


We identify downregulation of genes related to cell–ECM interactions and TGFβ signaling in FPRMS. We confirm that TGFβ signaling enhances cell–ECM interactions in FNRMS, utilizing confocal reflection microscopy to assess ECM remodeling, and a live-cell sensor to quantitatively assess TGFβ signaling. We also show that PAX3-FOXO1 increases NOS1 expression, stimulating nitric oxide synthesis, which inhibits TGFβ signaling and reduces cell–ECM interactions. We suggest that PAX3-FOXO1 reprograms ECM anchorage dependence by suppressing cell–ECM interactions. The fusion gene can determine sensitivity to growth inhibition via targeted disruption of cell–ECM interactions or TGFβ signaling. Reduced anchorage reliance by the gene may allow cells to survive in circulation and enhance FPRMS metastatic potential.
Introduction
Rhabdomyosarcoma (RMS) is an aggressive pediatric malignancy and shares histopathological features with developing skeletal muscle. RMS consists of two major subtypes: fusion-positive RMS (FPRMS), characterized by a chromosomal translocation that results in the oncogenic PAX3-FOXO1 fusion gene, and fusion-negative RMS (FNRMS), which is primarily driven by activating mutations in RTK/RAS pathways (Skapek et al., 2019). Despite distinct molecular differences and clinical outcomes between FPRMS and FNRMS, current treatment protocols do not differentiate between the two subtypes. This is largely due to the lack of understanding of their differential molecular features, which could potentially serve as vulnerable nodes for targeted therapies (Chen et al., 2019; Zarrabi et al., 2023).
RMS may arise due to abnormalities in developmental pathways that regulate skeletal muscle determination (Zarrabi et al., 2023). RMS cells typically originate from myogenic progenitors undergoing inappropriate differentiation arrest at specific developmental stages, leading to persistent proliferation (Wei et al., 2022). Specifically, there is a correlation between the developmental stage where differentiation arrest occurs and the emergence of subtype-specific core signatures, which shapes the different signaling pathways and functional characteristics between FPRMS and FNRMS. Furthermore, the aberrant transcription factor PAX3-FOXO1 may reshape the transcriptional landscape and hijack signaling pathways related to oncogenic transformation. The extracellular matrix (ECM), which provides a physical scaffold, plays a crucial role in regulating cell survival and growth through anchorage-dependent signaling pathways (Cox, 2021; Pickup et al., 2014; Winkler et al., 2020). In normal myogenic cells, cell–ECM interactions ensure that cells remain anchored to their niche, receiving signals that regulate myogenic differentiation (Choi et al., 2018; Melo et al., 1996; Osses and Brandan, 2002; Rozario and DeSimone, 2010; Thorsteinsdóttir et al., 2011). The cell-intrinsic differences between FNRMS and FPRMS prompt a reprogramming of their interactions with the ECM, which further fine-tunes maintenance of appropriate myogenic differentiation. Moreover, the anchorage dependence mediated by the ECM may hinder metastasis by impeding the dissemination of tumor cells through the circulatory system. This is achieved by inhibiting cell survival when there is loss of ECM attachment and weakening of cell–ECM interactions (Huh et al., 2023). It remains unclear whether and how PAX3-FOXO1 fusion status alters transcriptional and signaling networks to reprogram cell–ECM interactions and anchorage dependence, potentially leading to subtype-specific molecular vulnerabilities and metastatic propensity. Understanding the molecular mechanisms by which the fusion status of RMS may reprogram anchorage dependence is critical for discerning their different features associated with patient outcomes and developing targeted therapies for each RMS subtype.
Herein, we leverage single-cell RNA sequencing (scRNA-seq) to demonstrate that FNRMS and FPRMS signatures emerge in different cell populations at distinct stages in the trajectory of normal human skeletal myogenesis. Using a multimodal approach that combines transcriptomic and functional assays, we show differential cell–ECM interactions between FNRMS and FPRMS. We also demonstrate that PAX3-FOXO1 disrupts cell–ECM interactions to promote anchorage independence by suppressing TGFβ signaling through increased NOS1-mediated synthesis of nitric oxide (NO). We reveal that targeting cell–matrix interactions by disrupting focal adhesion proteins, such as FAK and Src, or TGFβ signaling could be sufficient to induce growth inhibition in the anchorage-dependent FNRMS subtype. The knockdown of PAX3-FOXO1 in FPRMS cells reprograms anchorage dependency, thereby resensitizing FPRMS to the growth-inhibitory effects of focal adhesion targeting of cell–ECM interactions. We also found that PAX3-FOXO1 may promote an “adhesion-to-suspension transition (AST)” (Huh et al., 2023), enhancing the survival capacity of circulating tumor cells (CTCs) and increasing metastatic potential, which may, at least partially, explain the higher metastatic propensity of FPRMS compared with FNRMS. Our findings could provide insights into how the PAX3-FOXO1 status shapes cell–ECM interactions to modulate metastatic potential and reveal molecular vulnerabilities that may inform subtype-specific targeted therapies to improve clinical outcomes.
Results
FPRMS- and FNRMS-like subtypes are enriched in distinct stages of skeletal muscle development
Wei et al. demonstrated that the distinct developmental stages at which myogenic differentiation becomes arrested exhibit RMS subtype-specific transcriptional programs (Wei et al., 2022). We hypothesized that these distinct stages of arrested development may reflect divergent characteristics, including differences in signaling pathways and phenotypes. To examine this hypothesis, we reanalyzed scRNA-seq data of human limb muscle tissues obtained from embryonic (6–8 wk), fetal (9–18 wk), and juvenile/adult stages (7–42 years) (GSE147457) (Fig. 1 a and Fig. S1 a) (Xi et al., 2020). The Leiden clustering of the scRNA-seq data (Traag et al., 2019) identified 10 distinct subgroups based on the similarities in gene expression (Fig. 1 b and Table S1) and differentially expressed genes (DEGs) of each subgroup (Table S2).

FPRMS and FNRMS mirror different subgroups in myogenic development. (a) t-SNE rendering scRNA-seq of skeletal muscle tissues obtained from human prenatal development (from 6 to 18 wk) and juveniles/adults (from 7 to 42 years). scRNA-seq data are from GSE147457. (b) Clusters identified by the Leiden algorithm, representing subgroups in myogenic development. (c) Population probability of cells in each subgroup of each week’s tissue sample. (d) Expression of FNRMS signatures (FNRMS score, top) and FPRMS signatures (FPRMS score, bottom) in the subgroups. (e) GSEA of highly expressed genes in Subgroups 3, 4, and 5 with FNRMS and FPRMS signatures as references, suggesting molecular similarity of Subgroup 3 with FPRMS and the similarity of Subgroups 4 and 5 with FNRMS. A positive ES and normalized ES indicate the similarity of inquired genes with the reference gene set. (f–h) GO analysis of (f) significantly low-expressed genes in Subgroup 3 compared with all other subgroups, (g) highly expressed genes in Subgroup 4 compared with all other groups, and (h) highly expressed genes in FNRMS compared with FPRMS, using GO Function gene sets as the reference gene set. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. Gene expression data of FPRMS and FNRMS are from GSE114621. t-SNE, t-distributed stochastic neighboring embedding; ES, enrichment score.
FPRMS and FNRMS mirror different subgroups in myogenic development. (a) t-SNE rendering scRNA-seq of skeletal muscle tissues obtained from human prenatal development (from 6 to 18 wk) and juveniles/adults (from 7 to 42 years). scRNA-seq data are from GSE147457. (b) Clusters identified by the Leiden algorithm, representing subgroups in myogenic development. (c) Population probability of cells in each subgroup of each week’s tissue sample. (d) Expression of FNRMS signatures (FNRMS score, top) and FPRMS signatures (FPRMS score, bottom) in the subgroups. (e) GSEA of highly expressed genes in Subgroups 3, 4, and 5 with FNRMS and FPRMS signatures as references, suggesting molecular similarity of Subgroup 3 with FPRMS and the similarity of Subgroups 4 and 5 with FNRMS. A positive ES and normalized ES indicate the similarity of inquired genes with the reference gene set. (f–h) GO analysis of (f) significantly low-expressed genes in Subgroup 3 compared with all other subgroups, (g) highly expressed genes in Subgroup 4 compared with all other groups, and (h) highly expressed genes in FNRMS compared with FPRMS, using GO Function gene sets as the reference gene set. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. Gene expression data of FPRMS and FNRMS are from GSE114621. t-SNE, t-distributed stochastic neighboring embedding; ES, enrichment score.

scRNA sequencing of skeletal muscle tissues reveals developmental alignment with RMS subtypes. ( a) t-SNE rendering scRNA-seq of skeletal muscle tissues obtained from human prenatal development and juveniles/adults from GSE147457, indicating cells from different samples. (b) PAGA, reconciling clustering with myogenic differentiation trajectory inference. (c) GO analysis of significantly highly expressed genes in Subgroup 3 compared with all other subgroups, indicating that Subgroup 3 is close to the skeletal–muscular system. (d) t-SNE rendering scRNA-seq of skeletal muscle tissues showing the expression level of PAX7 (myoblast marker) in each cell. (e) GO analysis of significantly highly expressed genes in Subgroup 4 compared with all other subgroups, indicating that Subgroup 4 is close to the vascular system. (f) t-SNE rendering scRNA-seq of skeletal muscle tissues showing the expression level of DLK1 (vascular marker) in each cell. In c and e, GO Process gene sets were used as the reference gene set. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. (g) Plot of Fetal Score versus Embryonic Score of the identified subgroups. Higher Fetal Scores and lower Embryonic Scores indicate a later stage in differentiation. (h) Decreasing Embryonic and increasing Fetal Scores from Subgroups 1–5. (i) Histogram of Embryonic (left) and Fetal (right) Scores in each subgroup. (j) Histogram of FNRMS (left) and FPRMS (right) scores in each subgroup. In i and j, dashed lines indicate averages of each score of each subgroup. t-SNE, t-distributed stochastic neighboring embedding; PAGA, partition-based graph abstraction.

