WB and quantification of DOC-insoluble and DOC-soluble FN in cell extracts and FN in the conditioned medium of cells treated with either scr RNA or siRNA. FN levels are shown relative to tubulin in the cell extract. Bars indicate mean SEM of three (n) independent experiments. Statistical significance was calculated using a two-tailed unpaired Student t test and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant. Scale bars in b, c, and f, 10 µm. au, arbitrary units; KD, knockdown.

FNRGD, FNRGE, and FNΔRGD assembly in the presence of heparin, Mn+2, or syndecan siRNA and syndecan expression after knockdown. (a) Quantification of FNRGD and FNΔRGD fibril content and number of branched points in absence and presence of heparin after 24 h. Bars indicate mean (n = 20–30 cells per condition) and SEM. (b) FN assembly by Fn1RGE/RGE fibroblasts seeded on laminin and treated without or with heparin. FN, green; F-actin, red; paxillin, white; and DAPI, blue. (c) Representative WB and quantification of DOC-insoluble and DOC-soluble FN from cell extracts and FN in the conditioned culture medium without and with heparin treatment for 24 h. FN levels are shown relative to total tubulin in the cell extract. (d) FNRGD, FNRGE, and FNΔRGD assembly by Fn1+/+, Fn1RGE/RGE and Fn1ΔRGD/ΔRGD fibroblasts, respectively, seeded on laminin and cultured in SRM with MnCl2 or with MnCl2 and heparin. FN, green; F-actin, red; paxillin, white; and DAPI, blue. (e and f) Representative WBs and quantification of syndecan 1–4 protein levels in Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts 96 h after transfection with siRNA to deplete syndecans. Syndecan protein levels were normalized to tubulin. (g) Quantification of FNRGD and FNΔRGD fibril content and number of branched points after syndecan knockdown (sdc-KD) or in the presence of scr RNA. Bars indicate mean and SEM of three independent experiments. Statistical significances were calculated using two-tailed Student t tests and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant. Scale bars, 10 µm. au, arbitrary units.
FNRGD, FNRGE, and FNΔRGD assembly in the presence of heparin, Mn+2, or syndecan siRNA and syndecan expression after knockdown. (a) Quantification of FNRGD and FNΔRGD fibril content and number of branched points in absence and presence of heparin after 24 h. Bars indicate mean (n = 20–30 cells per condition) and SEM. (b) FN assembly by Fn1RGE/RGE fibroblasts seeded on laminin and treated without or with heparin. FN, green; F-actin, red; paxillin, white; and DAPI, blue. (c) Representative WB and quantification of DOC-insoluble and DOC-soluble FN from cell extracts and FN in the conditioned culture medium without and with heparin treatment for 24 h. FN levels are shown relative to total tubulin in the cell extract. (d) FNRGD, FNRGE, and FNΔRGD assembly by Fn1+/+, Fn1RGE/RGE and Fn1ΔRGD/ΔRGD fibroblasts, respectively, seeded on laminin and cultured in SRM with MnCl2 or with MnCl2 and heparin. FN, green; F-actin, red; paxillin, white; and DAPI, blue. (e and f) Representative WBs and quantification of syndecan 1–4 protein levels in Fn1+/+ and Fn1ΔRGD/ΔRGD fibroblasts 96 h after transfection with siRNA to deplete syndecans. Syndecan protein levels were normalized to tubulin. (g) Quantification of FNRGD and FNΔRGD fibril content and number of branched points after syndecan knockdown (sdc-KD) or in the presence of scr RNA. Bars indicate mean and SEM of three independent experiments. Statistical significances were calculated using two-tailed Student t tests and corrected with a Bonferroni post-hoc test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant. Scale bars, 10 µm. au, arbitrary units.
Next, we directly addressed whether syndecans mediate FNΔRGD fibril assembly by depleting syndecan mRNAs in Fn1ΔRGD/ΔRGD fibroblasts using siRNAs. Depletion of all four syndecan mRNAs, with four specific siRNAs, compromised Fn1ΔRGD/ΔRGD fibroblast survival in SRM. Since the siRNA-mediated depletion of syndecan 1 (sdc1) reduced levels of sdc1 and sdc3 to 60% and the siRNA against syndecan 4 (sdc4) reduced levels of sdc2 and sdc4 to 13–30% in Fn1ΔRGD/ΔRGD fibroblasts (Fig. S5, e and f), we combined them to diminish syndecan expression in Fn1ΔRGD/ΔRGD and Fn1+/+ fibroblasts (Fig. 5 e). Whereas scrambled (scr) siRNA affected neither DOC-insoluble FN levels nor FNRGD and FNΔRGD fibril assembly (Fig. 5, f and g) in cell extracts from Fn1+/+-scr or Fn1ΔRGD/ΔRGD-scr fibroblasts, FNΔRGD assembly was severely compromised upon syndecan silencing in Fn1ΔRGD/ΔRGD-sdc-KD, but not in Fn1+/+-sdc-KD, fibroblasts, consolidating that syndecans mediate the aberrant FNΔRGD fibrillogenesis (Fig. 5, f and g; and Fig. S5 g).