scRNA sequencing of skeletal muscle tissues reveals developmental alignment with RMS subtypes. ( a) t-SNE rendering scRNA-seq of skeletal muscle tissues obtained from human prenatal development and juveniles/adults from GSE147457, indicating cells from different samples. (b) PAGA, reconciling clustering with myogenic differentiation trajectory inference. (c) GO analysis of significantly highly expressed genes in Subgroup 3 compared with all other subgroups, indicating that Subgroup 3 is close to the skeletal–muscular system. (d) t-SNE rendering scRNA-seq of skeletal muscle tissues showing the expression level of PAX7 (myoblast marker) in each cell. (e) GO analysis of significantly highly expressed genes in Subgroup 4 compared with all other subgroups, indicating that Subgroup 4 is close to the vascular system. (f) t-SNE rendering scRNA-seq of skeletal muscle tissues showing the expression level of DLK1 (vascular marker) in each cell. In c and e, GO Process gene sets were used as the reference gene set. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. (g) Plot of Fetal Score versus Embryonic Score of the identified subgroups. Higher Fetal Scores and lower Embryonic Scores indicate a later stage in differentiation. (h) Decreasing Embryonic and increasing Fetal Scores from Subgroups 1–5. (i) Histogram of Embryonic (left) and Fetal (right) Scores in each subgroup. (j) Histogram of FNRMS (left) and FPRMS (right) scores in each subgroup. In i and j, dashed lines indicate averages of each score of each subgroup. t-SNE, t-distributed stochastic neighboring embedding; PAGA, partition-based graph abstraction.
We mapped the distribution enrichment of the subgroups to the developmental trajectory from embryo to adult (Fig. 1 c). Subgroups 1–5, substantially spanning from embryonic and fetal status, share proximal connections within the trajectory (Fig. S1 b). We discovered that Subgroups 1 and 2, enriched in early embryonic stages (Weeks 6–7.25), demonstrated higher expression of genes indicative of early development—embryo (Subgroup 1) and heart/head/axon guidance (Subgroup 2)—through gene ontology (GO) analysis. Subgroups 3–5, which may represent later embryonic or fetal stages relative to Subgroups 1 and 2, exhibit molecular features indicating more differentiated tissues: muscle skeletal system (Subgroup 3) and fetus and cardiovascular system (Subgroups 4 and 5) (Fig. S1, c–f and Table S3). Additionally, for each subgroup we assessed their Fetal Score: the expression levels of highly expressed genes in Subgroup 5; and their Embryonic Score: those of highly expressed genes in Subgroup 1. Increased Fetal Scores and reduced Embryonic Scores of Subgroups 3–5 compared with Subgroups 1 and 2 suggest that they represent later developmental stages (Fig. S1, g–i).
Next, we assessed the enrichment of FNRMS and FPRMS core signatures, defined by Gryder et al. (2017) (Table S4), in the identified subgroups (Fig. 1 d). Interestingly, we discovered that the FPRMS core signature was enriched in Subgroup 3, while the FNRMS core signature was enriched primarily in Subgroups 4 and 5. Gene set enrichment analysis (GSEA) shows robust upregulation of FPRMS signatures in Subgroup 3 with a concurrent depletion of FNRMS signatures (Fig. 1 e and Fig. S1 j). A reverse trend is observed for Subgroup 4, showing depletion of FPRMS signatures and upregulation of FNRMS signatures. Subgroup 5 shows a more modest upregulation in FNRMS signatures compared with Subgroup 4 and, similarly, a less pronounced depletion of FPRMS signatures. Wei et al. discovered the enrichment of FPRMS core signatures at the stage where embryonic muscle transits to fetal muscle at Weeks 7–7.5 (Wei et al., 2022). Our analysis consistently exhibited that FPRMS-like Subgroup 3, expressing skeletal muscle–related genes, substantially started to emerge around Week 7.5 (Table S3 and Fig. 1 c). Furthermore, FNRMS-like Subgroups 4 and 5 exhibited the expression of genes defining the vascular system (Table S3), in line with previous reports indicating an endothelial cell-of-origin for FNRMS (Drummond et al., 2018). Conversely, higher expression of genes indicating skeletal muscle in Subgroup 3 may reflect myogenic reprogramming driven by the PAX3-FOXO1 fusion gene of FPRMS (Searcy et al., 2023). These results support the idea that the emergence of RMS subtype–specific transcriptional signatures coincides with distinct stages during normal skeletal muscle development.
To test whether the distinct molecular features of the most FPRMS-like Subgroup 3 and the most FNRMS-like Subgroup 4 mirror underlying differences between RMS subtypes, we investigated DEGs in each relevant subgroup (Tables S2, S3, S4, S5, and S6; and Fig. S1 j) followed by GO analysis. Intriguingly, functional annotation of inquired DEGs in Subgroups 3 and 4 revealed genes involved in cell–ECM interactions to be strongly downregulated for Subgroup 3 (Fig. 1 f). In contrast, cell–ECM interaction gene sets were strongly upregulated in Subgroup 4 (Fig. 1 g). Head-to-head comparison between the two RMS subgroups shows gene sets involved in cell–ECM interactions (e.g., ECM constituents, integrin binding, heparin binding) to be enriched in FNRMS relative to FPRMS (Fig. 1 h), consistent with the similarity of the subgroups corresponding to each RMS subtype. Thus, we hypothesize that FNRMS cells have higher cell–ECM interactions than FPRMS cells.
FNRMS shows higher cell–ECM interactions than FPRMS
To evaluate our hypothesis of higher cell–ECM interactions in FNRMS than in FPRMS, we performed immunofluorescence (IF) staining of F-actin cytoskeleton and phosphorylated focal adhesion kinase (pFAK) in FNRMS (RD and SMS-CTR) and FPRMS (Rh30 and Rh41) cell lines. We found that FNRMS exhibits prominent stress fibers necessary for establishing and maintaining cell–ECM interactions, as opposed to FPRMS, which is largely devoid of cytoskeletal stress fiber bundles. pFAK staining of FNRMS also showed increased focal adhesion formation, representing enhanced cell–ECM interactions (Fig. 2 a and Fig. S2 a). These differences could arise from cell-intrinsic differences between RMS subtypes in signaling pathways regulating cytoskeletal organization and focal adhesion formation, which determine cell–ECM interactions and, accordingly, related cell phenotypes (Berrier and Yamada, 2007; Wozniak et al., 2004; Zhao and Guan, 2009). Indeed, the higher cell–ECM interactions of FNRMS relative to FPRMS were further corroborated by an increased cell spreading area in FNRMS (Fig. 2 b).

FNRMS shows higher cell–ECM interaction than FPRMS. (a) IF images of FNRMS (RD and SMS-CTR) and FPRMS (Rh30 and Rh41) cell lines, stained for F-actin (with phalloidin) and pFAK, displaying focal adhesion. (b) Cell spreading areas for each RMS subtype cell line. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (minimum n = 36). (c) Initial 3 h collagen fiber displacement near spheroids, monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (d) Quantification of collagen fiber displacement (n = 5). (e) Collagen fiber alignment 16 h after gelation with spheroids. Dashed lines denote spheroid edges. Arrows point to the fibers aligned perpendicularly to FNRMS spheroid edges, while asterisks highlight the fibers aligned parallel to FPRMS spheroid edges. (f) Distribution of collagen fiber orientation around RMS cell spheroids. Angles at 90° represent perpendicular alignment to spheroid edges; 0° represents parallel alignment. Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Dots represent individual spheroid values (minimum n = 4). Asterisks (*) denote statistical differences from RD spheroid values, and pounds (#) indicate differences from SMS-CTR spheroid values. In b, d, and f, error bars represent SE and statistical significance was assessed using Wilcoxon’s rank sum test (b) or two-sided Student’s t test: *P < 0.05, **P < 0.01, ###P < 0.005, ***P < 0.005, and ****P < 0.001.
FNRMS shows higher cell–ECM interaction than FPRMS. (a) IF images of FNRMS (RD and SMS-CTR) and FPRMS (Rh30 and Rh41) cell lines, stained for F-actin (with phalloidin) and pFAK, displaying focal adhesion. (b) Cell spreading areas for each RMS subtype cell line. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (minimum n = 36). (c) Initial 3 h collagen fiber displacement near spheroids, monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (d) Quantification of collagen fiber displacement (n = 5). (e) Collagen fiber alignment 16 h after gelation with spheroids. Dashed lines denote spheroid edges. Arrows point to the fibers aligned perpendicularly to FNRMS spheroid edges, while asterisks highlight the fibers aligned parallel to FPRMS spheroid edges. (f) Distribution of collagen fiber orientation around RMS cell spheroids. Angles at 90° represent perpendicular alignment to spheroid edges; 0° represents parallel alignment. Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Dots represent individual spheroid values (minimum n = 4). Asterisks (*) denote statistical differences from RD spheroid values, and pounds (#) indicate differences from SMS-CTR spheroid values. In b, d, and f, error bars represent SE and statistical significance was assessed using Wilcoxon’s rank sum test (b) or two-sided Student’s t test: *P < 0.05, **P < 0.01, ###P < 0.005, ***P < 0.005, and ****P < 0.001.
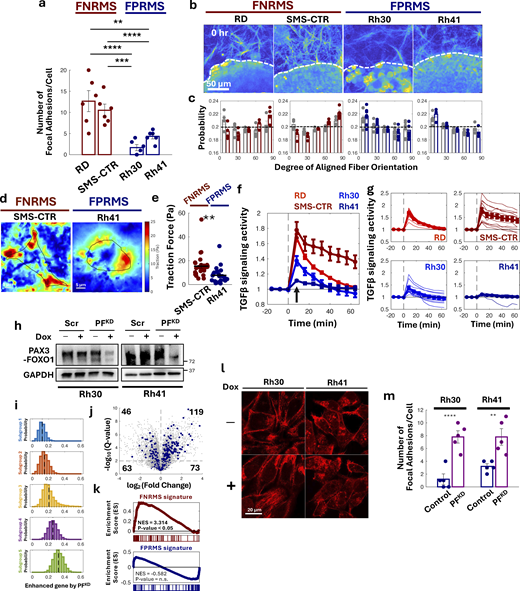
PAX3-FOXO1 reduces cell-ECM interaction.(a) Number of focal adhesions in FNRMS and FPRMS cells. Each dot indicates the number of focal adhesions in each cell (n = 6). (b) Collagen fiber alignment 0 h after gelation with spheroids. Dashed lines denote spheroid edges. (c) Distribution of collagen fiber orientation around RMS cell spheroids at 0 h (gray) and 16 h (color, Fig. 2 f). Angles at 90° represent perpendicular alignment to spheroid edges; 0° represents parallel alignment. Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Dots represent individual spheroid values (minimum n = 4). Error bars are SE. (d) Traction force microscopic images of SMS-CTR (FNRMS) and Rh41 (FPRMS) cells. (e) Traction force of SMS-CTR (FNRMS) and Rh41 (FPRMS) cells. Each dot indicates the value of each cell (n = 5). Time course TGFβ signaling activity of RMS cell lines, assessed by dynamic SMAD3 nuclear localization (n = 14). (f and g) Averages of each cell line and (g) faint lines indicating each cell in the cell line. Dashed lines indicate the time point of TGFβ treatment. An arrow indicates the value of Fig. 3 c. (h) PAX3-FOXO1 expression levels of scramble and PFKD cells with and without Dox induction. (i) Histogram of the average expression levels of genes enhanced by PFKD in each cell of each subgroup identified by scRNA-seq of developing skeletal muscle tissues. Dashed lines indicate averages of each subgroup. (j) Volcano plot illustrating gene expression changes induced by PFKD in FPRMS cells. Positive log2 (fold change) values indicate genes upregulated by PFKD. The horizontal dashed line represents the statistical threshold (P = 0.01), with genes above this line exhibiting statistically significant changes. Blue dots denote highly expressed genes in Subgroup 4, and the numbers indicate the count of these genes in each section. Among the 304 highly expressed genes in Subgroup 4, 119 genes showed a statistically significant increase in expression by PFKD. (k) GSEA of genes enhanced by PFKD with FNRMS (top) and FPRMS (bottom) signatures as the reference gene sets. The genes enhanced by PFKD exhibit statistically significant overlap with FNRMS signatures, not FPRMS signatures (n.s. = not significant). (l) F-actin stained by phalloidin in scramble FPRMS cells displayed cortical actin, indicating reduced cell–ECM interaction. (m) Number of focal adhesions in PFKD FPRMS and control cells. Each dot indicates the number of focal adhesions in each cell (n = 5). In a, e, and m, lines are averages of each group. Error bars are SE. Statistical significance was assessed using two-sided Student’s t tests or Wilcoxon’s rank sum test (e): **P < 0.01, ***P < 0.005, and ****P < 0.001. Dox, doxycycline. Source data are available for this figure: SourceData FS2.

PAX3-FOXO1 reduces cell-ECM interaction.(a) Number of focal adhesions in FNRMS and FPRMS cells. Each dot indicates the number of focal adhesions in each cell (n = 6). (b) Collagen fiber alignment 0 h after gelation with spheroids. Dashed lines denote spheroid edges. (c) Distribution of collagen fiber orientation around RMS cell spheroids at 0 h (gray) and 16 h (color, Fig. 2 f). Angles at 90° represent perpendicular alignment to spheroid edges; 0° represents parallel alignment. Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Dots represent individual spheroid values (minimum n = 4). Error bars are SE. (d) Traction force microscopic images of SMS-CTR (FNRMS) and Rh41 (FPRMS) cells. (e) Traction force of SMS-CTR (FNRMS) and Rh41 (FPRMS) cells. Each dot indicates the value of each cell (n = 5). Time course TGFβ signaling activity of RMS cell lines, assessed by dynamic SMAD3 nuclear localization (n = 14). (f and g) Averages of each cell line and (g) faint lines indicating each cell in the cell line. Dashed lines indicate the time point of TGFβ treatment. An arrow indicates the value of Fig. 3 c. (h) PAX3-FOXO1 expression levels of scramble and PFKD cells with and without Dox induction. (i) Histogram of the average expression levels of genes enhanced by PFKD in each cell of each subgroup identified by scRNA-seq of developing skeletal muscle tissues. Dashed lines indicate averages of each subgroup. (j) Volcano plot illustrating gene expression changes induced by PFKD in FPRMS cells. Positive log2 (fold change) values indicate genes upregulated by PFKD. The horizontal dashed line represents the statistical threshold (P = 0.01), with genes above this line exhibiting statistically significant changes. Blue dots denote highly expressed genes in Subgroup 4, and the numbers indicate the count of these genes in each section. Among the 304 highly expressed genes in Subgroup 4, 119 genes showed a statistically significant increase in expression by PFKD. (k) GSEA of genes enhanced by PFKD with FNRMS (top) and FPRMS (bottom) signatures as the reference gene sets. The genes enhanced by PFKD exhibit statistically significant overlap with FNRMS signatures, not FPRMS signatures (n.s. = not significant). (l) F-actin stained by phalloidin in scramble FPRMS cells displayed cortical actin, indicating reduced cell–ECM interaction. (m) Number of focal adhesions in PFKD FPRMS and control cells. Each dot indicates the number of focal adhesions in each cell (n = 5). In a, e, and m, lines are averages of each group. Error bars are SE. Statistical significance was assessed using two-sided Student’s t tests or Wilcoxon’s rank sum test (e): **P < 0.01, ***P < 0.005, and ****P < 0.001. Dox, doxycycline. Source data are available for this figure: SourceData FS2.
To verify the differential cell–ECM interactions more functionally, we utilized confocal reflection microscopy (CRM), a novel microscopic technique to characterize the spatial and temporal dynamics of cell–ECM interactions within a three-dimensional (3D) collagen matrix (Artym and Matsumoto, 2010; Brightman et al., 2000). Using the hanging drop method (Foty, 2011), we generated tumor spheroids of FPRMS and FNRMS cell lines and embedded them in 3D gels of collagen type I, the most abundant protein in the interstitial tissue, before initiating time-lapse CRM imaging. We quantified collagen fiber displacement and alignment as metrics to assess the extent to which the spheroids physically interact with and remodel the collagen matrix through particle image velocimetry (PIV) (Petitjean et al., 2010; Raffel et al., 2018). We found that collagen fibers near the FNRMS spheroids exhibit a higher displacement magnitude compared with those near FPRMS spheroids (Fig. 2, c and d; and Video 1). We next assessed collagen fiber alignment via matrix remodeling at the boundary edge of the tumor spheroids. Fiber orientation distribution analysis showed a higher proportion of perpendicularly aligned collagen fibers (relative to the edge) near FNRMS spheroids compared with FPRMS spheroids (Fig. 2, e and f; and Fig. S2, b and c). The perpendicular alignment of ECM collagen fibers implies that FNRMS cells exert pulling forces reorganizing ECM topographically, indicating stronger cell–ECM interactions (Provenzano et al., 2006). In contrast, a more parallel-like fiber alignment was noted for FPRMS resulting from the passive radial expansion of the spheroid, largely devoid of active pulling forces and reduced cell–ECM interactions. Additionally, we measured the traction forces of SMS-CTR (FNRMS) and Rh41 (FPRMS) cells using traction force microscopy (TFM) (Fig. S2, d and e) (Mittal and Han, 2021), through which SMS-CTR showed elevated traction force compared with Rh41 cells. Considering the traction force of cells is indicative of cell–ECM interaction (Liu et al., 2017; Style et al., 2014), these results align well with the higher cell–ECM interaction observed in FNRMS compared with FPRMS. Collectively, these results indicate that FNRMS exhibits stronger cell–ECM interaction relative to FPRMS.
Time-lapse CRM imaging of tumor spheroids of FNRMS (RD and SMS-CTR) and FPRMS (Rh30 and Rh41) cell lines embedded in 3D gels of collagen type I (related toFig. 2 c ). Images were taken every 3 h for 21 h (8 frames per second). Scale bar = 50 µm.
Time-lapse CRM imaging of tumor spheroids of FNRMS (RD and SMS-CTR) and FPRMS (Rh30 and Rh41) cell lines embedded in 3D gels of collagen type I (related toFig. 2 c ). Images were taken every 3 h for 21 h (8 frames per second). Scale bar = 50 µm.
TGFβ signaling stimulates cell–ECM interactions in FNRMS
To identify candidate pathways responsible for the differential cell–ECM interaction between the two RMS subtypes, we conducted GO analysis of the DEGs in Subgroups 3 and 4, using the KEGG database as a reference data set. Interestingly, the TGFβ signaling pathway was downregulated in FPRMS-like Subgroup 3 and upregulated in FNRMS-like Subgroup 4 (Fig. 3 a). TGFβ regulates ECM synthesis and remodeling, promoting cell–ECM interaction (Ignotz and Massagué, 1986; Verrecchia and Mauviel, 2002). The activation of TGFβ signaling involves the canonical pathway where TGFβ binds to its cognate receptor on the cell surface, leading to the activation of downstream SMAD proteins. These SMAD proteins, including SMAD3, translocate into the nucleus and regulate the transcription of target genes involved in cytoskeletal organization and ECM remodeling (Massagué, 2012; Richardson et al., 2023). A previous report demonstrated that FNRMS displayed a higher response to TGFβ than FPRMS, implicating higher TGFβ signaling activity in FNRMS (Schmitt-Ney and Camussi, 2015). Concomitantly, GO analysis on DEGs in FNRMS-like Subgroup 4 and FNRMS exhibited higher enrichment of genes associated with TGFβ signaling compared with each FPRMS counterpart (Fig. 3 a). Thus, we hypothesized that FNRMS has higher activity of TGFβ signaling relative to FPRMS, resulting in its heightened cell–ECM interaction.