Discussion
ECMs are organized into complex 3D structures that are essential for morphogenesis and postnatal homeostasis (Wilson et al., 2005). Knowledge of how these complex ECMs are assembled and how cells respond to them is pivotal for the understanding of development and disease and the successful design of bio-inspired materials. FN takes a central role in ECM assembly, as it is thought to serve as a binding platform for several unrelated ECM proteins such as collagens (Kadler et al., 2008), tenascins, Pln, etc. The assembly of FN is a cell- and force-driven process steered by α5β1 and αv-class integrins that bind, unfold, and permit FN cross-linking into a fibrillar network (Pankov et al., 2000; Mao and Schwarzbauer, 2005). A central question is whether αv-class integrins exclusively bind to the RGD motif or whether additional sites in FN (e.g., the NGR motif in FNI5 and other FN modules; Curnis et al., 2006; Takahashi et al., 2007) also serve as binding sites that mediate cell adhesion and drive FN assembly.
To address this question, we used an interdisciplinary approach whose results demonstrate that the RGD motif in FN is, like for α5β1 integrin, also the sole binding motif for αv-class integrins, and, unlike previously thought (Greenspoon et al., 1993; Beumer et al., 2000; Shiokawa et al., 1999; Leyton et al., 2001; Lee et al., 2019; Niederlechner et al., 2012), the substitution of the aspartate with a glutamate (RGD>RGE) in the FN-RGD motif impedes α5β1 integrin binding but leaves binding to αv-class integrins unaffected. Furthermore, we show that FNΔRGD is (poorly) assembled into short, thick, and dysfunctional fibrils by syndecans, which bind to the heparin II–binding site in FNIII12-14.
These results clearly indicate that the RGD motif in FN is essential not only for integrin binding and cell adhesion but also for the assembly of a functional FN matrix. SCFS and cell-spreading experiments using different RGD mutations in full-length FNs and pKO cells reconstituted to express either α5β1 and/or αv-class integrins confirmed this conclusion and showed that neither α5β1 nor αv-class integrins can bind to RGD-deleted FN. Whereas the RGD>RGE substitution abrogates binding of α5β1 integrin, αv-class integrins binding to and adhesion strengthening on FNIII7-10RGE occurs normally and is characterized by high on-rate binding events and rupture forces of single unbinding events that are also seen in experiments with FNIII7-10RGD fragments.
In line with our in vitro experiments, Fn1ΔRGD/ΔRGD mice develop pronounced morphogenetic defects between E7.5 and E8.5 that are similar to Itga5/Itgav double-null mice but differ substantially from those observed in Fn1RGE/RGE mice, which resemble the phenotype of Itga5-null mice (Takahashi et al., 2007; Girós et al., 2011; Yang et al., 1993). Together with the in vitro experiments, the phenotype of Fn1ΔRGD/ΔRGD mice confirms the FN-RGD motif to be the sole binding site in FN for both α5β1 and αv-class integrins in vivo. The selective loss of α5β1 binding to FNRGE observed in SCFS experiments provides an explanation why Itga5-null and Fn1RGE/RGE embryos share the same defects (Takahashi et al., 2007; Yang et al., 1993) and raises concerns regarding the RGE-containing FN fragments used in vitro and ex vivo as non–integrin-binding compounds (Beumer et al., 2000; Shiokawa et al., 1999; Leyton et al., 2001). In the Fn1ΔRGD/ΔRGD embryos, we observe a drastic reduction of FN in interstitial tissues that could, at least in part, be due to the defective development of mesoderm, which is also observed in Itga5/Itgav double-null embryos and associated with an abnormal development of the telencephalic vesicles and primitive heart. The assembly of FN in interstitial tissues is driven by cell surface receptors. In line with our previous report (Benito-Jardón et al., 2017), we confirm that the synergy site in FNIII9 is insufficient to associate with α5β1 in the absence of the RGD motif, and therefore, α5β1 integrin–synergy site interaction can be excluded as an RGD-independent assembly mechanism. Furthermore, the NGR/isoDGR motif in the FNI5 module, which was shown to bind αvβ3 integrins (Curnis et al., 2006; Takahashi et al., 2007; Curnis et al., 2010) clearly cannot compensate the absence of the RGD motif in FNΔRGD for cell adhesion, adhesion strengthening, cell spreading, and development. These findings, together with a report showing that substitution of asparagine (N) in the NGR motif of FNI5 and FNI7 with glutamine (Q) does not prevent fibril formation (Xu et al., 2010), strongly argue against a functional role of FN’s NGR motifs. Finally, α4β1, which binds to the variable region of FN and is also able to assemble FN fibrils (Sechler et al., 2000), is not expressed at this stage of development.