FNRMS exhibits higher TGFβ signaling activity than FPRMS. (a) GO analysis of significantly low-expressed genes in Subgroup 3, reflecting FPRMS (top); highly expressed genes in Subgroup 4, reflecting FNRMS (middle); and genes highly expressed in FNRMS compared with FPRMS (bottom), using KEGG gene sets as the reference gene sets. We highlight TGFβ signaling as a suppressed signaling pathway in Subgroup 3, an activated signaling pathway in Subgroup 4, and an enhanced signaling pathway in FNRMS compared with FPRMS. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. Gene expression data of FPRMS and FNRMS are from GSE114621. (b) Dynamic SMAD3 nuclear localization upon TGFβ treatment (4 ng/ml) in FNRMS and FPRMS cell lines, monitored using NG-Smad3 live-cell sensor. (c) TGFβ signaling activity, quantified by the ratio of intensity in nuclear SMAD3 after TGFβ treatment to before the treatment, of FNRMS and FPRMS cell lines. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (n = 14). (d) Higher expression of collagen type I (Col 1) and CTGF (TGFβ signaling downstream molecules) in FNRMS compared with FPRMS, analyzed by immunoblotting. (e) IF images of RD and SMS-CTR (FNRMS), with and without the treatment of a TGFβ inhibitor (LY2109761, 100 nM, 12 h), stained for F-actin (with phalloidin) and vinculin to show focal adhesion. (f) Cell spreading areas for FNRMS and FPRMS cell lines after treatment with TGFβ inhibitor in e. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (n = 30). (g) Collagen fiber displacement near FNRMS spheroids with and without the treatment of LY2109761 (100 nM), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by FNRMS spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (h) Quantification of the collagen fiber displacements surrounding FNRMS spheroids with and without TGFβ inhibition (n = 5). (i) Collagen fiber alignment 16 h after gelation with FNRMS spheroids, with and without the treatment of LY2109761. Dashed lines denote FNRMS spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges, while asterisks highlight the fibers aligned parallel to the spheroid edges. (j) Distribution of collagen fiber orientation around FNRMS spheroids with and without TGFβ inhibition. Dots represent individual spheroid values (minimum n = 4). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Asterisks (*) denote statistical differences of values between spheroids with and without LY2109761. In c, f, h, and j, error bars represent SE and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (h): *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. Source data are available for this figure: SourceData F3.
FNRMS exhibits higher TGFβ signaling activity than FPRMS. (a) GO analysis of significantly low-expressed genes in Subgroup 3, reflecting FPRMS (top); highly expressed genes in Subgroup 4, reflecting FNRMS (middle); and genes highly expressed in FNRMS compared with FPRMS (bottom), using KEGG gene sets as the reference gene sets. We highlight TGFβ signaling as a suppressed signaling pathway in Subgroup 3, an activated signaling pathway in Subgroup 4, and an enhanced signaling pathway in FNRMS compared with FPRMS. Color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. Gene expression data of FPRMS and FNRMS are from GSE114621. (b) Dynamic SMAD3 nuclear localization upon TGFβ treatment (4 ng/ml) in FNRMS and FPRMS cell lines, monitored using NG-Smad3 live-cell sensor. (c) TGFβ signaling activity, quantified by the ratio of intensity in nuclear SMAD3 after TGFβ treatment to before the treatment, of FNRMS and FPRMS cell lines. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (n = 14). (d) Higher expression of collagen type I (Col 1) and CTGF (TGFβ signaling downstream molecules) in FNRMS compared with FPRMS, analyzed by immunoblotting. (e) IF images of RD and SMS-CTR (FNRMS), with and without the treatment of a TGFβ inhibitor (LY2109761, 100 nM, 12 h), stained for F-actin (with phalloidin) and vinculin to show focal adhesion. (f) Cell spreading areas for FNRMS and FPRMS cell lines after treatment with TGFβ inhibitor in e. Each dot represents the value of a corresponding cell, and lines are averages of each cell line (n = 30). (g) Collagen fiber displacement near FNRMS spheroids with and without the treatment of LY2109761 (100 nM), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by FNRMS spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (h) Quantification of the collagen fiber displacements surrounding FNRMS spheroids with and without TGFβ inhibition (n = 5). (i) Collagen fiber alignment 16 h after gelation with FNRMS spheroids, with and without the treatment of LY2109761. Dashed lines denote FNRMS spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges, while asterisks highlight the fibers aligned parallel to the spheroid edges. (j) Distribution of collagen fiber orientation around FNRMS spheroids with and without TGFβ inhibition. Dots represent individual spheroid values (minimum n = 4). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). Asterisks (*) denote statistical differences of values between spheroids with and without LY2109761. In c, f, h, and j, error bars represent SE and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (h): *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. Source data are available for this figure: SourceData F3.
To confirm our hypothesis, we employed live-cell imaging and the NeonGreen (NG)-Smad3 live-cell sensor (Frick et al., 2017) to monitor the relative change in nuclear SMAD3 signal, indicative of TGFβ signaling activation, upon stimulation with recombinant TGFβ ligand (4 ng/ml) (Fig. 3 b). SMAD3 expression varied across cells, limiting our ability to assess cell-to-cell TGFβ signaling activity through comparison of overall SMAD3 expression (Zieba et al., 2012). However, assessing the relative change in nuclear SMAD3 signal in response to ligand stimulation allowed us to overcome this cell-to-cell variation. Intriguingly, the NG-Smad3 dynamics revealed a marked increase in TGFβ responsiveness across both FNRMS cell lines. In contrast, the FPRMS cell lines responded weakly to ligand stimulation compared with FNRMS cells (Fig. 3 c; and Fig. S2, f and g). Additionally, we discovered that FNRMS cells exhibited higher expression levels of the TGFβ signaling downstream markers collagen type I (Hillege et al., 2020) and CTGF (Schmitt-Ney and Camussi, 2015), compared with FPRMS cells (Fig. 3 d), supporting our hypothesis.
To evaluate whether increased TGFβ signaling activity in FNRMS affects cell–ECM interaction, we treated FNRMS cell lines with the selective TGFβ receptor type I/II (TβRI/II) dual inhibitor LY2109761, followed by IF staining for F-actin and vinculin at focal adhesions. Inhibition of TβRI/II disrupted stress fibers, characterized by a predominant localization of actin at the cell cortex. It also led to a marked decrease in the puncta of focal adhesion, indicated by vinculin staining (Fig. 3 e). TβRI/II inhibition additionally reduced the cell spreading area in FNRMS but not FPRMS, supporting our hypothesis that highly activated TGFβ signaling in FNRMS enhances its cell–ECM interaction (Fig. 3 f). LY2109761-treated FNRMS spheroids also exhibited a diminished capacity to physically interact with the surrounding collagen matrix, as indicated by reduced displacement and alignment of collagen fibers observed through CRM (Fig. 3, g–j and Video 2). These results suggest that TGFβ inhibition associated with the FNRMS subtype causes cytoskeletal and adhesive changes resulting in reduced cell–ECM interactions.
Time-lapse CRM imaging of FNRMS spheroids (RD and SMS-CTR) treated with the TGFβ inhibitor LY2109761 (10 µM, TGFBi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 3 g). Images were taken every 3 h for 21 h (8 frames per second).
Time-lapse CRM imaging of FNRMS spheroids (RD and SMS-CTR) treated with the TGFβ inhibitor LY2109761 (10 µM, TGFBi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 3 g). Images were taken every 3 h for 21 h (8 frames per second).
PAX3-FOXO1 inhibits TGFβ signaling to suppress cell–ECM interactions in FPRMS
The PAX3-FOXO1 fusion gene is the defining molecular alteration discriminating the two RMS subtypes. We hypothesized that the fusion gene inhibits TGFβ signaling to suppress cell–ECM interactions. To evaluate this hypothesis, we generated stable FPRMS cell lines expressing a conditional doxycycline-inducible lentivirus expressing shRNA directed against the PAX3-FOXO1 breakpoint (PFKD) (Hanna et al., 2018) (Fig. S2 h). We first identified DEGs in PFKD cells compared with control (Table S7). Interestingly, we discovered the positively enriched genes in PFKD cells overlap with highly expressed genes in FNRMS-like Subgroup 4, identified through scRNA-seq muscle biopsies (Fig. 4, a and b; and Fig. S2, i and j). These upregulated genes in PFKD also showed overlap with the FNRMS signatures, although there was no statistically significant negative enrichment in FPRMS signatures (Fig. S2 k). Furthermore, GO analysis of DEGs in PFKD revealed an upregulation of genes involved in cell–ECM interactions and cell adhesion (Fig. 4 c and Table S8).