These results raised the question of how FNΔRGD could be assembled. Syndecans are an alternative family of adhesion molecules that potentially initiate FN assembly. They directly bind to the heparin II domain (FNIII12-14) of FN (Woods et al., 2000; Mahalingam et al., 2007), are expressed on almost all cells (Morgan et al., 2007), colocalize with integrins in FAs, and have been proposed to make the initial contacts with FN (Woods and Couchman, 1994; Bloom et al., 1999). Syndecan 1, for example, has been shown to promote FN assembly through activation of αv-class integrins (Beauvais and Rapraeger, 2010; Yang and Friedl, 2016) or α5β1 cosignaling (Wang et al., 2010), and syndecan 2 to facilitate the assembly of FN fibrils (Galante and Schwarzbauer, 2007; Klass et al., 2000). Our cell culture experiments also reveal the involvement of syndecans in the formation of FNΔRGD fibrils. Both addition of heparin to block the syndecan binding site in FN and the depletion of syndecan expression almost completely abrogated FNΔRGD fibril assembly. It is possible that the weak interactions of syndecans with the actomyosin cytoskeleton and, hence, transmission of weak pulling forces to the syndecan–FN bonds are sufficient to partially unfold FNΔRGD and assemble FNΔRGD fibrils that require the input of integrins for further maturation into thick, long, and functional fibrils.
In addition to the small FNΔRGD deposits in interstitial tissues of Fn1ΔRGD/ΔRGD embryos, we also observed aberrant FNΔRGD deposits in BMs. FN deposition at BMs has recently been shown to be assembled differently from interstitial FN fibrils (Lu et al., 2020). Although the study did not identify how cells initiate FN recruitment, cells generate pulling forces induced by integrins binding to the BM components laminin and collagen IV, initiate FN unfolding, and induce fibril formation, and, as FAs mature, switch to α5β1 integrin binding, which will elongate and move the FN fibrils into the cell center. Interestingly, we also observed that fibroblasts seeded on laminin recruit cFNΔRGD to the cell periphery, where it remains and, probably due to the lack of α5β1–FNΔRGD interactions, never matures further, elongates, and moves into the cell center. Interestingly, discontinuous FNΔRGD deposits also predominate in BMs of Fn1ΔRGD/ΔRGD embryos. Syndecans, which make first contacts with FN, crosstalk with integrins, and modulate integrin functions (Woods and Couchman, 1994; Bloom et al., 1999; Wang et al., 2010; Chronopoulos et al., 2020; Bass et al., 2007), may also serve as initiators of the FN assembly process in BMs.
FN peptides containing an RGE motif are frequently used in control experiments to show that they, in contrast to RGD-containing peptides, do not impair, for example, cell adhesion to FN, or even other ECM proteins containing cell binding RGD motifs (Ruoslahti and Pierschbacher, 1987; Cherny et al., 1993; Greenspoon et al., 1993). It is not clear why the robust binding of αv-class integrins to the RGE motif went unnoticed. A possible explanation is that the FN-RGE peptide–mediated blocking of αvβ3 integrin for FN binding was masked by the high coexpression of other FN-binding integrins such as α5β1 or α4-class integrins, which do not bind FN-RGE and, hence, mask the inhibitory effect for αv integrins. Alternatively, the molecular properties of the used peptides, including their structure and sequences flanking the RGD (or RGE) motif, may have abrogated the recognition by αv-class integrins, which would also explain why the RGD>RGE mutation in VN eliminates αvβ3 and αvβ5 integrin binding (Cherny et al., 1993). Finally, it is also possible that due to the overwhelming literature on a loss of function associated with the RGE motif, potential functional hints of RGE have been overlooked. Nevertheless, our observation reported here opens the opportunity to dictate whether α5β1 and/or αv-class integrins bind FN by substituting the aspartate to glutamate in FN’s RGD motif, and to expand the toolbox for designing FN-RGE–containing biomaterials that only allow αv-class integrin engagement or prevent αv-class integrin and favor α5β1 integrin binding to FN-RGD by applying soluble RGE-containing peptidomimetica. This strategy, for example, could direct the differentiation of bipotent pancreatic progenitor cells (Mamidi et al., 2018) into either the duct lineage, by inhibiting αv-class integrins with soluble RGE peptidomimetica, or the hormone-producing cell lineage, by exclusively engaging αv-class integrins. Further interesting applications include regulating osteoclasts and bone resorption (Nakamura et al., 2007), angiogenesis (Lovett et al., 2009; Korntner et al., 2019), or cancer progression (Kozlova et al., 2001) through the specific engagement or inhibition of αv-class integrins to FN.