PAX3-FOXO1 suppresses TGFβ signaling and cell–ECM interaction. (a) Higher expression of genes enhanced by PAX3-FOXO1 knockdown (PFKD) in FNRMS-like Subgroups 4 and 5, than in FPRMS-like Subgroup 3. The color indicates the average cellular expression level of genes highly expressed in PFKD FPRMS cells compared with control cells. (b) GSEA indicates that genes highly expressed in Subgroup 4, compared with other subgroups, show statistically significant positive enrichment of genes upregulated by PFKD in FPRMS cells. (c) GO analysis of highly expressed genes in PFKD FPRMS cells (Rh30 and Rh41) compared with control cells, using GO Function gene sets as the reference gene set. (d and e) Immunoblotting and (e) its quantification show higher pMLC of PFKD FPRMS cells than control cells, implying their higher cell–ECM interaction (n = 3). (f) IF images of control and PFKD Rh30 and Rh41, stained for F-actin (with phalloidin) and vinculin, showing focal adhesion in PFKD cells. (g) GO analysis of highly expressed genes in PFKD FPRMS cells (Rh30 and Rh41) compared with control cells, using KEGG gene sets as the reference gene set. In c and g, color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. (h) Dynamic SMAD3 nuclear localization upon TGFβ treatment (4 ng/ml) in PFKD FPRMS cell lines, monitored using NG-Smad3 live-cell sensor. (i) TGFβ signaling activity, quantified by the ratio of intensity in nuclear SMAD3 after TGFβ treatment to before the treatment, in PFKD FPRMS cells. Each dot represents the value of a cell, and lines are averages of each cell line (minimum n = 8). (j) Immunoblotting of nuclear and cytoplasmic fractionized proteins for SMAD2/3, lamin B1 (housekeeping control for nuclear proteins), and GAPDH (housekeeping control for cytoplasmic proteins). SMAD2/3, lamin B1, and GAPDH blots are the same as in Fig. S3 c. (k) Quantification of nuclear proteins exhibits higher SMAD3 nuclear localization of PFKD FPRMS cells than control cells, implying their higher TGFβ signaling activity (n = 3). In e, i, and k, error bars represent SE and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (i): *P < 0.05, **P < 0.01, and ***P < 0.005. Source data are available for this figure: SourceData F4.
PAX3-FOXO1 suppresses TGFβ signaling and cell–ECM interaction. (a) Higher expression of genes enhanced by PAX3-FOXO1 knockdown (PFKD) in FNRMS-like Subgroups 4 and 5, than in FPRMS-like Subgroup 3. The color indicates the average cellular expression level of genes highly expressed in PFKD FPRMS cells compared with control cells. (b) GSEA indicates that genes highly expressed in Subgroup 4, compared with other subgroups, show statistically significant positive enrichment of genes upregulated by PFKD in FPRMS cells. (c) GO analysis of highly expressed genes in PFKD FPRMS cells (Rh30 and Rh41) compared with control cells, using GO Function gene sets as the reference gene set. (d and e) Immunoblotting and (e) its quantification show higher pMLC of PFKD FPRMS cells than control cells, implying their higher cell–ECM interaction (n = 3). (f) IF images of control and PFKD Rh30 and Rh41, stained for F-actin (with phalloidin) and vinculin, showing focal adhesion in PFKD cells. (g) GO analysis of highly expressed genes in PFKD FPRMS cells (Rh30 and Rh41) compared with control cells, using KEGG gene sets as the reference gene set. In c and g, color represents the count of inquired genes overlapping with each specific reference gene set. Circle size indicates adjusted P value, and strength means the ratio of inquired to reference gene counts, implying the enhanced expression of the corresponding reference. (h) Dynamic SMAD3 nuclear localization upon TGFβ treatment (4 ng/ml) in PFKD FPRMS cell lines, monitored using NG-Smad3 live-cell sensor. (i) TGFβ signaling activity, quantified by the ratio of intensity in nuclear SMAD3 after TGFβ treatment to before the treatment, in PFKD FPRMS cells. Each dot represents the value of a cell, and lines are averages of each cell line (minimum n = 8). (j) Immunoblotting of nuclear and cytoplasmic fractionized proteins for SMAD2/3, lamin B1 (housekeeping control for nuclear proteins), and GAPDH (housekeeping control for cytoplasmic proteins). SMAD2/3, lamin B1, and GAPDH blots are the same as in Fig. S3 c. (k) Quantification of nuclear proteins exhibits higher SMAD3 nuclear localization of PFKD FPRMS cells than control cells, implying their higher TGFβ signaling activity (n = 3). In e, i, and k, error bars represent SE and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (i): *P < 0.05, **P < 0.01, and ***P < 0.005. Source data are available for this figure: SourceData F4.
Next, we examined whether PFKD FPRMS cells demonstrate features of enhanced cell–ECM interactions as suggested by sequencing analysis. PFKD cells had significantly increased phosphorylation of myosin light chain (pMLC), which establishes cytoskeletal stress fibers (Kassianidou et al., 2017) and mature focal adhesions (Totsukawa et al., 2004), and is suggestive of higher cell–ECM interactions (Fig. 4 e). Furthermore, PFKD cells had stress fiber bundles and a notably increased number of vinculin-enriched focal adhesions relative to controls, indicating stronger cell–matrix interactions (Fig. 4 f; and Fig. S2, l and m). GO analysis on DEGs in PFKD cells also showed significant upregulation of the TGFβ pathway (Fig. 4 g and Table S8), mirroring the phenotype of FNRMS and FNRMS-like Subgroup 4 shown in Fig. 3 a. Using the NG-Smad3 live-cell sensor, we noted increased TGFβ responsiveness for both PFKD Rh30 and Rh41 cell lines relative to controls (Fig. 4, h and i; and Fig. S3, a and b). Immunoblotting of the nuclear fraction also confirmed elevated SMAD2/3 protein expression in PFKD FPRMS cell lines, indicative of higher TGFβ signaling activation (Fig. 4, j and k; and Fig. S3 c).

PAX3-FOXO1 deactivates TGFβ signaling by enhancing NO synthesis. ( a) Averaged time course TGFβ signaling activity of PFKD (purple) and control (blue) FPRMS cells, assessed by dynamic SMAD3 nuclear localization in each cell line (minimum n = 8). An arrow indicates the value of Fig. 4 i. (b) Faint lines indicate each cell in the cell line. Dashed lines indicate the time point of TGFβ treatment. (c) Higher SMAD2/3 nuclear localization of PFKD FPRMS cells compared with control scramble cells. SMAD2/3 levels in cytoplasmic proteins were similar between control and PFKD Rh30 cells, and PFKD Rh41 cells exhibited lower cytoplasmic SMAD2/3 levels compared with controls. SMAD2/3, lamin B1, and GAPDH blots are the same with Fig. 4 j. (d) Pronounced cell spreading areas of PFKD cells compared with control scramble cells (n = 30). (e) Displacement of collagen fibers around spheroids of scramble and PFKD FPRMS cells, Rh30 (top) and Rh41 (bottom), assessed by CRM. In d and e, each dot denotes the value of a corresponding cell and lines are the averages of each cell line (minimum n = 3). (f) Distribution of collagen fiber orientation around the spheroids of scramble FPRMS cells at 0 and 16 h (n = 3). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). At 16 h, statistical significance between Dox-induced scramble and PFKD cells (Fig. 5 e) was assessed by two-sided Student’s t tests: *P < 0.05, **P < 0.01, and ***P < 0.005. (g) DAF-2 DA staining of scramble Rh30 and Rh41 cells and PFKD Rh41 cells, with and without induction, indicating their intracellular NO concentration. (h) Quantification of DAF-2 DA, corresponding to NO concentration, in scramble and PFKD FPRMS cells (Fig. 6, d and e). Dots represent individual cell values (minimum n = 21). The values were normalized to the average of non–Dox-induced PFKD Rh30 cells (for other Rh30 cells) and Rh41 cells (for other Rh41 cells). (i) Increased number of focal adhesions in Rh41 cells after treatment with ARL17477. Each dot indicates the number of focal adhesions in each cell (n = 7). In d, e, h, and i, lines are averages of each group. Error bars are SE. Statistical significance was assessed using two-sided Student’s t tests: *P < 0.05, ***P < 0.005, and ****P < 0.001. Dox, doxycycline.

PAX3-FOXO1 deactivates TGFβ signaling by enhancing NO synthesis. ( a) Averaged time course TGFβ signaling activity of PFKD (purple) and control (blue) FPRMS cells, assessed by dynamic SMAD3 nuclear localization in each cell line (minimum n = 8). An arrow indicates the value of Fig. 4 i. (b) Faint lines indicate each cell in the cell line. Dashed lines indicate the time point of TGFβ treatment. (c) Higher SMAD2/3 nuclear localization of PFKD FPRMS cells compared with control scramble cells. SMAD2/3 levels in cytoplasmic proteins were similar between control and PFKD Rh30 cells, and PFKD Rh41 cells exhibited lower cytoplasmic SMAD2/3 levels compared with controls. SMAD2/3, lamin B1, and GAPDH blots are the same with Fig. 4 j. (d) Pronounced cell spreading areas of PFKD cells compared with control scramble cells (n = 30). (e) Displacement of collagen fibers around spheroids of scramble and PFKD FPRMS cells, Rh30 (top) and Rh41 (bottom), assessed by CRM. In d and e, each dot denotes the value of a corresponding cell and lines are the averages of each cell line (minimum n = 3). (f) Distribution of collagen fiber orientation around the spheroids of scramble FPRMS cells at 0 and 16 h (n = 3). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). At 16 h, statistical significance between Dox-induced scramble and PFKD cells (Fig. 5 e) was assessed by two-sided Student’s t tests: *P < 0.05, **P < 0.01, and ***P < 0.005. (g) DAF-2 DA staining of scramble Rh30 and Rh41 cells and PFKD Rh41 cells, with and without induction, indicating their intracellular NO concentration. (h) Quantification of DAF-2 DA, corresponding to NO concentration, in scramble and PFKD FPRMS cells (Fig. 6, d and e). Dots represent individual cell values (minimum n = 21). The values were normalized to the average of non–Dox-induced PFKD Rh30 cells (for other Rh30 cells) and Rh41 cells (for other Rh41 cells). (i) Increased number of focal adhesions in Rh41 cells after treatment with ARL17477. Each dot indicates the number of focal adhesions in each cell (n = 7). In d, e, h, and i, lines are averages of each group. Error bars are SE. Statistical significance was assessed using two-sided Student’s t tests: *P < 0.05, ***P < 0.005, and ****P < 0.001. Dox, doxycycline.
PFKD cells exhibited increased spreading area relative to their control counterparts (Fig. 5 a and Fig. S3 d). However, upon pharmacological inhibition of TβRI/II in PFKD cells, this effect was nullified. In line with these findings, CRM imaging of PFKD spheroids embedded in 3D collagen gels revealed increased cell–ECM interactions, as indicated by greater collagen fiber displacement compared with control (Fig. 5, b and c; Fig. S3 e; and Video 3). However, this effect was also nullified upon pharmacological inhibition of TβRI/II with LY2109761. The same trend was observed for cell-mediated alignment of collagen fibers, where PFKD possessed a greater ability to reorganize the matrix by aligning the surrounding fibers relative to controls, and this effect was similarly abolished with LY2109761 treatment (Fig. 5, d and e; and Fig. S3 f). These results suggest that the PAX3-FOXO1 fusion gene disrupts cell–ECM interactions by inhibiting TGFβ signaling.

Knockdown of PAX3-FOXO1 enhances cell–ECM interaction via TGFβ signaling. (a) Cell spreading areas of PFKD FPRMS cell lines upon treatment with the TGFβ inhibitor LY2109761 (100 nM, 12 h) (n = 30). (b) Collagen fiber displacement near spheroids of control FPRMS cells (left), PFKD FPRMS cells (middle), and LY2109761-treated PFKD FPRMS cells (100 nM, right), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (c) Quantification of the collagen fiber displacement of PFKD FPRMS cell lines upon TGFβ inhibition (n = 5). In a and c, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. (d) Collagen fiber alignment 16 h after gelation with control FPRMS cells (left), PFKD FPRMS cells (middle), and LY2109761-treated PFKD FPRMS cells (100 nM, right). Dashed lines denote the spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges, while asterisks (*) highlight the fibers aligned parallel to the edges. (e) Distribution of collagen fiber orientation around the spheroids with and without TGFβ inhibition. Dots represent individual spheroid values (minimum n = 4). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). In a, c, and e, error bars represent SE and statistical significance was assessed using the Wilcoxon rank sum test (a) or two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s = not significant.
Knockdown of PAX3-FOXO1 enhances cell–ECM interaction via TGFβ signaling. (a) Cell spreading areas of PFKD FPRMS cell lines upon treatment with the TGFβ inhibitor LY2109761 (100 nM, 12 h) (n = 30). (b) Collagen fiber displacement near spheroids of control FPRMS cells (left), PFKD FPRMS cells (middle), and LY2109761-treated PFKD FPRMS cells (100 nM, right), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (c) Quantification of the collagen fiber displacement of PFKD FPRMS cell lines upon TGFβ inhibition (n = 5). In a and c, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. (d) Collagen fiber alignment 16 h after gelation with control FPRMS cells (left), PFKD FPRMS cells (middle), and LY2109761-treated PFKD FPRMS cells (100 nM, right). Dashed lines denote the spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges, while asterisks (*) highlight the fibers aligned parallel to the edges. (e) Distribution of collagen fiber orientation around the spheroids with and without TGFβ inhibition. Dots represent individual spheroid values (minimum n = 4). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). In a, c, and e, error bars represent SE and statistical significance was assessed using the Wilcoxon rank sum test (a) or two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s = not significant.
Time-lapse CRM imaging of FPRMS (Rh30 and Rh41) and PFKDspheroids treated with the TGFβ inhibitor LY2109761 (10 µM, TGFBi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 5 b). Images were taken every 3 h for 18 h (8 frames per second).
Time-lapse CRM imaging of FPRMS (Rh30 and Rh41) and PFKDspheroids treated with the TGFβ inhibitor LY2109761 (10 µM, TGFBi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 5 b). Images were taken every 3 h for 18 h (8 frames per second).
PAX3-FOXO1-mediated NO synthesis inhibits TGFβ signaling
We then sought to investigate how the PAX3-FOXO1 fusion gene regulates the TGFβ signaling pathway. The fusion gene acts as a rogue transcription factor that can reprogram myogenic development. Gryder et al. identified the genes regulated by PAX3-FOXO1 through chromatin immunoprecipitation (ChIP) sequencing (Gryder et al., 2017). Among the target genes, NOS1, which is translated into the NO synthase 1 enzyme that is crucial for NO synthesis, exhibited the most significant positive-fold change in expression in FPRMS compared with FNRMS (Fig. 6 a and Table S9). In line with this, we also discovered the greatest decrease in the expression of NOS1 in PFKD Rh30 and Rh41 cells compared with controls (Fig. 6 a). Interestingly, NO, apart from its role in vasodilation, has also been implicated in suppressing ECM synthesis (Kim et al., 1999) and serving as a negative regulator of focal adhesion assembly (Goligorsky et al., 1999; Takahashi et al., 1996). Furthermore, NO can suppress TGFβ signaling in various contexts (Hovater et al., 2014; Kanno et al., 2008; Lee et al., 2011). Thus, we hypothesized that NO, regulated by PAX3-FOXO1, inhibits TGFβ signaling activity and disrupts cell–ECM interactions.

PAX3-FOXO1 stimulates the production of NO. (a) Higher NOS1 expression in FPRMS than FNRMS (top) and decreased NOS1 expression by PFKD in Rh30 (middle) and Rh41 (bottom). DEGs among PAX3-FOXO1 target genes between FPRMS and FNRMS (data were from GSE114621), between PFKD Rh30 and control Rh30, and between PFKD Rh41 and control Rh41. PAX3-FOXO1 target genes are identified in Gryder et al. (2017) (PMID: 28446439). NOS1 is the gene that commonly exhibits the greatest fold change increase in FPRMS versus FNRMS and decrease by PFKD. (b and c) DAF-2 DA staining and its pseudocolored images that probe NO in FNRMS and FPRMS cell lines and (c) its intensity indicating NO concentration in each cell line, suggesting higher NO concentration in FNRMS than FPRMS cells (n = 40). The values were normalized to the average of RD cells. (d and e) DAF-2 DA staining of PFKD Rh30 and control Rh30 (FPRMS) and (e) its intensity indicating their NO concentrations, implying higher NO concentration in PFKD FPRMS than control cells. The values were normalized to the average of control cells. Each dot represents the intensity of a corresponding cell, and lines are averages of each cell line (minimum n = 21). In c and e, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. Error bars represent SE, and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (c): *P < 0.05, ***P < 0.005, and ****P < 0.001.
PAX3-FOXO1 stimulates the production of NO. (a) Higher NOS1 expression in FPRMS than FNRMS (top) and decreased NOS1 expression by PFKD in Rh30 (middle) and Rh41 (bottom). DEGs among PAX3-FOXO1 target genes between FPRMS and FNRMS (data were from GSE114621), between PFKD Rh30 and control Rh30, and between PFKD Rh41 and control Rh41. PAX3-FOXO1 target genes are identified in Gryder et al. (2017) (PMID: 28446439). NOS1 is the gene that commonly exhibits the greatest fold change increase in FPRMS versus FNRMS and decrease by PFKD. (b and c) DAF-2 DA staining and its pseudocolored images that probe NO in FNRMS and FPRMS cell lines and (c) its intensity indicating NO concentration in each cell line, suggesting higher NO concentration in FNRMS than FPRMS cells (n = 40). The values were normalized to the average of RD cells. (d and e) DAF-2 DA staining of PFKD Rh30 and control Rh30 (FPRMS) and (e) its intensity indicating their NO concentrations, implying higher NO concentration in PFKD FPRMS than control cells. The values were normalized to the average of control cells. Each dot represents the intensity of a corresponding cell, and lines are averages of each cell line (minimum n = 21). In c and e, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. Error bars represent SE, and statistical significance was assessed using two-sided Student’s t test or Wilcoxon’s rank sum test (c): *P < 0.05, ***P < 0.005, and ****P < 0.001.
To validate this hypothesis, we first confirmed that both FPRMS cell lines show increased intracellular NO relative to FNRMS counterparts using the fluorescent probe DAF-2 DA (Zhou and He, 2011) (Fig. 6, b and c). Correspondingly, PFKD cells show a reduction in NO relative to control FPRMS cells (Fig. 6, d and e; and Fig. S3, g and h). These results suggest that PAX3-FOXO1 is an upstream positive regulator of NOS1-mediated NO synthesis. To investigate whether PAX3-FOXO1-mediated NO synthesis via NOS1 has a downstream inhibitory effect on TGFβ signaling and cell–ECM interactions, we performed IF staining for nuclear SMAD3 in Rh41 FPRMS cells treated with the NOS1 inhibitor ARL17477. We demonstrated a marked increase in nuclear SMAD3 intensity upon pharmacological NOS1 inhibition relative to controls (Fig. 7, a and b), further corroborated by immunoblotting of SMAD2/3 in the nuclear fraction (Fig. 7, c and d). These results support our hypothesis by confirming that NOS1 inhibition stimulates TGFβ signaling. NOS1 inhibition also increased F-actin stress fiber assembly and vinculin-enriched focal adhesions (Fig. 7 e and Fig. S3 i). Moreover, NOS1 inhibition led to increased pMLC and cell spreading area relative to untreated FPRMS controls (Fig. 7, f–h). NOS1 inhibition in FPRMS spheroids embedded within collagen type I gels also enhanced their ability to physically interact with the matrix, resulting in higher displacement and alignment of collagen fibers as visualized by CRM (Fig. 7, i–l and Video 4). Taken together, these findings imply that PAX3-FOXO1 disrupts cell–ECM interaction by inhibiting TGFβ signaling via the PAX3-FOXO1-stimulated synthesis of NO.

NO stimulated by PAX3-FOXO1 suppresses TGFβ signaling and cell–ECM interaction. (a) IF and its pseudocolored images of Rh41 (FPRMS) cells stained for SMAD3 with and without the treatment of NOS inhibitor ARL17477 (10 µM, 12 h). (b) Intensity of nuclear SMAD3 in Rh41 cells with and without the treatment of ARL17477 (minimum n = 50). (c and d) Immunoblotting of nuclear and cytoplasmic fractionized proteins and (d) its quantification suggest that NOS inhibition increases nuclear localization of SMAD3. Lamin B1 is a control of nuclear fractionized proteins (n = 3). (e) IF images of Rh41 cell lines with and without the treatment of ARL17477 (10 µM, 12 h), stained for F-actin (with phalloidin) and vinculin, showing focal adhesion. (f and g) Immunoblotting and (g) its quantification suggest that NOS inhibition increases pMLC, implying higher cell–ECM interaction (n = 3). (h) Cell spreading areas of Rh41 with and without NOS inhibition (n = 40). (i) Collagen fiber displacement near Rh41 spheroids with and without the treatment of ARL17477 (10 µM), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (j) Quantification of collagen fiber displacement with and without ARL17477 (n = 3). (k) Collagen fiber alignment 16 h after gelation with Rh41 spheroids, with and without the treatment of ARL17477. Dashed lines denote the spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges. (l) Distribution of collagen fiber orientation around Rh41 spheroids with and without NOS inhibition (n = 5). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). In b, d, g, h, j, and l, error bars represent SE and statistical significance was assessed using Wilcoxon’s rank sum test (b and h) or two-sided Student’s t test: *P < 0.05, ***P < 0.005, and ****P < 0.001. In b, h, j, and l, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. Source data are available for this figure: SourceData F7.
NO stimulated by PAX3-FOXO1 suppresses TGFβ signaling and cell–ECM interaction. (a) IF and its pseudocolored images of Rh41 (FPRMS) cells stained for SMAD3 with and without the treatment of NOS inhibitor ARL17477 (10 µM, 12 h). (b) Intensity of nuclear SMAD3 in Rh41 cells with and without the treatment of ARL17477 (minimum n = 50). (c and d) Immunoblotting of nuclear and cytoplasmic fractionized proteins and (d) its quantification suggest that NOS inhibition increases nuclear localization of SMAD3. Lamin B1 is a control of nuclear fractionized proteins (n = 3). (e) IF images of Rh41 cell lines with and without the treatment of ARL17477 (10 µM, 12 h), stained for F-actin (with phalloidin) and vinculin, showing focal adhesion. (f and g) Immunoblotting and (g) its quantification suggest that NOS inhibition increases pMLC, implying higher cell–ECM interaction (n = 3). (h) Cell spreading areas of Rh41 with and without NOS inhibition (n = 40). (i) Collagen fiber displacement near Rh41 spheroids with and without the treatment of ARL17477 (10 µM), monitored by CRM and analyzed by PIV. Top: pseudocolored images showing the displacement degree of collagen fibers caused by spheroids. Bottom: arrows indicate the direction and magnitude of collagen fiber displacement. (j) Quantification of collagen fiber displacement with and without ARL17477 (n = 3). (k) Collagen fiber alignment 16 h after gelation with Rh41 spheroids, with and without the treatment of ARL17477. Dashed lines denote the spheroid edges. Arrows point to the fibers aligned perpendicularly to the spheroid edges. (l) Distribution of collagen fiber orientation around Rh41 spheroids with and without NOS inhibition (n = 5). Dashed lines indicate a probability of 0.2 indicating an equal distribution of fiber orientations (random orientation). In b, d, g, h, j, and l, error bars represent SE and statistical significance was assessed using Wilcoxon’s rank sum test (b and h) or two-sided Student’s t test: *P < 0.05, ***P < 0.005, and ****P < 0.001. In b, h, j, and l, each dot represents the value of a corresponding cell or spheroid. Lines are averages of each condition. Source data are available for this figure: SourceData F7.
Time-lapse CRM imaging of FPRMS spheroids treated with the NOS1 inhibitor ARL17477 (10 µM, NOSi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 7 i). Images were taken every 3 h for 18 h (8 frames per second).
Time-lapse CRM imaging of FPRMS spheroids treated with the NOS1 inhibitor ARL17477 (10 µM, NOSi). Spheroids are embedded in 3D gels of collagen type I (related to Fig. 7 i). Images were taken every 3 h for 18 h (8 frames per second).
PAX3-FOXO1 status determines anchorage-dependent tumor cell survival
We then examined whether suppressed TGFβ signaling and cell–ECM interactions correlate with the clinical outcomes of FPRMS patients. Intriguingly, analysis of FPRMS patient data (Missiaglia et al., 2012) revealed that patients with higher expression of ECM-related genes (GOMF_Extracellular_Matrix_Structural_Constituent gene set) and TGFβ signaling genes (GO HALLMARKS_TGF_BETA_SIGNALING) exhibited longer survival periods than those with lower expression (Fig. 8 a). We additionally identified that the gene COL1A1 in the ECM structural constituent gene set shows the most statistically significant increase in expression by PFKD, overlapping with the significantly downregulated genes in FPRMS-like Subgroup 3. The expression of COL18A1, also in the ECM structural constituent gene set, shows the most statistically significant enhancement by PFKD among the genes decreased in FPRMS compared with FNRMS. Additionally, ID1 exhibits the most significant increase in the expression level by PFKD among the TGFβ signaling gene set, commonly overlapping with downregulated genes in both Subgroup 3 and FPRMS. Our analysis revealed that patients with higher expression of COL1A1, COL18A1, and ID1 exhibited longer survival periods than those with lower expression (Fig. S4 a). These results suggest that reduced cell–ECM interaction by PAX3-FOXO1 via TGFβ signaling may lead to unfavorable patient outcomes.