Materials and methods
Production of the FNIII7-10 fragments
We subcloned the human cDNA encoding FNIII7-10 in the expression vector pET-15b (Takahashi et al., 2007). The substitution of D to E or deletion of the entire RGD motif was achieved by site-directed mutagenesis and allowed expression of FNIII7-10RGE or FNIII7-10ΔRGD fragments, respectively, in the Escherichia coli strain Rosetta T1R. The fragments were purified using TALON Metal Affinity chromatography (Clontech) and gel filtration chromatography using Superdex 200 10/300 GL columns (GE Healthcare) and eluted in PBS.
SCFS
For cell attachment, cantilevers were plasma cleaned (PDC-32G; Harrick Plasma) and then incubated overnight at 4°C in PBS containing concanavalin A (2 mg/ml; Sigma-Aldrich; Friedrichs et al., 2013). For substrate coatings, a 200-µm-thick four-segmented polydimethylsilane mask fused to the surface of glass-bottom Petri dishes (WPI) was used (Yu et al., 2015). Each of the four polydimethylsilane-framed glass surfaces were incubated overnight at 4°C with cFNRGD, cFNΔRGD, FNIII7-10RGD, FNIII7-10ΔRGD, or FNIII7-10RGE (all 50 µg/ml in PBS). For SCFS, we mounted a Nanowizard II atomic force microscope (AFM) equipped with a CellHesion module (both from JPK Instruments; Puech et al., 2006) onto an inverted fluorescence microscope (Observer Z1; Zeiss). The temperature was kept at 37°C throughout the experiment by a PetriDish heater (JPK Instruments). 200-µm-long V-shaped silicon nitride tip-less cantilevers having nominal spring constants of 0.06 N/m (NP-0; Bruker) were used. Each cantilever was calibrated prior the measurement by determining its sensitivity and spring constant using the thermal noise analysis of in-build routines (Hutter and Bechhoefer, 1993). To adhere a single fibroblast to the AFM cantilever, overnight serum-starved fibroblasts with confluency up to ∼80% were washed with PBS, detached with 0.25% (wt/vol) trypsin/EDTA for 2 min, suspended in SCFS media (DMEM supplemented with 20 mM Hepes) containing 1% (vol/vol) FCS, pelleted, and finally resuspended in serum-free SCFS media. Fibroblasts were allowed to recover for at least 30 min from trypsin treatment (Schubert et al., 2014). Attachment of a single fibroblast to the free end of the cantilever was achieved by pipetting the fibroblast suspension onto functionalized Petri dishes. The functionalized cantilever was lowered onto a single fibroblast with a speed of 10 µm/s until a force of 5 nN was recorded. After 5 s contact, the cantilever was retracted at 10 µm/s for 100 µm, and the cantilever-bound fibroblast was incubated for 10–15 min for binding probability experiments and 7–10 min for adhesion force measurements to assure firm binding to the cantilever. Using differential interference contract microscopy, the morphological state of the fibroblast was monitored. For binding probability experiments, the fibroblast bound to the cantilever was lowered onto the coated substrate with a speed of 1 µm/s until a contact force of 200 pN, and the cantilever was immediately, or after a contact time of 250 ms, retracted at 1 µm/s for ≥13 µm until the fibroblast and substrate were fully separated. This resulted in a total contact time between fibroblasts and substrate of ∼50 ms or ∼300 ms, respectively. After the experimental cycle, the fibroblast was allowed to recover for 0.5 s, and the contact area on the substrate was altered. This experimental cycle was repeated 100–200 times for each fibroblast used. FD curves were analyzed for unbinding events using in-build JPK software to determine binding probability as the ratio of FD curves showing unbinding events and total number of attempts. For adhesion force experiments, the rounded fibroblast bound to the cantilever was lowered onto the coated substrate with a speed of 5 µm/s until a contact force of 2 nN was recorded. For contact times of 5, 20, 50, or 120 s, the cantilever height was maintained constant. Subsequently, the cantilever was retracted at 5 µm/s and for 100 µm until the fibroblast and substrate were fully separated. After the experimental cycle, the fibroblast was allowed to recover for a time period equal to contact time before measuring the adhesion force for a different contact time. A single fibroblast was used to probe the adhesion force for all contact times or until morphological changes (that is, spreading) was observed. The sequence of contact time measurements and area of the substrate were varied. The adhesion of at least 10 fibroblasts was measured per condition to obtain statistically firm results. Adhesion forces were determined after baseline correction of FD curves with JPK software (JPK Instruments). Single unbinding forces were determined using an in-house-written Igor code (Igor 6; WaveMetrics). Two-tailed Mann-Whitney tests were applied to determine significant differences between the binding probability of fibroblast lines at different conditions. Mann-Whitney tests and statistical analysis of slope differences for linear fits were done using Prism (GraphPad) in-build routines.
Transgenic mouse strains
To generate the Fn1ΔRGD/ΔRGD mouse strain, part of the mouse FN gene was isolated from a phage library and subcloned, and the RGD sequence CGTGGGGAC, encoded by exon 30, was deleted by site-directed mutagenesis to generate the targeting vector (Fig. S2) for establishing mutant ES cells and finally Fn1ΔRGD/ΔRGD mice. The targeting vector contained a floxed neo cassette, which was removed by breeding the Fn1ΔRGD/ΔRGD mouse strain with a delete-Cre line. Mice and embryos were genotyped by Southern blot and genomic PCR (see Fig. S2) with DNA from tail or yolk sac biopsies using the primers 5′-CAAAGAAGACCCCAAGAGCA-3′ (forward) and 5′-ACAAGCCCTGGCCTTTAGTT-3′ (reverse). The Fn1RGE/RGE strain was previously established and genotyped as described previously (Takahashi et al., 2007). Time mattings were performed to recover E7.5, E8.5, and E9.5 embryos.
Fn1flox/+ mice (Sakai et al., 2001) were mated with Cre-deleter line to generate Fn1−/+ mice, which were subsequently intercrossed to generate Fn1−/− embryos.
Histology
Embryos were isolated and fixed in 4% phosphate-buffered PFA, embedded in tissue freezing medium (Leica), and sectioned at 7-µm thickness. Sections were immunostained and imaged with a Zeiss fluorescence microscope equipped with an Axiocam MRm (monochrome charge-coupled device) using AxioVision software and a Zeiss confocal LSM 780.
Cell lines
To establish Fn1ΔRGD/ΔRGD and Fn1+/+ fibroblast cell lines, blastocysts were flushed from the oviduct of heterozygous Fn1+/ΔRGD intercrosses at E3.5 and cultured in DMEM containing 20% FCS (Thermo Fisher Scientific), 1,000 U/ml leukemia inhibitory factor, and 7 × 10−4 β-mercaptoethanol to generate ES cells as described previously (Fässler et al., 1995). To differentiate ES cells into fibroblasts, medium was changed to DMEM supplemented with 10% FCS and 1% DMSO. Finally, the fibroblasts were immortalized by retroviral transduction of the SV-40 large T antigen and cloned.
Fn1-null fibroblasts, Fn1RGE/RGE fibroblasts, and pKO, pKO-β1, pKO-αv, and pKO-β1/αv integrin–expressing fibroblasts have previously been described and characterized (Schiller et al., 2013; Takahashi et al., 2007; Sakai et al., 2001). All fibroblast cell lines were grown in DMEM containing 10% FCS and 1% penicillin-streptomycin (Gibco).
Quantitative real-time PCR
RNA was isolated using RNeasy Kit (Qiagen). The sequences of the oligonucleotides used were 5′-TCCAAATCAGAAGCTCCAACAA-3′ for Itga8 forward, 5′-CGCTCACGAAATTGCTGTCA-3′ for Itga8 reverse, 5′-ACCCAGAAGACTGTGGATGG-3′ for GAPDH forward, and 5′-ACACATTGGGGGTAGGAACA-3′ for GAPDH reverse.