PAX3-FOXO1 modulates distinct cell–ECM interaction of FPRMS from FNRMS, determining their response to drugs targeting cell–ECM interaction and TGFβ signaling. (a) Overall survival probability in FPRMS was evaluated based on the differential expression of ECM- and TGFβ signaling–related gene sets. Kaplan–Meier survival curves of FPRMS data (minimum n = 8) revealed significantly improved clinical outcomes in patients with elevated expression of these gene sets to the low-expression cohort (P < 0.05). Clinical data of FPRMS patients were obtained from PMID: 22454413. (b) Higher resistance of FPRMS cell lines than FNRMS cell lines to pharmaceutical cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by the CCK8 assay (n = 3). (c) Pronounced resistance of FPRMS spheroids compared with FNRMS spheroids to pharmaceutical cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by CellTiter-Glo 3D cell viability assay (minimum n = 3). The treatment of ARL17477 (NOS inhibitor, 10 µM) recovered the responses of FPRMS cells. Negative values demonstrate reduced viability from the drugs. (d) The PAX3-FOXO1 knockdown decreases the resistance of RMS cells to cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by the CCK8 assay (minimum n = 3). In b and d, relative survival of cells treated with drugs means the ratio of viability of drug-treated cells to that of untreated cells. We treated dasatinib (Src inhibitor, 10 nM), defactinib (FAK inhibitor, 10 µM), and LY2109761 (TGFβ inhibitor, 1 µM) for 1 day. In c, drugs were treated for 2 days. In b–d, error bars represent SE. In b and d, statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. In c, statistical significance between cells (denoted by *) and between FPRMS cells with and without ARL17477 (denoted by #) was assessed using two-sided Student’s t test: *P < 0.05, ***P < 0.005, ****P < 0.001, #P < 0.05, ####P < 0.001, and n.s. = not significant.
PAX3-FOXO1 modulates distinct cell–ECM interaction of FPRMS from FNRMS, determining their response to drugs targeting cell–ECM interaction and TGFβ signaling. (a) Overall survival probability in FPRMS was evaluated based on the differential expression of ECM- and TGFβ signaling–related gene sets. Kaplan–Meier survival curves of FPRMS data (minimum n = 8) revealed significantly improved clinical outcomes in patients with elevated expression of these gene sets to the low-expression cohort (P < 0.05). Clinical data of FPRMS patients were obtained from PMID: 22454413. (b) Higher resistance of FPRMS cell lines than FNRMS cell lines to pharmaceutical cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by the CCK8 assay (n = 3). (c) Pronounced resistance of FPRMS spheroids compared with FNRMS spheroids to pharmaceutical cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by CellTiter-Glo 3D cell viability assay (minimum n = 3). The treatment of ARL17477 (NOS inhibitor, 10 µM) recovered the responses of FPRMS cells. Negative values demonstrate reduced viability from the drugs. (d) The PAX3-FOXO1 knockdown decreases the resistance of RMS cells to cell–ECM interaction perturbation (dasatinib and defactinib) and TGFβ inhibition (LY2109761), assessed by the CCK8 assay (minimum n = 3). In b and d, relative survival of cells treated with drugs means the ratio of viability of drug-treated cells to that of untreated cells. We treated dasatinib (Src inhibitor, 10 nM), defactinib (FAK inhibitor, 10 µM), and LY2109761 (TGFβ inhibitor, 1 µM) for 1 day. In c, drugs were treated for 2 days. In b–d, error bars represent SE. In b and d, statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. In c, statistical significance between cells (denoted by *) and between FPRMS cells with and without ARL17477 (denoted by #) was assessed using two-sided Student’s t test: *P < 0.05, ***P < 0.005, ****P < 0.001, #P < 0.05, ####P < 0.001, and n.s. = not significant.

FPRMS clinical outcome may be independent of cell-ECM perturbation. ( a) Overall survival probability of FPRMS based on the differential expression of genes associated with the ECM (COL1A1 and COL18A1) and TGFβ signaling (ID1). The Kaplan–Meier survival curves of FPRMS data (minimum n = 8) indicate significantly improved clinical outcome in patients with elevated expression of the selected genes of interest compared with the low-expression cohort (P < 0.05 and Bonferroni-adjusted P > 0.4). Clinical data were obtained from PMID: 22454413. (b) Higher resistance of FPRMS than FNRMS to pharmaceutical cell–ECM perturbation (Src inhibitor, dasatinib; FAK inhibitor, defactinib) and TGFβ inhibition (TGFβ inhibitor, LY2109761), assessed by the CCK8 assay (n = 3). (c) Treatment of ARL17477 does not affect FPRMS spheroid survivals (minimum n = 3). We assessed spheroid survival using the CellTiter-Glo 3D cell viability assay. In b and c, statistical significance of cell survival was assessed by two-sided Student’s t tests: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. Arrows indicate the concentration used in main figures.

FPRMS clinical outcome may be independent of cell-ECM perturbation. ( a) Overall survival probability of FPRMS based on the differential expression of genes associated with the ECM (COL1A1 and COL18A1) and TGFβ signaling (ID1). The Kaplan–Meier survival curves of FPRMS data (minimum n = 8) indicate significantly improved clinical outcome in patients with elevated expression of the selected genes of interest compared with the low-expression cohort (P < 0.05 and Bonferroni-adjusted P > 0.4). Clinical data were obtained from PMID: 22454413. (b) Higher resistance of FPRMS than FNRMS to pharmaceutical cell–ECM perturbation (Src inhibitor, dasatinib; FAK inhibitor, defactinib) and TGFβ inhibition (TGFβ inhibitor, LY2109761), assessed by the CCK8 assay (n = 3). (c) Treatment of ARL17477 does not affect FPRMS spheroid survivals (minimum n = 3). We assessed spheroid survival using the CellTiter-Glo 3D cell viability assay. In b and c, statistical significance of cell survival was assessed by two-sided Student’s t tests: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant. Arrows indicate the concentration used in main figures.
We hypothesized that the distinct cell–ECM interactions associated with the presence or absence of the PAX3-FOXO1 fusion gene would exhibit varying sensitivities to growth inhibition upon cell–ECM perturbation. FAK and Src are essential for formation of focal adhesions, which link the cell cytoskeleton to the ECM (Bouchard et al., 2007; Mitra and Schlaepfer, 2006; Sulzmaier et al., 2014; Westhoff et al., 2004). These complexes facilitate bidirectional cell–ECM communication, which is crucial for regulating cell survival signaling. Disruption of cell–ECM interactions by Src inhibition (dasatinib) or FAK inhibition (defactinib) resulted in diminished cell and spheroid (3D cell culture) survival for FNRMS, which exhibits pronounced cell–ECM interactions, compared with FPRMS (Fig. 8, b and c; and Fig. S4 b). Similarly, inhibiting TGFβ signaling (LY2109761), an upstream regulator of cell–ECM interactions in our study, also resulted in growth inhibition for FNRMS but not FPRMS. Interestingly, we discovered that NOS inhibition recovered the sensitivity of FPRMS spheroids to the drugs although the inhibition alone did not affect viability (Fig. S4 c). Knockdown of PAX3-FOXO1 also resensitized FPRMS cells to the growth-inhibiting effects of Src, FAK, and TGFβ inhibition (Fig. 8 d; and Fig. S5, a and b). These results suggest that achieving growth inhibition in FPRMS may require a combined strategy involving genetic perturbation of the PAX3-FOX1 fusion gene or NOS inhibition and simultaneous disruption of cell–ECM interactions.

PAX3-FOXO1 may determine the independence of cell-ECM perturbation. ( a) Cell survival of scramble and PFKD FPRMS cells, Rh30 (top) and Rh41 (bottom), upon the treatment of dasatinib (10 µM) and LY2109761 (10 µM) (n = 3). (b) Higher sensitivity of PFKD FPRMS cells than control cells to pharmaceutical cell–ECM perturbation (Src inhibitor, dasatinib; FAK inhibitor, defactinib) and TGFβ inhibition (TGFβ inhibitor, LY2109761) (minimum n = 3). In a and b, arrows indicate the concentration used in main figures. (c) Suspended spheroids of scramble FPRMS cells in media for 2 days via the hanging drop method, resulting in no ECM interaction. Dashed lines indicate the size of the spheroid on day 0. (d and e) Growth of the suspended spheroids of scramble and PFKD FPRMS (Fig. 8, d–f) assessed by the size of spheroids (d) (n = 4) and by the CellTiter-Glo 3D cell viability assay (e) (n = 5). Relative spheroid sizes (the ratios of spheroid sizes on day 2 to those on day 0) in d and differences of log10 (intensity values) from day 2 to day 0 in e mean the degrees of their growth. Dots represent individual spheroid values. In d and e, error bars represent SE. In a, b, d, and e, statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant.

PAX3-FOXO1 may determine the independence of cell-ECM perturbation. ( a) Cell survival of scramble and PFKD FPRMS cells, Rh30 (top) and Rh41 (bottom), upon the treatment of dasatinib (10 µM) and LY2109761 (10 µM) (n = 3). (b) Higher sensitivity of PFKD FPRMS cells than control cells to pharmaceutical cell–ECM perturbation (Src inhibitor, dasatinib; FAK inhibitor, defactinib) and TGFβ inhibition (TGFβ inhibitor, LY2109761) (minimum n = 3). In a and b, arrows indicate the concentration used in main figures. (c) Suspended spheroids of scramble FPRMS cells in media for 2 days via the hanging drop method, resulting in no ECM interaction. Dashed lines indicate the size of the spheroid on day 0. (d and e) Growth of the suspended spheroids of scramble and PFKD FPRMS (Fig. 8, d–f) assessed by the size of spheroids (d) (n = 4) and by the CellTiter-Glo 3D cell viability assay (e) (n = 5). Relative spheroid sizes (the ratios of spheroid sizes on day 2 to those on day 0) in d and differences of log10 (intensity values) from day 2 to day 0 in e mean the degrees of their growth. Dots represent individual spheroid values. In d and e, error bars represent SE. In a, b, d, and e, statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and n.s. = not significant.
FPRMS has been reported to have a higher metastatic potential compared with FNRMS (Nguyen and Barr, 2018). During metastasis, cancer cells intravasate into blood or lymphatic vessels, spreading through the circulatory system. These CTCs benefit from reprogramming their anchorage dependency, suppressing cell–ECM interactions by deactivating YAP signaling and undergoing AST (Huh et al., 2023). This reprogramming enables CTCs to resist anoikis, the death of anchorage-dependent cells upon detachment from ECM, within the circulatory system. We assessed the growth of spheroids in the absence of ECM attachment using the hanging drop method. This setup effectively interrogates the ability of cells to survive and grow independently of substrate attachment, which is one of the prerequisites for metastasis. Indeed, the growth of suspended FNRMS spheroids was inhibited in the hanging drop environment without ECM contact (Fig. 9, a–c). In contrast, suspended FPRMS spheroids showed significant expansion over 48 h, bypassing the need for cell–ECM interactions, whereas PFKD nullified this effect (Fig. 9, d–f and Fig. S5, c–e). Furthermore, PFKD FPRMS cells exhibited higher nuclear localization of YAP, indicating YAP activation, and reduced expression of AST signature genes (Fig. 9, g and h).

PAX3-FOXO1 may modulate metastatic potentials. (a) Suspended spheroids of FNRMS and FPRMS cells in media for 2 days via the hanging drop method, resulting in no ECM interaction. (b and c) Growth of the suspended spheroids assessed by the size of spheroids and (c) by CellTiter-Glo 3D cell viability assay (n = 4). (d) Suspended spheroids of control and PFKD FPRMS cells for 2 days in media via the hanging drop method. (e and f) Growth of the suspended spheroids of PFKD and control FPRMS cells assessed by the size of spheroids (n = 4) and (f) by the CellTiter-Glo 3D cell viability assay (n = 5). (g) IF images of PFKD and control FPRMS cells stained for YAP and vinculin. Higher nuclear localization of YAP in PFKD cells indicates higher YAP activation than in control cells. (h) GSEA suggests the reduced expression of AST markers in PFKD FPRMS cells compared with control cells. The list of AST markers from PMID: 36991428 was used as the reference gene set. (i) Enhanced intravasation capacity of FPRMS cells compared with FNRMS cells assessed by the transwell assay (n = 4). The capacity of FPRMS cells is reduced by the treatment of ARL17477 (10 µM). All values were normalized to the average of RD cells. (j) Reduced intravasation capacity by PFKD assessed by the transwell assay (n = 3). The values were normalized to the average of non–Dox-induced PFKD cells. In a and d, dashed lines indicate the size of the spheroid on day 0. Relative spheroid sizes (the ratios of spheroid sizes on day 2 to those on day 0) in b and e and differences of log10 (intensity values) from day 3 or 2 to day 0 in c and f mean the degrees of their growth. Dots represent individual spheroid values. In b, c, e, f, i, and j, error bars represent SE and statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. (k) Proposed model of PAX3-FOXO1 regulated suppression of cell–ECM interaction via TGFβ signaling and NOS1. Dox, doxycycline.
PAX3-FOXO1 may modulate metastatic potentials. (a) Suspended spheroids of FNRMS and FPRMS cells in media for 2 days via the hanging drop method, resulting in no ECM interaction. (b and c) Growth of the suspended spheroids assessed by the size of spheroids and (c) by CellTiter-Glo 3D cell viability assay (n = 4). (d) Suspended spheroids of control and PFKD FPRMS cells for 2 days in media via the hanging drop method. (e and f) Growth of the suspended spheroids of PFKD and control FPRMS cells assessed by the size of spheroids (n = 4) and (f) by the CellTiter-Glo 3D cell viability assay (n = 5). (g) IF images of PFKD and control FPRMS cells stained for YAP and vinculin. Higher nuclear localization of YAP in PFKD cells indicates higher YAP activation than in control cells. (h) GSEA suggests the reduced expression of AST markers in PFKD FPRMS cells compared with control cells. The list of AST markers from PMID: 36991428 was used as the reference gene set. (i) Enhanced intravasation capacity of FPRMS cells compared with FNRMS cells assessed by the transwell assay (n = 4). The capacity of FPRMS cells is reduced by the treatment of ARL17477 (10 µM). All values were normalized to the average of RD cells. (j) Reduced intravasation capacity by PFKD assessed by the transwell assay (n = 3). The values were normalized to the average of non–Dox-induced PFKD cells. In a and d, dashed lines indicate the size of the spheroid on day 0. Relative spheroid sizes (the ratios of spheroid sizes on day 2 to those on day 0) in b and e and differences of log10 (intensity values) from day 3 or 2 to day 0 in c and f mean the degrees of their growth. Dots represent individual spheroid values. In b, c, e, f, i, and j, error bars represent SE and statistical significance was assessed using two-sided Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. (k) Proposed model of PAX3-FOXO1 regulated suppression of cell–ECM interaction via TGFβ signaling and NOS1. Dox, doxycycline.
Finally, we performed a transwell assay to assess cell intravasation ability. In this setup, a membrane filter with 8-µm pores models the barrier that cancer cells must penetrate during intravasation and establishes a serum concentration gradient between the upper chamber (containing serum-free media) and the lower chamber (containing serum-supplemented media), thereby driving cell migration (Justus et al., 2023; Moutasim et al., 2011). Cells that successfully migrate to the lower chamber may reflect higher intravasation capacity. FPRMS cells demonstrated a higher number of migrating cells to lower chambers than FNRMS cells. Additionally, NOS inhibition and PFKD diminished the number of migrating cells from FPRMS (Fig. 9, i and j). Altogether, these results indicate that the PAX3-FOXO1 status differentially regulates anchorage-dependent growth via distinct cell–matrix interactions, potentially contributing to the higher metastatic potential of FPRMS compared with FNRMS. The PAX3-FOXO1 fusion gene may thus confer metastatic competence by reprogramming anchorage dependence to sustain cancer cell growth and survival in ectopic environments, such as the circulatory system (Huh et al., 2023; Wachtel and Schäfer, 2018).
Discussion
We have characterized how the fusion gene status of the two RMS subtypes modulates differential cell–ECM interactions and anchorage dependency to inform subtype-specific therapies. Through scRNA-seq data analysis, we confirmed that FPRMS and FNRMS signatures emerge in distinct cell populations at distinct stages during myogenesis. These variations in the stage where differentiation arrest occurs likely mirror their divergent genetic underpinnings and signaling pathways. Additionally, we showed that FNRMS exhibits higher expression of genes involved in cell–ECM interactions compared with FPRMS. Molecular and functional assays, including CRM, confirmed FNRMS cells also exhibit heightened cell–ECM interactions compared with FPRMS. Intriguingly, Gizaw et al. have recently reported that silencing the master transcription factor PROX1 inhibited RMS cell growth and resulted in significant upregulation of cell–matrix and cell–cell adhesion pathways (Gizaw et al., 2022).
The oncogenic effect of PAX3-FOXO1 is thought to result from the aberrant transcriptional activation of multiple downstream target genes whose expression dysregulates signaling pathways. Several of these downstream effectors have activities that promote cancer development, such as stimulating proliferation and inhibiting apoptosis. Some of these direct transcriptional targets of PAX3-FOXO1 include various receptor tyrosine kinases, notably FGFR4 (Taylor et al., 2009) and MET (Ginsberg et al., 1998). PAX3-FOXO1 also acts as a pioneer transcription factor that alters the cellular epigenetic state to co-opt normal myogenic differentiation programs (Sunkel et al., 2021). Our findings expand on how PAX3-FOXO1 transcriptionally reprograms anchorage dependency in RMS by downregulating genes involved in cell–matrix interactions. Specifically, we elucidated that PAX3-FOXO1 regulates a NO-mediated inhibition of TGFβ signaling (Fig. 9 k), leading to changes in the cytoskeleton and focal adhesions, thereby disrupting cell–matrix interactions.
We speculate that the higher metastatic propensity of FPRMS may be attributed in part to the PAX3-FOXO1-mediated reprogramming of anchorage dependency. Conceivably, CTCs of FPRMS may benefit from this reprogramming, allowing them to survive in circulation, as opposed to those of FNRMS that are more reliant on cell–ECM interactions for their growth. However, the higher cell–ECM interactions may endow FNRMS with greater competency during the initial invasion through the adjacent stroma of the primary tumor (Lambert et al., 2017). Recently, Regina et al. demonstrated that primary FPRMS cultures exhibit marked heterogeneity in PAX3-FOXO1 expression levels (Regina et al., 2021). FPRMS cells with lower expression levels of PAX3-FOXO1 display higher ECM adhesion, reminiscent of FNRMS, which could facilitate local invasion. Thus, we need a better understanding of how FPRMS may modulate the expression levels of PAX3-FOXO1 and the plasticity of cell–ECM interactions at the single-cell level to potentially acquire different competencies during the complex, multistep metastatic journey.
Our study also underscores the therapeutic implications of targeting cell–ECM interactions in RMS subtypes. The differential sensitivity of FNRMS and FPRMS to inhibition of either focal adhesion formation (FAK and Src) or TGFβ signaling suggests that disrupting cell–ECM interactions could be a viable strategy for FNRMS treatment. Given the clinical challenges in directly targeting the chimeric transcription factor PAX3-FOXO1 in FPRMS, a combined approach focusing on the downstream effector NOS1/NO, along with focal adhesion inhibition, may show greater therapeutic benefit. These proposed strategies may have clinical promise as targeted therapies, maximizing the curative effects and minimizing long-term sequelae resulting from current conventional chemotherapies.
Materials and methods
Cell culture
The FNRMS cell line, RD, was purchased from the ATCC (CCL 136). The FNRMS cell line SMS-CTR and the FP-RMS cell lines, Rh30 and Rh41, were all obtained from the Childhood Oncology Group. All cell lines were grown in high glucose DMEM supplemented with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin. FPRMS cells Rh30 and Rh41 were transduced with lentiviruses expressing shRNA PAX3-FOXO1 (5′-CCGGTCTCACCTCAGAATTCAATTCCTCGAGGAATTGAATTCTGAGGTGAGATTTTTG-3′ and 5′-AATTCAAAAATCTCACCTCAGAATTCAATTCCTCGAGGAATTGAATTCTGAGGTGAGA-3′) and TET-pLKO-puro-scrambled (5′-CCGGCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTTG-3′ and 5′-AATTCAAAAACCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′) (kind gifts from Dr. Mark E. Hatley, St. Jude Children’s Research Hospital, Memphis, TN, USA) used as a control. Both plasmids were cotransfected with packaging plasmids pMD2.G VSV-G envelope (#12259; Addgene) and psPAX2 packaging vector (#12260; Addgene) into 293T cells with Lipofectamine 3000 Transfection Kit (#L3000001; Invitrogen) according to the manufacturer’s protocol. FPRMS cells were transduced with viral particles after filtering (0.45 µM). Selection of efficiently transduced cells was achieved by treatment with puromycin (2 μg/ml final concentration). To induce shRNAs, we incubated cells with doxycycline (50 ng/ml) for 2 days.
Single-cell sequencing analysis
scRNA-seq data were obtained from GSE147457, Wk6 to Yr41, and stored in the AnnData format, which is optimized for handling large-scale annotated data matrices. We used the scanpy library in Python for subsequent analysis. Initial quality control steps involved filtering out low-quality cells and genes. Cells with <500 genes were excluded to remove potential debris and doublets, respectively. To account for variations in sequencing depth between individual cells, the data were normalized by scaling the total counts for each cell to a fixed value. Following normalization, a log transformation was applied to stabilize the variance across genes, making the data more amenable to downstream analysis. Then, we identified highly variable genes, which are the most informative for distinguishing between different cell types.
We performed t-distributed stochastic neighbor embedding to reduce the dimensionality of the data set, capturing the most significant sources of variation. Cells were clustered using the Leiden algorithm with a resolution of 0.4, which detects communities in the data based on their nearest neighbors in the reduced dimensional space. Additionally, we performed partition-based graph abstraction analysis to examine the connectivity between clusters. Student’s t tests and false discovery rates were performed to identify DEGs in each cluster, or, in other words, subgroup.
RNA-sequencing analysis
Total RNA was extracted from PFKD and control FPRMS cells using an RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. A NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) was used for DNA and mRNA quantification and qualification. RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies), and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies). The RNA-sequencing libraries were prepared using NEBNext Ultra II RNA Library Prep Kit for Illumina using the manufacturer’s instructions (New England Biolabs). Briefly, mRNAs were initially enriched with Oligo d(T) beads. Enriched mRNAs were fragmented for 15 min at 94°C. The first and second strand cDNAs were subsequently synthesized. cDNA fragments were end-repaired and adenylated at 3′ ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies) and quantified by Qubit 2.0 Fluorometer (Life Technologies) and by quantitative PCR (KAPA Biosystems). The sequencing libraries were clustered on one flowcell lane. After clustering, the flowcell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to the manufacturer’s instructions. The samples were sequenced using a 2x150 bp paired-end configuration.
Raw sequence data (.bcl files) generated from the sequencer were converted into fastq files and demultiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. After investigating the quality of the raw data, sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner uses a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step. Unique gene hit counts were calculated by the Counts feature from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis.
GSEA and GO analysis
We identified DEGs in each subgroup from scRNA-seq results (GSE147457) and in each RMS subtype (GSE114621). Additionally, we normalized the expression levels of each gene in the FPRMS cell lines for both control and PFKD conditions. We used the normalized values from Rh30 and Rh41 cell lines to calculate fold change and adjusted P values of each gene using Student’s t test and the false discovery rate. Thresholds of fold change >0.5 or less than −0.5 and adjusted P <0.05 were applied to identify DEGs. We used string version 12.0 (https://string-db.org/) for GO analysis and GSEA (https://www.gsea-msigdb.org/) to explore enriched signaling pathways in FNRMS-like clusters and PFKD cells. For GSEA, we ranked DEGs based on fold changes.
Kaplan–Meier survival analysis of clinical data
RNA assay of RMS patients was downloaded from https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-TABM-1202 ([Missiaglia et al., 2012]). Raw reads were imported using the affy R package. Raw reads were normalized using the rma function in the affy package, and gene names were mapped to probe ID using the mapIDs function and the hgu133plus2.db package. For multiple probes mapping to the same gene, the probe with the highest value was kept. Samples with PAX3 translocation (n = 34) were kept for downstream analysis. The pathway score for each sample was calculated by the mean normalized expression level of all genes in the pathway gene set, and cohorts for survival analysis were stratified by the survival cutpoint analysis using a minimum sample number of 8. KM plot and corresponding P value were generated using the survminer package in R.
NG-Smad3 live-cell sensor assays
NG-Smad3 live-cell sensor was thankfully gifted from Dr. Lea Goentoro (Caltech, Pasadena, CA, USA). We transfected the plasmid using a Lipofectamine 3000 Transfection kit (#L3000001; Invitrogen) according to the manufacturer’s protocol and selected the transfected cells using puromycin at a 2 µg/ml concentration. The transfected cells were replated on 6-well glass-bottomed plates and imaged on a Nikon Eclipse Ti2-E inverted microscope equipped with a live-cell chamber that maintained humidity, 37°C temperature, and 5% CO2, using a Nikon CFI Plan Apochromat λ 20× (NA 0.75, W.D. 1.0 mm) objective lens. Images were acquired with Nikon NIS-Elements software using the integrated Nikon DS-Qi2 camera every 16 min for 1 h, at least 10 min before TGFβ treatment and 50 min after the treatment. Using a MATLAB-based algorithm, we assessed the SMAD3 intensity in the nucleus over time and quantified TGFβ signaling activity.
Immunoblotting