Surface integrin analysis by flow cytometry
Cells were detached with trypsin, and trypsinization was stopped with 100 µg/ml trypsin inhibitor (Calbiochem). Cells were washed with PBS and finally resuspended in FACS buffer (3% BSA in TBS). Cells were counted, and ∼5 × 105 cells were distributed in round-bottom wells of 96-well plates or in a round-bottom polystyrene tube, spun down, and resuspended in primary antibody or isotype control dilution (1:200) in FACS buffer. Cells were stained for 30 min on ice followed by two cycles of centrifugation and resuspension in FACS buffer to wash out the unbound antibody. When nonlabeled antibodies were used, an extra incubation step with labeled secondary antibody diluted 1:200 in FACS buffer was done (30 min on ice), followed by two cycles of washes. Stained cells were then resuspended in 300 µl FACS buffer and analyzed in a FACSCanto (BD Bioscience). During analysis, the same settings of forward scatter and side scatter were used to select size and granularity for each cell line, which were established using and nonlabeled sample of each cell line. These settings were maintained among replicates and experiments. Also, for comparison of different cell lines, the same laser and detector settings were used. Data were evaluated using FlowJo (BD Bioscience), and fluorescence intensity measurements were normalized using the intensity values of the isotope control for each antibody. Experiments were done three times with technical triplicates in each experiment.
FN purification
To purify cFN, wild-type and mutant fibroblast cells were grown to 100% confluence in DMEM containing 10% FCS. The medium was removed, and cells were washed three times with SRM 46.5% AIM-V (Life Technologies), 5% RPMI (Life Technologies), and 1% non-essential amino acid solution (Sigma-Aldrich) and left overnight. The SRM was discarded, replaced by fresh SRM, and collected every alternative day. cFNΔRGD, cFNRGE, and cFNRGD were purified from the conditioned SRMs using gelatin-Sepharose columns as described previously (Akiyama, 2013). Briefly, media were centrifuged to pellet down cell debris. Media were incubated with gelatin-Sepharose (GE Healthcare Life Sciences), columns were washed with TBS (0.15 M NaCl in 10 mM Tris-HCl, pH 7.4), and cFN was eluted with 4 M urea in TBS and dialyzed against TBS. Purified FN was analyzed by 8% SDS-PAGE and stained with Coomassie brilliant blue and by WB.
Antibodies and F-actin labels
For flow cytometry, the following antibodies conjugated to PE were used: hamster anti-mouse β1 integrin (1:200; 102207; BioLegend), rat anti-mouse α5 integrin (1:200; 557447; PharMingen), hamster anti-mouse β3 integrin (1:200; 12–0611; BD Biosciences), rat anti-mouse αv integrin (1:200; 551187; PharMingen), rat anti-mouse α4 integrin (1:200; 01271D; PharMingen), and unconjugated rat anti-mouse syndecan 4 (1:200; 550350; PharMingen) together with mouse anti-rat FITC (10094D; PharMingen).
The following primary antibodies were used for immunostaining (IF) and WB: rabbit anti-FN (1:600 for IF and 1:5,000 for WB; AB2033; Millipore), rabbit anti-Nidogen (1:1,500; generous gift from Dr. Rupert Timpl, Max Planck Institute of Biochemistry, Martinsried, Germany), mouse monoclonal anti-paxillin (1:300; 610051; BD Transduction Laboratories), rat anti-tubulin (1:3,000; MAB1318; Millipore), mouse monoclonal anti-mouse syndecan 1 (1:2,000; sc-12765; Santa Cruz Biotechnology); mouse monoclonal anti-mouse syndecan 2 (1:2,000; sc-376160; Santa Cruz Biotechnology); mouse monoclonal anti-mouse syndecan 3 (1:2,000; sc-398194; Santa Cruz Biotechnology); and mouse monoclonal anti-mouse syndecan 4 (1:2,000; sc-12766; Santa Cruz Biotechnology).
The secondary antibodies used for IF were: goat anti-rabbit conjugated with Cy3 (1:5,000; ab6939; Abcam), goat anti-rabbit conjugated with Alexa488 (1:500; A11008; Molecular Probes), donkey anti-mouse conjugated with Alexa647 (1:500; A31571; Molecular Probes), goat anti-rabbit Alexa647 (1:500; ab150083; Abcam), goat anti-mouse Alexa488 (1:500; A32723; Molecular Probes). The secondary antibodies used for WB were all diluted 1:1,000: goat anti-rabbit conjugated with horseradish peroxidase (172-1019; Bio-Rad), goat anti-mouse conjugated with horseradish peroxidase (170-6516; Bio-Rad) and goat anti-rat conjugated with horseradish peroxidase (5204-2504; Bio-Rad).
To label actin filaments, we used phalloidin labeled with either tetramethylrhodamine B isothiocyanate (1:500; P1951; Sigma-Aldrich) or fluorescein isothiocyanate (1:500; P5282; Sigma-Aldrich).
Adhesion assays to cFN
Glass coverslips of 18 × 18 mm were poly-maleic anhydride-1-octadecene treated (Prewitz et al., 2013), coated with 20 µg/ml purified mouse cFN for 1 h at 37°C, blocked for 30 min at RT with 3% BSA in PBS, and then seeded with 3 × 104 fibroblasts starved overnight in SRM, detached with 2 mM EDTA in PBS, and resuspended in DMEM. After 90 min of culture in DMEM, cells were fixed 10 min with 4% PFA, washed with PBS, permeabilized with 0.1% Triton X-100, and processed for IF. Images were taken at 22°C with a confocal Olympus FV1000 microscope with 40× (NA 1.30 oil) and 63× (NA 1.35 oil) objectives using the 488-, 594-, and 635-nm laser lines for fluorochrome detection. Spreading areas were calculated using the polygonal selection tool of ImageJ. Between 20 and 30 cells from three independent experiments were analyzed.
FN fibrillogenesis assay
Fn1ΔRGD/ΔRGD and Fn1+/+ ES cells were induced to grow in DMEM high glucose supplemented with 6% FCS (FN depleted), 1.4% methylcellulose, 450 µM monothioglycerol (Sigma-Aldrich), and 10 µg/ml human insulin (Sigma-Aldrich). Approximately 2.5 × 103 ES cells in 2 ml medium were seeded in a 35-mm dish and grown for 11 d without changing the medium, allowing formation of cell aggregates and assembly of their own secreted FN. Afterward, the ES cell aggregates were centrifuged and included into 15% gelatin to form a block. The gelatinized block was fixed with 4% PFA at 4°C for 1 h, washed with PBS, and cryoprotected with 7.5% (3 h) and 15% (overnight) sucrose. Blocks were embedded in tissue freezing medium (Leica) for cryosectioning and IF.
For FN assembly with Fn1+/+, Fn1ΔRGD/ΔRGD, and Fn1RGE/RGE fibroblasts, cells were prestarved overnight with SRM containing 1% FN-depleted calf serum and detached with 2 mM EDTA, and 15 × 103 cells were seeded onto glass coverslips coated with 10 µg/ml laminin (from Engelbreth-Holm-Swarm murine sarcoma BM; L2020; Sigma-Aldrich), VN (07180; Stem Cell), or collagen type I (PureCol, 5005; Advanced BioMatrix) for 1 h at RT and blocked for 1 h with 1% BSA-PBS. Cells were cultured for indicated times in SRM. Cells were then fixed with 4% paraformaldehyde and immunostained.
To block the FN heparin-binding sites, cells were seeded for 3 h in SRM, which was then replaced by SRM containing 0.1 mg/ml heparin (Sigma-Aldrich) for 24 or 72 h. MnCl2 was added to the cell culture medium for 3 h at a concentration of 100 µM. Slides were then mounted with Elvanol No-Fade mounting medium. Images were taken at 22°C with a Zeiss confocal LSM 780 microscope using a 100× (NA 1.46 oil) objective and ZEN2010 software for acquisition. For detection of Alexa Fluor, the 488-, 561-, and 633-nm laser lines were used. Images were processed, merged, and gamma adjusted in Fiji ImageJ (version 1.37).
Image analysis of fibrillar FN
Pictures were processed, analyzed, and merged in Fiji ImageJ. To measure the FN network in Fig. S5 a, the area of extracellular FN stained was quantified and normalized to the total area of each image and cell numbers. The amount of FN branches was quantified as described in Zhang et al. (2020). Briefly, pictures were set to binary 8 bits, the same threshold was set for all of them, noise was removed with a despeckle function and Skeleton plugin, and analysis was applied by Fiji ImageJ. The amount of FN branches was summarized and normalized to the total number of cells and image area.
DOC extraction of cell lysates
To differentiate between cell-soluble FN and FN assembled into an insoluble network, ∼5 × 103 cells were seeded on laminin (10 µg/ml)-coated 24-well plates and starved overnight with SRM. For heparin-treated cells, SRM was replaced after 3 h with SRM containing 0.1 mg/ml heparin. Cells and medium were collected after 24, 72, and 120 h of incubation and processed for WB. To this end, cells were washed with PBS twice, lysed with DOC buffer (2% DOC, 2 mM Tris-HCl, pH 8.8, and 2 mM EDTA) containing protease inhibitors (cOmplete; 05892970001; Roche) for 10 min on ice and then passed through a 25-gauge syringe and centrifuged at 14,000 rpm for 20 min. The clear supernatant was collected and kept as the soluble FN fraction (intracellular), while the pellet was washed with DOC buffer and centrifuged again to obtain the final pellet containing the cross-linked (fibrillar) FN network, which was resuspended in 1% SDS, 2 mM Tris-HCl, pH 8.8, and 2 mM EDTA containing protease inhibitors. Half of the DOC-soluble and the whole insoluble fractions were separated in an 8% SDS gel. The culture medium was centrifugated to pellet cell debris and directly loaded in the SDS gel to determine the soluble FN remaining in the culture medium. Proteins were transferred onto a polyvinylidene difluoride membrane and processed with antibodies. FN levels were measured using Fiji ImageJ and normalized to tubulin levels present in the cellular fraction.