RMS cells were lysed from their respective drug-treated and knockdown cell lines and collected in RIPA buffer containing protease/phosphatase inhibitors (Halt Protease/Phosphatase Inhibitor Cocktail 100x). After centrifugation at 20,000 g for 15 min to remove cellular debris, the supernatant was quantified using a BCA assay. Subsequently, the protein samples underwent SDS-PAGE using Mini-PROTEAN TGX Gels (4–20%; Bio-Rad) and were transferred onto polyvinylidene fluoride membranes. Membranes were then blocked in 3% BSA in TBST for 1 h before being incubated overnight at 4°C with primary antibodies, including FOXO1, SMAD2/3, MLC2, pMLC2 (Ser 19), lamin B1, collagen I, CTGF, and GAPDH, all diluted at 1:1,000. Next, membranes were washed three times in TBST before exposure to HRP-conjugated IgG secondary antibodies (diluted at 1:2,000–1:5,000) for 1 h at room temperature. Membranes were washed again three times with TBST for 5 min each to remove unbound secondary antibody. Protein bands were visualized using Clarity Western ECL Substrate (Bio-Rad) using the iBright FL1500 imaging system, and quantification was performed in ImageJ (National Institutes of Health). A full list of antibodies and their specifications can be found in Data S1.
IF and DAF-2 DA staining
RMS cells were seeded on collagen type I (10 µg/ml for 1 h, #A1048301; Thermo Fisher Scientific)–coated coverslips. Following pertinent treatments with drug inhibitors, cells were fixed with 4% paraformaldehyde in PBS for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 20 min, and blocked with 1% BSA in PBS for 45 min. Coverslips were incubated for 2 h at room temperature with primary antibodies diluted in blocking solution (1:100–1:250 dilution range) and then washed three times with PBS to remove unbound antibodies. For IF staining, coverslips were incubated with Alexa 488 or Alexa 594 secondary antibodies (Invitrogen) at a concentration of 5 µg/ml in blocking solution for 1 h at room temperature. For DAF-2 DA staining, coverslips were incubated with 10 µM (4.46 µg/ml) DAF-2 DA (Cayman Chemical) in blocking solution for 1 h at room temperature. The coverslips were then washed three times with PBS to remove unbound secondary antibodies and mounted on the cover glass after counterstaining with DAPI (Prolong Gold Antifade Reagent; Invitrogen). Epifluorescence and confocal imaging were performed in NIS-Elements software on a Nikon Eclipse Ti2-E inverted microscope with an integrated DS-Qi2 camera using the CFI Plan Apochromat λ 60× Oil (NA 1.40, W.D. 0.13 mm) objective lens in Nikon Type F immersion oil. We analyzed the images using a customized MATLAB script to read out the intensity score of each pixel in regions of interest, indicating the expression levels of corresponding proteins. Statistical analysis was performed using two-sided Student’s t tests. Focal adhesions, visualized as puncta in pFAK or vinculin staining, were manually counted.
Traction force microscopy
The TFM experiment was performed on 0.6- and 12.7-kPa high refractive index silicone gel by mixing two parts of Q-gel 920 A and B (CHT Silicones) in ratios 1:1 and 1:2, respectively (Han et al., 2015; Mittal et al., 2024). To fabricate the TFM substrate, 300 μl of the silicone Q-gel was spin-coated (1,000 rpm, 30 s) on a 35-mm glass-bottom dish (#1 cover glass; MatTek Corporation), cured at 80°C for 2 h, and stored in PBS. To visualize gel deformation, the TFM gel surface was functionalized with (3-aminopropyl)triethoxysilane (#440140; Sigma-Aldrich) and covalently bonded with 40-nm-diameter carboxylated far-red fluorescent beads (excitation/emission 690/720 nm; Invitrogen) at a density of 1 bead/µm2 using 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (#39391; Sigma-Aldrich). As an adhesion protein, collagen II (#35209; Southern Biotech) was coated in two different quantities, 0.5 µg/ml for sparse and 100 µg/ml for dense collagen density.
Cells were plated on the sparsely and densely collagen II–coated gel substrates. After seeding for 4 h, single-shot bright-field images of live cells and bead images at 642 nm were taken using a 60× objective total internal reflection fluorescence microscope (optoTIRF; CAIRN Research, Faversham ME13 8UP, UK) housed in a Nikon Ti-S microscope. The relaxed bead images as reference images for TFM were obtained after removing the cells using 0.5 ml of 10% bleach.
Using the bead and cell images, traction force reconstruction was performed as previously described (Han et al., 2015; Mittal et al., 2024). Detailed equations can be found in Han et al. (2015). To calculate the displacement field, an image cross-correlation–based particle tracking velocimetry method was used by comparing bead images before and after the cell is present on the substrate. The force reconstruction was done using the Fast Boundary Element Method with L2-norm–based regularization. The regularization parameter was chosen based on the L-curve, L-corner method.
Confocal reflection microscopy
RMS single-cell suspension was used for hanging drop cultures by detaching adherent cancer cell cultures at ∼70% confluence, followed by exposure to a 0.25% trypsin/EDTA solution. A dilution was then performed to allow for the seeding of 20,000 cells per 20 μl drop of cell culture media, which were deposited across the lid of a 60-mm petri dish. The lid was carefully inverted and placed over the PBS-filled bottom chamber of the petri dish. The hanging drops were maintained in a 37°C, 5% CO2 incubator for 48 h until compact aggregated spheroids were formed. For the embedding of spheroids into a collagen-based ECM matrix, a 3 mg/ml stock of rat tail collagen I solution (Gibco) was used to prepare a total volume of 1 ml of pH-buffered collagen gel solution at a final concentration of 2 mg/ml. In a sterile tube, 100 μl of 1 M HEPES, 100 μl of 10x DMEM, and 17 μl of 1N NaOH were mixed with 800 μl of collagen I solution. The collagen I solution was kept on ice to prevent polymerization. The spheroids formed in hanging drops were then collected in growth media, centrifuged at low speed (100 g for 2 min), resuspended in growth media, and embedded in the collagen I solution at a 1:3 ratio (cells: gel mixture). LY2109761 (10 µM) and ARL17477 (10 µM) were also added to the mixture when investigating the effect of these drugs. The resulting cell-encapsulating 3D gel mixture (200 μl) was deposited in wells of a glass-bottom 24-well plate. The plate was incubated for 1 h to allow the 3D gel: cell mixture to polymerize, and media were then added on top.
Live-cell imaging with CRM for 16 h was conducted to visualize collagen reorganization and remodeling of the collagen fiber network by RMS spheroids as a proxy for cell–ECM interactions. CRM imaging relies on the reflection (back-scattering) of light from structures within a biological sample that have differences in refractive indices. Collagen, being a fibrous protein with a higher refractive index than surrounding medium or cells, can produce a strong reflectance signal in confocal microscopy. This reflectance signal can be used to visualize dynamic collagen reorganization, morphology, and remodeling as the RMS tumor spheroids interact with the 3D matrix. In short, CRM was used to capture images every 3 h. Samples of RMS tumor spheroids within the 3D matrix were imaged via NIS-Elements software using an inverted Nikon Eclipse Ti2-E microscope with a Plan Apochromat λ 20× (NA 0.75, W.D. 1.0 mm) objective lens and integrated DS-Qi2 camera. A continuous wave laser at 488 nm was employed for imaging the spheroids. Despite collecting the 475–495 nm bandwidth centered on the 488 reflection using 488 laser sources for CRM, the guiding mirror (45° reflection mirror) was eliminated to enable light reflection on the main beam splitter (80/20) at a 90° angle, maximizing excitation light of the same wavelength that illuminated the samples. The pinhole size for image acquisition was set at 1 AU (5 µm). To improve signal-to-noise ratio and image quality, a dwelling time of 22.3 μs was used.
Collagen alignment analysis
We utilized PIV to analyze collagen displacement for 3 h within the CRM images. Using the matpiv function in MATLAB, we extracted two time-series images. The interrogation window size was set to 25 µm, and the analysis was performed with a 50% overlap ratio to ensure dense vector fields. A single-pass correlation method was employed to compute the displacement vectors. The outputs included the grid coordinates x and y, and the corresponding velocity components u and v, corresponding to the magnitude of the displacement in x and y directions.
Furthermore, we developed a MATLAB-based algorithm to analyze the directions of collagen fibers in CRM images. First, we applied a Gaussian filter to smooth the image and reduce noise. For collagen segmentation, we utilized the Sobel filter in both horizontal and vertical directions. Horizontal edge detection was achieved by directly applying the Sobel filter, while vertical edge detection was obtained by the transposed Sobel filter. To isolate the fibers in the images, a threshold was determined based on the mean and standard deviation of the image intensities, and a binary mask was created to highlight regions exceeding this threshold. The masked image was then used to calculate the angles of collagen alignment with respect to the x axis, corresponding to the edges of nearby spheroids.
Cell survival assay (CCK8 assay)
Cell viability was quantitatively assessed with a colorimetric assay using Cell Counting Kit-8 (CCK8, #96992; Sigma-Aldrich). In brief, 100 μl of cell suspension (5,000 cells/well) was dispensed in a 96-well plate. The plate was preincubated for 24 h in a humidified incubator (37°C, 5% CO2) followed by the addition of drug inhibitor treatments into the media at the appropriate concentrations and further incubation for 48 h. Subsequently, cell viability was assessed by adding 10 μl of CCK8 solution into each well, incubating the plate for 4 h, and measuring the absorbance at 450 nm using a microplate reader. Control wells with media only were used for normalization by subtracting the background signal of media-only wells from each absorbance measurement in cell-containing wells. Triplicates were used for each control and drug-treated condition, e.g., dasatinib (Src inhibitor), defactinib (FAK inhibitor), and LY2109761 (TGFβ inhibitor). Relative survival is reported as the average normalized absorbance measurement.
3D cell survival assays (CellTiter-Glo assay)
Single-cell suspensions of FNRMS, FPRMS, PFKD, and scramble cells were prepared for spheroid formation by detaching adherent cell cultures at ∼70% confluence using 0.25% trypsin–EDTA, followed by dilution to a final density of 5,000 cells per well in culture media. The cells were then plated into round-bottom multiwell plates (#4515; Corning) and incubated at 37°C with 5% CO2 for 48 h to allow the formation of compact spheroid aggregates. Drug treatments were initiated 48 h after spheroid formation, using dasatinib (Src inhibitor), defactinib (FAK inhibitor), and LY2109761 (TGFβ inhibitor). All conditions, including controls and drug-treated groups, were performed in triplicate. For experiments employing hanging drop cultures, suspended spheroids were directly collected from the cultures, as described previously, and transferred to the plate for further assays.
To assess 3D cell viability, we used the CellTiter-Glo 3D Cell Viability Assay (#G9681; Promega) with a luminometer. After spheroid formation and drug treatment for 48 h, 100 μl of CellTiter-Glo 3D reagent was added to 100 μl of media containing spheroid cells in each well. The plates were vigorously mixed for 5 min to induce cell lysis, releasing ATP from the spheroids. The plate was then incubated at room temperature for 30 min, and luminescence was recorded using a microplate reader with an integration time of 1 s per well. Control wells containing only media were used for background normalization, and the absorbance of media-only wells was subtracted from cell-containing wells. Triplicates were used for each control and drug-treated condition, including dasatinib, defactinib, and LY2109761. Relative survival was reported as the average normalized absorbance measurement.
Transwell assay
Cell invasion was assessed using transwell chambers (Millicell 24-well Cell Culture Insert Plate PCF, 8 μm pore size). FNRMS and FPRMS cells were trypsinized, resuspended in serum-free DMEM at 20,000 cells per well, and added to the upper chamber. Cells were added in media with and without ARL17477 (10 µM) treatment to assess its effect on invasion. The inserts were placed in the lower chamber containing 300 μl of DMEM supplemented with 20% FBS. After 24 h of incubation at 37°C and 5% CO2, cell media and non-migrated cells were removed from the upper chamber using cotton swabs. At the same time, migrated cells in the lower chamber were fixed with 4% paraformaldehyde in PBS and stained with Hoechst dye for nuclear visualization. Stained cells were imaged using epifluorescence microscopy (Nikon Eclipse Ti2-E inverted microscope, DS-Qi2 integrated camera, CFI Plan Apochromat λ 20× [NA 0.75, W.D. 1.0 mm] objective lens, NIS-Elements acquisition software), and the number of cells per field of view at 20× magnification was quantified. Statistical analyses were conducted using unpaired two-sided Student’s t tests with triplicates included for both control and ARL17477-treated conditions.
Statistical analysis
We performed paired Student’s t test if the data sets were normally distributed. If the normality assumption was not met, we performed Wilcoxon’s rank sum tests. Normality was assessed using the Jarque–Bera test.
Online supplemental material
Fig. S1 and Table S1 present supportive scRNA-seq analysis of skeletal muscle tissue, highlighting distinct RMS subtypes during skeletal development. Table S2 lists DEGs in each subgroup, and Table S3 shows GO analysis of DEGs for Subgroups 3 and 4. Table S4 provides a list of FNRMS and FPRMS core signatures identified by Gryder et al. (2017). Fig. S2 demonstrates enhanced cell–ECM interactions in FNRMS cells compared with FPRMS cells. Table S5 lists DEGs between FPRMS and FNRMS, and Table S6 presents GO analysis of these DEGs. Fig. S3 supports a role of the PAX3–FOXO1 fusion gene in suppressing TGFβ signaling by modulating NOS1 expression and NO synthesis, thereby reducing cell–ECM interactions. Table S7 lists DEGs between PFKD and FPRMS cells, Table S8 presents GO analysis of these DEGs, and Table S9 shows DEG analysis results identifying ChIP targets. Fig. S4 illustrates distinct responses of RMS subtypes to cell–ECM perturbations, and Fig. S5 suggests that PAX3–FOXO1 mediates these differential responses and promotes anchorage-independent spheroid growth. Time-lapse CRM imaging includes spheroids in collagen type I gels from FNRMS and FPRMS cell lines (Video 1), FNRMS spheroids in collagen type I gels treated with the TGFβ inhibitor LY2109761 (Video 2), PFKD FPRMS spheroids in collagen type I gels treated with LY2109761 (Video 3), and FPRMS spheroids in collagen type I gels treated with the NOS1 inhibitor ARL17477 (Video 4). Data S1 shows list of antibodies and drugs used in experiments. Data S2 shows raw data of immunoblotting experiments. Data S3 shows raw data of graphs.
Data availability
Raw data are available in the article itself and its supplementary materials. Also, publicly available data used in the analysis are noted in each figure legend. Additionally, the data are available from the corresponding author ([email protected]) upon reasonable request.
Acknowledgments
This work was supported by the following funds: CHLA start-up support, The Saban Research Institute Intramural Fund, Kure It Cancer Research, Concern Foundation (to A. Chronopoulos, I. Chavez, C.K. Vemula, V. Zamloot, Y. Pan, and J. Park), and NIH 2R15GM135806-02 (to N. Mittal and S.J. Han).
Author contributions: A. Chronopoulos: conceptualization, data curation, formal analysis, investigation, and writing—original draft, review, and editing. I. Chavez: investigation and resources. C.K. Vemula: data curation, investigation, methodology, validation, and visualization. N. Mittal: data curation, formal analysis, and visualization. V. Zamloot: data curation and writing—original draft, review, and editing. Y. Pan: data curation and writing—review and editing. S.J. Han: data curation, investigation, methodology, resources, software, visualization, and writing—original draft. J. Park: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, and writing—original draft, review, and editing.