Syndecan 1–4 knockdown
Syndecan mRNA translation was blocked in Fn1ΔRGD/ΔRGD cells by gene silencing with siRNA. 150 nM of predesigned siRNA pools (SMARTpool; Dharmacon) against syndecan 1 (Sdc1; reference no. 20969) and syndecan 4 (Sdc4; reference no. 20971) were lipofected into subconfluent, overnight-starved fibroblasts (Lipofectamine 3000; Invitrogen) seeded on laminin-coated coverslips in SRM during a 3-h period before transfection. The SRM was replaced 12 h after transfection, and cells were incubated for 96 h in SRM and then processed for IF. Syndecan levels were analyzed by WB to check gene silencing and compare with cells transfected with siRNAs containing an scr sequence.
Statistical analysis
In Fig. 1, Fig. S1, and Table S1, two-tailed Mann-Whitney tests were performed to test for significance using Prism (GraphPad). The n (number of cells or analyzed FD curves) and P values are indicated in the figure. In Figs. 2, 3, 4, and 5 and corresponding supplementary figures, data are presented as means ± SD of the mean (SEM). Each experiment was independently performed at least three times. The sample number (n) used for statistical testing is indicated in the corresponding figure legend. P values are indicated in each figure (*, P < 0.05; **, P < 0.01; and ***, P < 0.001). All P values are from two-tailed unpaired t tests, and Bonferroni correction was performed for tests with multiple pairwise comparisons. Statistical analysis was performed using Prism (GraphPad Software). Data distribution was assumed to be normal, but this was not formally tested.
Online supplemental material
Fig. S1 shows the AFM-based SCFS setup that we used to quantify adhesion of fibroblasts to different full-length cFNs and FNIII7-10 fragments. It also shows the relative frequency distribution of unbinding events in experiments with a contact time of ~300 ms and the adhesion strengthening dynamics for each condition. Fig. S2 illustrates the strategy to generate Fn1ΔRGD/ΔRGD mice and their histological analysis by immunofluorescence to different ECM proteins (FN, laminin, and collagen type IV). Fig. S3 shows surface β1, β3, α5, αv, and α4 integrins and syndecan 4 levels and Itga8 expression in the different cell lines. Fig. S4 presents FNRGD and FNΔRGD assembly by cells cultured on VN- or collagen I–coated glass. Fig. S5 shows FNRGD, FNRGE, and FNΔRGD assembly and quantification in the presence of heparin and/or Mn+2. It also shows the levels of syndecans in Fn1ΔRGD/ΔRGD and Fn1+/+ cells after knockdown with siRNAs and the quantification of FN fibrils assembled by the cells on each condition. Table S1 displays the statistical comparison of adhesion-strengthening dynamics of pKO-αv, pKO-β1, pKO-αv/β1, and pKO cell lines to FNIII7-10ΔRGD, FNIII7-10RGD, or FNIII7-10RGE fragments.
Acknowledgments
We thank Dr. Roy Zent for critically reading and commenting on the manuscript.
This work was supported by the Max Planck Society (M. Costell and R. Fässler), the European Research Council (grant 810104 to R. Fässler), and the Swiss National Science Foundation (grant 31003A_182587/1 to D.J. Müller). M. Benito-Jardón was supported by a contract from the Conselleria Valenciana d’Educació i Ciència, Spain.
The authors declare no competing financial interests.
Author contributions: Conceptualization, R. Fässler and M. Costell; Supervision, M. Moser, D.J. Müller, R. Fässler, and M. Costell; Investigation, M. Benito-Jardón, N. Strohmeyer, S. Ortega-Sanchís, M. Bharadwaj, M. Moser, and M. Costell; Formal analysis, M. Benito-Jardón, N. Strohmeyer, M. Moser, and M. Costell; Writing – original draft, M. Benito-Jardón, N. Strohmeyer, R. Fässler, and M. Costell; Writing – review and editing, M. Benito-Jardón, N. Strohmeyer, M. Moser, D.J. Müller, R. Fässler, and M. Costell; Project administration and funding acquisition, D.J. Müller, R. Fässler, and M. Costell.

